

Background: The mechanism(s) of G3-based TGS in E. histolytica is largely unknown.
Results: 5′-polyphosphate antisense sRNAs are identified; mechanistic insights linking these sRNAs with TGS are provided by IFA, FISH, IP, and ChIP assays.
Conclusion: TGS in E. histolytica G3 strain is mediated by an siRNA pathway, which utilizes antisense 5′-polyphosphate sRNAs.
Significance: This is the first demonstration of (a) 5′-polyphosphate antisense sRNAs mediating TGS and (b) RNAi-mediated TGS in protozoan parasites.
Keywords: Gene Expression, Gene Silencing, Histones, Parasite, siRNA, Entamoeba histolytica, Transcriptional Gene Silencing, 5′-Polyphosphate Antisense Small RNA
Abstract
In the deep-branching eukaryotic parasite Entamoeba histolytica, transcriptional gene silencing (TGS) of the Amoebapore A gene (ap-a) in the G3 strain has been reported with subsequent development of this parasite strain for gene silencing. However, the mechanisms underlying this gene silencing approach are poorly understood. Here we report that antisense small RNAs (sRNAs) specific to the silenced ap-a gene can be identified in G3 parasites. Furthermore, when additional genes are silenced in the G3 strain, antisense sRNAs to the newly silenced genes can also be detected. Characterization of these sRNAs demonstrates that they are ∼27 nucleotides in size, have 5′-polyphosphate termini, and persist even after removal of the silencing plasmid. Immunofluorescence analysis (IFA) and fluorescence in situ hybridization (FISH) show that both the Argonaute protein EhAGO2-2 and antisense sRNAs to the silenced genes are localized to the parasite nucleus. Furthermore, α-EhAGO2-2 immunoprecipitation confirmed the direct association of the antisense sRNAs with EhAGO2-2. Finally, chromatin immunoprecipitation (ChIP) assays demonstrate that the loci of the silenced genes are enriched for histone H3 and EhAGO2-2, indicating that both chromatin modification and the RNA-induced transcriptional silencing complex are involved in permanent gene silencing in G3 parasites. In conclusion, our data demonstrate that G3-based gene silencing in E. histolytica is mediated by an siRNA pathway, which utilizes antisense 5′-polyphosphate sRNAs. To our knowledge, this is the first study to show that 5′- polyphosphate antisense sRNAs can mediate TGS, and it is the first example of RNAi-mediated TGS in protozoan parasites.
Introduction
RNA interference (RNAi) is a well conserved mechanism of gene regulation in most eukaryotes (1). Small RNA molecules of 20–30-nt2 size can be classified into three categories: small interfering RNAs (siRNAs), microRNAs (miRNAs) and PIWI-associated small RNAs (piRNAs), all of which are linked to both transcriptional gene silencing (TGS) and post-transcriptional gene silencing (2, 3). The small RNAs (sRNAs) are found in association with Argonaute proteins and along with other factors form an effector complex, either RISC (RNA-induced silencing complex, involved in post-transcriptional gene silencing) in the cytoplasm or RITS (RNA-induced transcriptional silencing complex, involved in TGS) in the nucleus. The guide strand of the sRNA leads the RISC/RITS to target homologous sequences by base-pairing interactions. The silencing effect can be at the level of messenger RNA translation blockage (miRNA) or destabilization (siRNA) or at the DNA and chromatin level (miRNA, siRNA, and piRNA) (4, 5).
siRNAs, miRNA, and piRNA have been indicated in modulation of chromatin structure and TGS in plants, fungi, and animal cells (5, 6). An siRNA pathway for epigenetic inheritance of heterochromatin in Schizosaccharomyces pombe and chromatin modification in Arabidopsis thaliana are the best known examples of RNAi-involved TGS (7, 8). In addition, RNAi has been demonstrated to trigger long term, heritable gene silencing in Drosophila melanogaster and in Caenorhabditis elegans (9, 10). Recently, it has also become known that promoter-targeted siRNA can cause long term TGS in human cells (11, 12). To date, siRNA species involved in the TGS pathways are either Dicer-dependent (as in S. pombe, A. thaliana, and humans) or Dicer-independent (as in D. melanogaster). Recently, an abundant population of 22-nt 5′-triphosphorylated sRNAs was identified in C. elegans (13). Although these small RNAs in nematode cells are widespread, their biogenesis and functions in gene silencing are still in the early stages of study. Two recent papers have suggested different pathways involving different RNA-dependent RNA polymerase (RdRP) for somatic and germ line cells; modulation of gene expression through targeting of cognate mRNAs has been indicated (14, 15).
The RNAi pathway and endogenous sRNAs have been demonstrated to exist in some protozoan parasites, including Trypanosoma brucei, Toxoplasma gondii, Giardia lamblia, Trichomonas vaginalis, and Entamoeba histolytica (16–18). In these systems, when the mechanism of gene regulation by sRNAs has been identified, it is at the post-transcriptional (rather than transcriptional) level (19). In E. histolytica, a population of 27-nt sRNAs was identified by our previous sequencing efforts (20). These ∼27-nt sRNAs have features reminiscent of secondary siRNAs in C. elegans, including 5′-polyphosphate termini and antisense orientation to genes. We have also shown an inverse correlation between sRNA abundance and gene expression, suggesting that these sRNAs could mediate gene silencing (20).
E. histolytica is an intestinal protozoan parasite, which affects 50 million people worldwide (21). The study of this parasite has been hindered by the lack of genetic tools for functional gene knock-out studies. Recently, a TGS method using G3 parasites was established and has been shown to be reliable for silencing several important genes in Entamoeba (22, 23). This G3 parasite strain was originally generated by transfecting E. histolytica strain HM-1:IMSS with a plasmid (psAp-2), which contained only the 5′ region of the ap-a gene promoter along with a truncated segment of a repetitive retrotransposon element and an adjacent short T-rich stretch. The resulting parasite strain was found to have ap-a permanently silenced, even after curing the plasmid. A single cloned parasite from this transfectant line is named G3 (24). Importantly, it was later found that a second unrelated gene of interest could be transcriptionally silenced by transfecting G3 with a plasmid in which a second gene was cloned directly after the ap-a promoter fragment (23, 25, 26). The resulting secondary gene silencing is also inheritable and can be maintained after removal of the plasmid. The silencing mechanism in G3 parasites has been studied, and histone H3 Lys-4 methylation was noted to be reduced at the ap-a locus; the reduction of histone methylation was also noted for additional genes silenced in the G3 strain (24, 27). However, efforts to demonstrate DNA methylation and to identify siRNAs to the silenced loci have been negative to date (24), leading to the hypothesis that G3 silencing is largely controlled at the level of chromatin modification (27).
In this report, we systematically examined G3 parasites and the G3-based gene silencing method for sRNAs. In contrast to previous studies, we found abundant antisense sRNAs to the silenced genes in the G3 strain. Further characterization of these antisense sRNAs showed that they have 5′-polyphosphate termini, similar to what we have previously identified for endogenous antisense sRNAs in E. histolytica (20). We further identified the E. histolytica Argonaute protein (EhAGO2-2) was localized in the nucleus of amebic trophozoites. Using RNA-FISH, we detected antisense sRNAs to the silenced ap-a gene in the parasite nucleus. To obtain direct evidence that these antisense sRNAs are associated with EhAGO2-2, we demonstrated the presence of these sRNAs specifically in material immunoprecipitated with EhAGO2-2 from G3 lysate but not from controls. Finally, ChIP assay and polymerase chain reaction (PCR) analysis demonstrated that the genomic loci of the silenced genes targeted by sRNAs are enriched for histone 3 and EhAGO2-2. In conclusion, our results show that the mechanism of gene silencing in the G3 strain in E. histolytica is linked to the RNAi pathway and mediated by 5′-polyphosphorylated antisense sRNAs, which are localized to the nucleus. Given the features of inheritable long term gene silencing, nuclear localization, direct association of EhAGO2-2 with antisense sRNAs and the specific enrichment to the silenced loci by ChIP analysis, we propose a model in which these sRNA mediate TGS; thus, the 5′-polyphosphorylated antisense sRNAs guide Argonaute for target recognition and induce chromatin modifications required for permanent gene silencing. Our data therefore contribute two novel findings to the field. First, they demonstrate a role of 5′-polyphosphorylated antisense sRNAs in regulating TGS. Second, this is the first study to identify that RNAi-mediated TGS is functional in a protozoan parasite.
EXPERIMENTAL PROCEDURES
Parasite Cultures
E. histolytica trophozoites (HM-1:IMSS and Rahman strains) were grown under standard conditions as described previously (28, 29). The G3 parasites were a gift from Dr. David Mirelman, and G3-based secondary gene silenced parasites were generated by transfection of G3 parasites with psAP-2/5′STIRP plasmid or psAP-2/3′STIRP plasmid (see “Plasmid Construction for EhSTIRP Gene Silencing”). EhROM(KD) parasites were generated as described previously (26), with separate cultures having been maintained with or without drug at 2 μg/ml G418. The N-terminal Myc-tagged EhAGO2-2 parasites were previously generated (20) and have been maintained at 24 μg/ml G418.
Plasmid Construction for EhSTIRP Gene Silencing
To construct the silencing plasmid for EhSTIRP1, the first 966 bp from the 5′-end of the EhSTIRP1 gene were amplified by PCR and cloned into the plasmid vector psAP-2 downstream of the 5′ upstream segment (473 bp) of the ap-a gene; this plasmid is named psAP-2/5′STIRP. To construct the silencing plasmid for all EhSTIRP genes, a 822-bp fragment of the 3′-end EhSTIRP1 (from bp 6188 to 6966) was cloned into the plasmid vector psAP-2; this plasmid is named psAP-2/3′STIRP. The PCR primers are listed in supplemental Table S-II.
Transfection of G3 Parasites
G3 parasites were transfected using previously published protocols (26). Briefly, G3 trophozoites were seeded in 35-mm Petri dishes and transfected with 20 μg of plasmid DNA using SuperFect (Qiagen) reagent. The transfected parasites were allowed to grow for 24 h, and drug selection started at 1 μg/ml G418 and maintained until a stable cell line was achieved. Drug concentration was then increased in a stepwise manner to 6 μg/ml G418. EhSTIRP expression levels were tested using Northern blot analysis. After EhSTIRP down-regulation was confirmed, parasite cultures were maintained with or without drug (6 μg/ml G418), and EhSTIRP silencing was confirmed in drug-free parasites at 2, 5, and 10 months after drug removal.
Total RNA and Small RNA-enriched RNA Preparation
Total RNA was isolated from parasites by TRIzol (Invitrogen) according to the protocol provided by the manufacturer. Small RNA-enriched RNA was extracted using the mirVana kit (Ambion) according to the manufacturer's protocol.
Northern Blot Analysis
Standard Northern blot analysis was performed on a Hybond N+ (Amersham Biosciences) nylon membrane with RNAs transferred from a denaturing agarose gel at 0.7% (for EhSTIRP) or 1.5% (for ap-a), loaded with 15 μg of total RNA for each sample, probed with either PCR (for EhSTIRP, ap-a, and actin) or oligonucleotide probes (for ap-a, ap-b, and SAPLIP 1) (all PCR primers and oligonucleotide probes are listed in supplemental Tables S-I and S-III). PCR probes were labeled with [α-32P]dATP using Klenow (New England Biolabs) and hybridized overnight at 65 °C in perfectHyb buffer (Sigma). Oligonucleotide probes were labeled with [γ-32P]ATP using T4 PNK (New England Biolabs) and hybridized overnight at 37 °C. Washing conditions are for 20 min each using a wash solution (2× SSC, 0.05% SDS) followed by (0.1× SSC, 0.1% SDS) at 50 °C for PCR probes and 37 °C for oligonucleotide probes. Small RNA Northern blot analysis was done as previously described (20). Briefly, 20–50 μg of sRNA-enriched samples were separated on a denaturing 12% polyacrylamide gel, transferred to a membrane, probed with end-labeled 32P-labeled oligonucleotides in perfectHyb buffer (Sigma) at 37 °C, and washed using low (2× SSC, 0.1% SDS at 37 °C for 15 min) and medium (1× SSC, 0.1% SDS at 37 °C for 15 min) stringency conditions.
Terminator and Capping Assays
Assays were done as described previously (20). Briefly, 10 μg of sRNA-enriched RNA sample was spiked with a control sample (a synthetic 21-mer RNA with 5′-end labeled with 32P). For the Terminator assay, the sample mixture was treated with Terminator enzyme (Epicenter), following the provided protocol from the manufacturer. For the capping assay, the ScriptCap m7G capping system (Epicenter) was used, with a provided alternate cap zero capping protocol. After enzymatic treatment, samples were phenol/chloroform-extracted and resolved on a 12% polyacrylamide gel, and Northern blot analysis was performed using a radiolabeled probe to detect the sRNA of interest.
EhAGO2-2 Antibody Production, IFA, and FISH
EhAGO2-2 peptides (amino acids 12–25 and peptide sequence C-DQTIDKPVRERIDK-NH2) were used as the antigen to generate polyclonal antibodies in rabbits (EZBiolab). For IFA, the parasites were fixed with methanol/acetone (50:50), followed by blocking in 3% BSA-PBS for 30 min. Primary antibody was added at a 1:100 dilution in 1% BSA-PBS and incubated at room temperature for 1 h. After rinsing cells in PBS, fluorescent secondary antibody, anti-Rabbit Alexa 488 (Cell Signaling), was added at a 1:1000 dilution and incubated in the dark for 30 min at room temperature. After staining, cells were rinsed in PBS and mounted on chamber slides using Vectashield mounting medium (Vector Laboratories). For FISH, a protocol was developed based on previously published methods (30–32). The parasites were fixed with 4% ultrapure formaldehyde diluted in PBS for 10 min. Cells were permeabilized by the addition of 70% ethanol (EtOH) overnight. On the next day, cells were treated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide for further cross-linking, followed by overnight incubation in hybridization buffer (25% formamide, 0.05 m EDTA, 4× SSC, 10% dextran sulfate, 1× Denhardt's solution, yeast tRNA, and salmon sperm DNA) with an oligonucleotide probe that was labeled at the 5′-end with fluorescein (Integrated DNA Technologies) at 37 °C (supplemental Table S-IV). After three washes in 4× SSC, 2× SSC, and 1× SSC, the cells were blocked with the Image-iT signal enhancer for 30 min (Invitrogen), followed by a further incubation in blocking buffer (Roche Applied Science) for 1 h at room temperature. Then, 2 μg/ml anti- fluorescein-POD antibody (Roche Applied Science) in blocking buffer was added to the cells and incubated at room temperature for 1 h. After three washes in 1× wash buffer, signals were amplified by the TSA Plus fluorescein kit (PerkinElmer Life Sciences) for 10–15 min. The slides were then mounted in mounting solution. All imaging was performed on a Leica CTR6000 microscope, using a BD CARVII confocal unit. Image analysis and deconvolution were performed using the LAS-AF program (using default settings) from Leica.
Cell Fractionation
The cell fractionation protocol was adapted from Ref. 33. Parasites were chilled to 4 °C for 10 min, and cells were collected by centrifugation at 900 rpm at 4 °C for 5 min. The cell pellet was resuspended in Buffer A (10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.6% Nonidet P-40) (plus protease inhibitors E64d and leupeptin). After incubation on ice for 20 min, samples were pelleted at 4 °C for 10 min at 1200 rpm. Supernatant (cytosolic fraction) and pellet (nuclear fraction) were both collected, and each was used for total RNA preparation.
Immunoprecipitation Experiments
The protocol as described previously (20) was used with some modifications. Total lysate was split into equal pools; one was used for IP with α-EhAGO2-2 antibody, and the other pool was incubated with beads only (as a control). Briefly, α-EhAGO2-2 antibody was incubated with parasite lysate (overnight), and protein A beads were added and rotated for 4 h. After incubation, the beads were pelleted and washed twice (all done at 4 °C). RNA was isolated using the mirVANA kit (Ambion) from the washed pellet. Samples were resolved on a 12% denaturing polyacrylamide gel, and Northern blot was probed with the ap-a S1 probe.
Chromatin Isolation and ChIP Assay
A modified protocol was adapted from methods described previously (33–35). Parasites of HM-1:IMSS strain, G3 strain, G3:ROM(KD), and G3:5′STIRP were grown in 25-cm2 T flasks; cells were cross-linked in PBS + 0.5% formaldehyde for 10 min at room temperature; and the cross-linking reaction was stopped by the addition of glycine to 0.125 m for 5 min. Cells were iced, pelleted, and washed once in PBS. A ChIP assay utilized 2 × 106 cells, which were first lysed in nuclei isolation buffer (10 mm Hepes-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.6% Nonidet P-40) plus protease inhibitor mixture (Pierce) and 1 μm E-64 (Sigma). Nuclei were pelleted by spinning for 10 min at 1200 rpm, resuspended in sonication buffer (34), and sonicated for 12 × 16 s. Whole nuclei and cell membranes were removed by centrifugation at 20,000 relative centrifugal force, and supernatant was saved and cleared with Protein A beads (Upstate Biotechnology) for 1 h at 4 °C. Antibodies were then added and mixed overnight at 4 °C with gentle agitation. Polyclonal anti-EhAGO2-2 antibodies (EZBiolab) were used along with control antibodies, rabbit polyclonal to H3 (ab1791; Abcam), and rabbit anti-Myc (Cell Signaling). After antibody incubation, the beads were added and incubated for 3 h at 4 °C and were washed according to Ref. 34. Elution was done by incubation for 30 min at 65 °C in 0.1 m NaHCO3, 1% SDS, and cross-linking was reversed by incubation for 2 h at 65 °C, followed by proteinase K digestion. The final DNA was collected by a Qiagen PCR purification column, and from 50 μl of eluate, 4 μl was used for PCR amplification. Primer sequences and conditions are listed in supplemental Table S-V. A minimum of two independent ChIP assays were done for each parasite strain/transfectant; quantitation of the PCR bands was done by using net intensity as determined from analysis with Eastman Kodak Co. molecular imaging software (version 4.0.5f7). The enrichment was calculated as (density of test band in IP/density of reference band in IP)/(density of test band in whole cell extract/density of reference band in whole cell extract).
RESULTS
Antisense sRNAs Are Detected to Silenced ap-a Gene in G3 Parasites
In order to verify the identity of G3 parasites, we confirmed the silencing of ap-a and the closely related genes amoebapore B (ap-b) and saposin-like protein (SAPLIP 1) (supplemental Fig. S1A). All results matched previously published results for the G3 strain (22, 23). In order to probe for sRNAs, we designed strand-specific oligonucleotide probes to regions of the ap-a gene as well as selected regions upstream and downstream of ap-a (Fig. 1, top) (supplemental Table S-I). Northern blot analysis detected specific antisense sRNAs using ap-a sense probes S1, S5, and S7 in the G3 sample but not in the control strain, HM-1:IMSS (Fig. 1, bottom). Two populations of antisense sRNA were detected at ∼27 and ∼32-nt, with the 27-nt fraction having the stronger signal. The small RNA size was determined using a sRNA ladder and is in agreement with the sizes of endogenous sRNA populations previously cloned and identified in E. histolytica trophozoites (20). The abundance of these antisense sRNAs was greatest toward the 5′-end of the ap-a gene and decreased toward the 3′-end of the gene (signal intensity S1 > S5 > S7). As a loading control, we probed these blots using an endogenous sRNA (probe EHS-D-A16-1), which showed equal signal on three blots (supplemental Fig. S1C). There was no detectable sRNA signal from probes in the untranslated region (UTR) and downstream region (DS) (probes 5UTRS, 5UTRAS, 3UTRS, and DS1) (data not shown for probes 3UTRS and DS1) or for probes in the intergenic region (US1S and US1AS). A probe (SINE1-AS) located in the ap-a upstream SINE1 element region had a weak signal at the 27-nt size range, whereas its complementary probe (SINE1-S) identified no detectable signal (data not shown). Of note, the same SINE1-AS probe had previously been shown by another group to detect a 140-nt transcript in the G3 strain (24). We also observed some differences between HM-1:IMSS and G3 parasites in the 80–150-nt range, but this was probably due to variability in sRNA sample quality from two sample preparation methods (between the mirVana kit used in this study and the PEG 8000 procedure that was used for the previous study). In contrast to the strong antisense sRNA signal, all probes designed to detect sense sRNA (AS1 and AS3) gave no signal (data not shown). In previous studies, no sRNAs were detected to the silenced loci in the G3 strain (24), but given our data with an abundance of antisense sRNAs, the previous lack of detection was probably a technical issue (sample preparation, probe design, amount of sRNA used in the Northern blot, or probe labeling).
FIGURE 1.

Abundant antisense sRNAs to the silenced ap-a gene in G3 parasites are detected by sense probes. Top, the ap-a genomic locus and adjacent regions and the location of strand-specific oligonucleotide probes are shown. The genomic locus is drawn to scale, including SINE1, a T-rich region, an intergenic region (IGR), ap-a, and its 5′- and 3′-UTRs. The essential region for ap-a silencing in the psAP-2 plasmid (the 140-bp SINE1 and ap-a upstream region before the ATG) is depicted below. The oligonucleotide probes and their strand specificity are represented by arrows (the probes with Northern blots pictured are in black; the probes without Northern blots that are pictured but are mentioned under “Results” as data not shown are in gray). Sense probes for detecting ap-a antisense sRNAs are US1S (upstream sense), 5UTRS (5UTR sense), S1 (sense probe 1), S3, S5, S7, 3UTRS (3UTR sense), and DS1 (downstream sense). Antisense probes for detecting ap-a sense sRNAs are US1AS (upstream antisense), 5UTRAS, AS1, and AS3. Probes for detecting SINE1 sRNAs are SINE1-AS (SINE1 antisense primer) and SINE1-S (SINE1 sense primer). Bottom, Northern blot analysis detects antisense sRNAs for ap-a in G3 parasites. Identical Northern blots (each blot was loaded with 20 μg of sRNA-enriched sample from E. histolytica trophozoites of HM-1:IMSS or G3 strain) were probed with oligonucleotide probes to detect sRNAs of interest (the name of the probe is shown below each blot). A representative loading control is also shown (methylene blue staining).
It has previously been reported that in addition to ap-a silencing, the related genes ap-b and SAPLIP 1 are also down-regulated in G3 parasites, probably due to their high sequence homology to ap-a (23). In order to determine whether these genes were also silenced by an sRNA mechanism, we probed for antisense sRNA for these two genes. Due to high sequence homology in the ORF region to ap-a, we designed one gene-specific sense probe for each of these genes. Northern blot analysis revealed antisense sRNAs to both ap-b and SAPLIP 1 (supplemental Fig. S1, B, D, and E). These data indicate that ap-a, ap-b, and SAPLIP 1 genes have antisense sRNAs and are probably silenced via an RNAi mechanism in the G3 parasite strain.
Antisense sRNAs Detected for the Silenced EhROM1 Gene in G3:ROM1 Knockdown Parasites
Previously, we reported the down-regulation of EhROM1 using the G3-based gene silencing approach (26). The plasmid used vector psAP-2 with the first 447 bp from the 5′-end of the EhROM1 coding regions as a trigger. The G3 parasites transfected with this plasmid showed almost complete down-regulation of EhROM1 expression by RT-PCR and Northern blot analysis; the resulting parasites were named G3:ROM(KD) (26). To test if there were antisense sRNAs associated with EhROM1 in G3:ROM(KD) parasites, oligonucleotide probes were designed to the EhROM1 trigger region (Fig. 2, top) (supplemental Table S-I). Northern blot analysis using probe RS-86 showed specific antisense sRNA signal at 27–32-nt for G3:ROM(KD) parasites but not for G3 and other controls in which the EhROM1 gene is expressed (Fig. 2, bottom). The abundance of the EhROM1 gene-specific antisense sRNAs was lower (∼5-fold) when compared with ap-a-specific antisense sRNAs in G3. Other probes, RS1 and RS168, also detected specific antisense sRNA signal for G3:ROM(KD) parasites, but their signal was much less than that of probe RS-86 (data not shown). Of note, one probe (RS471) outside the transgene was also utilized but failed to detect any signal (data not shown), presumably due to the low abundance of small RNAs toward the 3′-region of the gene.
FIGURE 2.

Antisense sRNAs are detected for the silenced EhROM1 gene in G3:ROM(KD) parasites. Top, strand-specific oligonucleotide probes to the EhROM1 gene (arrows in black are probes with Northern blots pictured; arrows in gray are the probes without Northern blots pictured but mentioned under “Results” as data not shown). Four sense probes for detecting antisense sRNAs are shown as RS-86 (ROM1 sense probe starting at position −86), RS1 (ROM1 sense probe starting at position 1), RS168 (ROM1 sense probe starting at position 168), and RS471 (ROM1 sense probe starting at position 471). The trigger region used for psAP-2 cloning is shown below (between positions −86 to 447). Bottom, Northern blot analysis detects antisense sRNAs for EhROM1 in G3:ROM(KD) parasites. A Northern blot (loaded with 50 μg of sRNA-enriched sample from E. histolytica trophozoites of strain HM-1:IMSS, Rahman, G3, and G3:ROM(KD)) was probed with oligonucleotide probe RS-86 to detect antisense sRNAs; the blot was stripped and probed with ap-a probe S1. Methylene blue staining is shown as a loading control.
Antisense sRNAs Detected for EhSTIRP in G3:STIRP Knockdown Parasites
To strengthen our finding that specific antisense sRNAs are associated with the G3-based gene silencing cell lines, we applied the G3-based gene silencing approach on another gene, the E. histolytica serine-, threonine-, isoleucine-rich protein (EhSTIRP). This gene was previously identified as a putative virulence determinant due to its high expression in E. histolytica virulent strains and lack of expression in nonvirulent strains (36). Two plasmid constructs were generated based on psAP-2 plasmid using either a trigger region selected at the 5′-end of EhSTIRP1 at positions 1–966 (which should be specific to EhSTIRP1) or at the 3′-end of EhSTIRP1, at positions 6188–6966 (which should target the entire EhSTIRP gene family due to a 99% nucleotide similarity in this region among the three EhSTIRP genes) (36) (Fig. 3A, top) (supplemental Table S-I). After transfection of the plasmid into G3, both plasmids caused down-regulation of the EhSTIRP gene family, as shown by Northern blot analysis (Fig. 3B). We noticed that G3:3′STIRP had almost complete silencing for all three EhSTIRP genes, whereas G3:5′STIRP had some remaining signal, which may come from incomplete silencing of the EhSTIRP gene family. To detect antisense sRNAs, strand-specific oligonucleotide probes were designed at both 5′- and 3′-trigger regions as well as regions outside the triggers (Fig. 3A, top). Northern blot analysis detected antisense sRNAs to the silenced EhSTIRP gene in both transfectant lines (Fig. 3A, bottom). Interestingly, we observed antisense sRNAs from both the region that was included in the construct and from the rest of the gene. The probes (SS6188 and SS6342) that are included in the psAP-2/3′STIRP construct showed the antisense sRNAs for G3:3′STIRP but not for G3 and G3:5′STIRP. In contrast, probes (SS1 and SS624) that are included in the psAP-2/5′STIRP construct resulted in antisense sRNAs for both G3:5′STIRP and G3:3′STIRP but with a stronger signal intensity for G3:5′STIRP. The probe SS1028, which is in neither silencing construct resulted in antisense sRNAs in both G3:5′STIRP and G3:3′STIRP transfectants at an equal level. These results indicate that there are probably two pools of antisense sRNAs in the silenced EhSTIRP transfectants. The first pool is probably processed directly from the trigger transcript. After the trigger region initiates the silencing phenomenon, antisense sRNAs recognize the endogenous target gene; subsequently, an additional pool of antisense sRNAs to the entire gene appears to be generated (see “Discussion”).
FIGURE 3.

Antisense sRNAs are detected for EhSTIRP in G3:STIRP knockdown parasites. A, top, location of the strand-specific oligonucleotide probes on the EhSTIRP1 gene (arrows in black are probes with Northern blots pictured in the figure(s); arrows in gray are the probes without Northern blots pictured but mentioned under “Results” as data not shown). Sense probes for detecting antisense sRNAs are designed for three regions: for the 5′-trigger region (SS1 and SS624), for the 3′-trigger region (SS6188, SS6342, SS6501, and SS6801), and for the region outside the triggers (SS1028 and SS2520). The trigger region cloned into psAP-2 is shown below the EhSTIRP1 schematic; for 5′STIRP, it is between positions 1 and 984; for 3′STIRP, it is between positions 6188 to 6966. Bottom, Northern blot detects antisense sRNAs for EhSTIRP in G3:STIRP knockdown parasites. A Northern blot (loaded with 50 μg of sRNA-enriched sample from E. histolytica trophozoites of G3, G3:5′STIRP, and G3:3′STIRP) were probed with oligonucleotide probes shown in the top to detect antisense sRNAs. The blot was sequentially probed, stripped, and reprobed; probes used are shown below each blot. Methylene blue staining is shown as a loading control. B, Northern blot analysis shows gene silencing of EhSTIRP transcript. A Northern blot loaded with 15 μg of total RNA for each sample was probed with PCR probes for EhSTIRP, ap-a, and actin (as a loading control). The down-regulation of EhSTIRP is shown for G3:3′STIRP and G3:5′STIRP. Due to the high degree of similarity between genes in the EhSTIRP gene family, it is unclear whether low signal in the G3:5′STIRP lane represents transcript from EhSTIRP1 or if there is cross-hybridization with other EhSTIRP gene family members.
Antisense sRNAs to Silenced Gene Persist Despite Removal of Trigger Plasmid
The G3 parasites have maintained ap-a silencing for many years without drug selection and despite removal of the trigger plasmid (24). Thus, the abundance of antisense sRNAs to the silenced ap-a gene in G3 parasites indicates that these sRNAs are inheritable from one generation to the next. We wanted to test if this inheritable feature is also true for a second gene silenced by the G3-based method. The G3:ROM(KD) parasites have been kept with and without drug selection for 2 years. RT-PCR analysis showed that the plasmid was cured after 6 months without drug selection (data not shown). Northern blot analysis showed that the antisense sRNAs specific to the silenced EhROM1 gene are present even after loss of the trigger plasmid (Fig. 4A). However, it does appear that the parasites maintained in drug selection had more antisense sRNAs to EhROM1 than those maintained without drug. This can be potentially explained by the on-going expression of the trigger transcript keeping high level production of the first pool of antisense sRNAs. For both G3:5′STIRP and G3:3′STIRP parasites, after initial drug selection and confirmation of EhSTIRP silencing, cultures were maintained with or without drug selection; RT-PCR analysis confirmed loss of the plasmid after 6 months without drug selection (data not shown). Northern blot analysis of G3:3′STIRP parasites with or without drug selection showed that without plasmid present, we could still detect EhSTIRP antisense sRNAs either in the 3′-end trigger region or in the non-trigger region (Fig. 4B). However, once again parasites with continuous drug selection had more antisense sRNAs than those under no drug selection. We therefore conclude that once gene silencing is established, antisense sRNAs persist as a permanent inheritable genetic marker for gene-specific silencing, albeit at lower levels than when the silencing plasmid trigger is maintained.
FIGURE 4.

Antisense sRNAs to the silenced gene in G3-based transfectants are persistent in the absence of plasmid. A, Northern blot analysis of sRNAs in EhROM1 silenced parasites with and without selection plasmid. The G3:ROM(KD) parasites have been kept as separate cultures with and without drug selection for 2 years. RT-PCR analysis showed that the plasmid was cured after 6 months without drug selection (data not shown); the RNA sample was prepared after confirming the loss of plasmid. ap-a probe S1 Northern and methylene blue staining are shown as loading controls. B, Northern blot analysis of sRNAs in EhSTIRP silenced parasites with and without plasmid. After confirmation of EhSTIRP gene silencing, cultures were maintained with or without drug selection; RT-PCR analysis confirmed the loss of plasmid after 6 months without drug selection (data not shown). Probes are shown below each blot. ap-a probe S1 Northern and methylene blue staining are used as loading controls.
Antisense sRNAs to Silenced Genes Have 5′-Polyphosphate Termini
We previously demonstrated the existence of an abundant endogenous sRNA population in E. histolytica trophozoites, which are 27-nt in size and have 5′-polyphosphate termini (20), a feature similar to the secondary siRNAs from C. elegans that are involved in an amplified silencing mechanism (13). We wanted to address if antisense sRNAs to the silenced ap-a gene in G3 parasites contain 5′-polyphosphate termini. For this purpose, both a Terminator exonuclease assay and a 5′-end capping assay were applied to the G3 RNA sample with a “spiked” control (a synthetic 21-mer RNA species, which is labeled to have a 5′-monophosphate structure). The data show that the control RNA species were degraded by Terminator treatment and unaffected by a 5′-end capping assay, as would be expected due to its 5′-monophosphate structure (Fig. 5). In contrast, the antisense sRNAs to ap-a (both 27- and 32-nt populations) were resistant to Terminator cleavage and migrated one nucleotide higher on the gel after treatment with capping enzyme; both analyses indicate that these sRNA populations have 5′-polyphosphate termini (Fig. 5). We further extended the analysis to the RNA samples from G3:ROM(KD) and G3:3′STIRP parasites, and in each case the antisense sRNAs to the silenced gene were resistant to Terminator exonuclease and able to be capped with capping enzyme (supplemental Fig. S2). Therefore, the antisense sRNAs that are generated to the silenced gene in the G3-based gene silencing method have a 5′-polyphosphate structure. This structure is reminiscent of the structural features of the endogenous 27-nt population in E. histolytica, which are associated with EhAGO2-2 (20).
FIGURE 5.
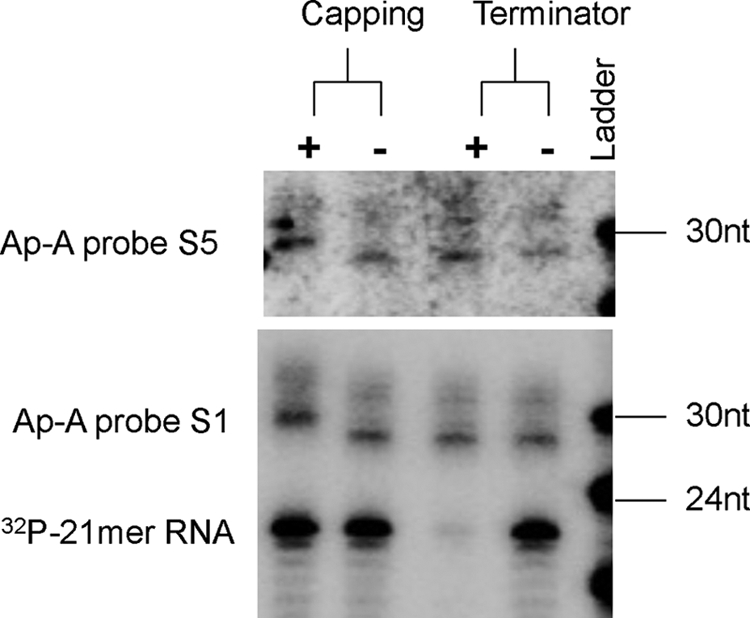
Antisense sRNAs to the silenced ap-a gene in G3 parasites have 5′-polyphosphate termini. Biochemical analysis of the 5′ termini of antisense sRNAs to the silenced ap-a gene. Probes used for detection are ap-a S1 and S5. A 21-nt 5′-monophosphate and 3′-OH radiolabeled probe is included as a control. Both 27- and 32-nt sRNAs are resistant to treatment with Terminator enzyme, indicating that they do not have a 5′-monophosphate structure. Both sRNA populations migrated one nucleotide higher on the gel after treatment with capping enzyme, indicating they are capped with a single G and hence probably have 5′-polyphosphate termini. The control probe was susceptible to Terminator enzyme and did not cap with capping enzyme, confirming its 5′-monophosphate structure.
Key RNAi Component, Argonaute Protein EhAGO2-2, Is Localized to Parasite Nucleus
All sRNAs have to be loaded into Argonaute family proteins for their proper function (37). Analysis of the E. histolytica genome predicts three Argonaute family protein genes, EHI_186850, EHI_125650, and EHI_177170, which we have named Ehago2-1, Ehago2-2, and Ehago2-3 (18). Only Ehago2-2 is highly expressed in both the trophozoite and cyst stages by microarray analysis, whereas the other two genes are only detectable by RT-PCR (38). Previous work from our laboratory (20) has shown that an N-terminal Myc-tagged EhAGO2-2 (also referred to in that paper as EhPiwi-rp) associates with 27-nt 5′-polyphosphate sRNAs in E. histolytica trophozoites, indicating that this protein functions in the amebic RNAi pathway. To further characterize the EhAGO2-2, we generated a peptide antibody to its N-terminal sequence, and Western blot analysis revealed a band at the expected size at 110 kDa (supplemental Fig. S3). To determine the precise cellular localization of EhAGO2-2, IFAs were performed on HM-1:IMSS parasites. Anti-EhAGO2-2 antibody showed specific signal in the parasite nucleus (Fig. 6). We then performed the same IFA experiments on an N-terminal Myc-tagged EhAGO2-2-transfected amebic cell line and identified a stronger nuclear signal with the anti-EhAGO2-2 antibody (Fig. 6). IFAs using an anti-Myc antibody on stable transfectants expressing an N-terminal Myc-tagged EhAGO2-2 confirmed signal in the nucleus (supplemental Fig. S4). In summary, IFA experiments using both anti-EhAGO2-2 antibody and anti-Myc antibody revealed that EhAGO2-2 is primarily localized in the parasite nucleus. Argonaute localization may be indicative of its function and mechanism of action. For instance, in A. thaliana, only AGO4 is involved in TGS and is a nucleus- localized protein (39). Another Argonaute example of nuclear localization for TGS is Drosophila PIWI, which binds piRNAs (40). However, some Argonaute proteins have both nuclear and cytoplasmic localization, such as the fission yeast AGO1, which is responsible for both TGS and post-transcriptional gene silencing (41). As shown by a recent study in C. elegans, one of the Argonautes, NRDE-3, can shuttle between the cytoplasm and nucleus (42). Our finding of nuclear EhAGO2-2 is supportive of its functioning in TGS, although further studies would be needed to rule out any cytoplasmic activity.
FIGURE 6.

Argonaute protein EhAGO2-2 is primarily localized in the parasite nucleus. IFA using anti-EhAGO2-2 antibody on HM-1:IMSS parasites showed specific signal in the parasite nucleus; no signal is seen from the control prebleed serum. An IFA on parasites stably transfected with N-terminal Myc-tagged EhAGO2-2 (pKT3M-MycAGO2-2) revealed a strong nuclear signal but not the control prebleed serum.
Antisense sRNAs to Silenced ap-a Gene Are in Parasite Nucleus, Where They Directly Associate with EhAGO2-2
FISH was used to identify the localization of antisense sRNAs to the silenced ap-a gene in G3 parasites. The ap-a sense probe S1 was 5′-end-labeled with fluorescein. Hybridization of the probe to HM-1:IMSS, G3, and G3:ROM(KD) parasites showed an enriched signal in the nucleus in G3 and G3:ROM(KD) parasites but not in the control strain, HM-1:IMSS (Fig. 7A). Confocal microscopy further substantiated this observation with significant nuclear enrichment noted (Fig. 7B). These data are in good agreement with EhAGO2-2 localization. We further attempted to probe EhROM1-specific sRNAs using fluorescein-labeled EhROM1 sense probe ROM1-FAM and found that the nuclear signal was significantly weaker (data not shown). We reason that this is probably due to the significantly lower abundance of EhROM1 sRNAs compared with the ap-a sRNAs (as noted in Fig. 2 (bottom)).
FIGURE 7.

Antisense sRNAs are enriched in the parasite nucleus. A, FISH localization of antisense sRNA for G3 and G3:ROM(KD) parasites and control HM-1:IMSS using FAM-labeled ap-a S1 probe. Signal from G3 and G3:ROM(KD) parasites is enriched in the nucleus, whereas there is no nuclear signal for HM-1:IMSS; DAPI stains the nucleus. B, confocal images of HM-1:IMSS and G3 trophozoites. For the selected parasites, 100-slice planes on the z axis were analyzed using conditions outlined under “Experimental Procedures.” Images shown are phase, DAPI, nuclear-only three-dimensional projection (between slice 45–80), and a single slice crossing the nucleus. C, antisense sRNAs are enriched in the parasite nuclear fraction. Total RNA from HM-1:IMSS and G3 parasites was extracted after separation of parasite nuclei and cytoplasm. 10 μg of RNA is loaded for each lane. Northern blot analysis detects enriched antisense sRNA signal for the G3 nuclear lane using ap-a sense probe S1. As a control, the blot was probed with a U5 snRNA probe, which showed enriched signal in nuclear sample lanes, and with a tRNA probe, which showed the enriched signal in cytoplasm sample lanes.
In order to further demonstrate enrichment of antisense sRNAs in the nucleus, we fractionated parasite material into cytosolic and nuclear fractions. We isolated total RNA from each fraction and performed Northern blot analysis using sense probe ap-a S1 (Fig. 7C). The results indicate that the antisense sRNA is greatly enriched in nuclear fraction compared with the cytosolic fraction. As a control for cell fractionation, tRNA was probed on the same blot and showed increased signal for the cytosolic fraction relative to the nuclear fraction. In agreement with this, the small nuclear RNA U5 had higher signal for the nuclear fraction than the cytosol fraction. Equal loading was shown by SYBR Gold staining of the gel before membrane transfer (Fig. 7C).
We have previously shown that EhAGO2-2 is in direct association with the endogenous 27-nt 5′-polyphosphate sRNA population in E. histolytica (20). To determine if the ap-a-specific antisense sRNAs are also in association with EhAGO2-2, we performed anti-EhAGO2-2 IP experiments for parasite G3 lysate along with beads-only IP control. RNA was prepared from IP experiments, and Northern blot detected an enriched signal for the ap-a S1 probe in the α-EhAGO2-2 IP sample but not the control IP, indicating that the AGO2–2 pull-down is specific (Fig. 8). As a control for nonspecific IP pull-down of RNA, tRNA was probed on the same blot, which was reduced ∼3000-fold after IP. Thus, we have demonstrated that EhAGO2-2 is in direct association with the antisense small RNAs associated with gene silencing in the G3 strain.
FIGURE 8.
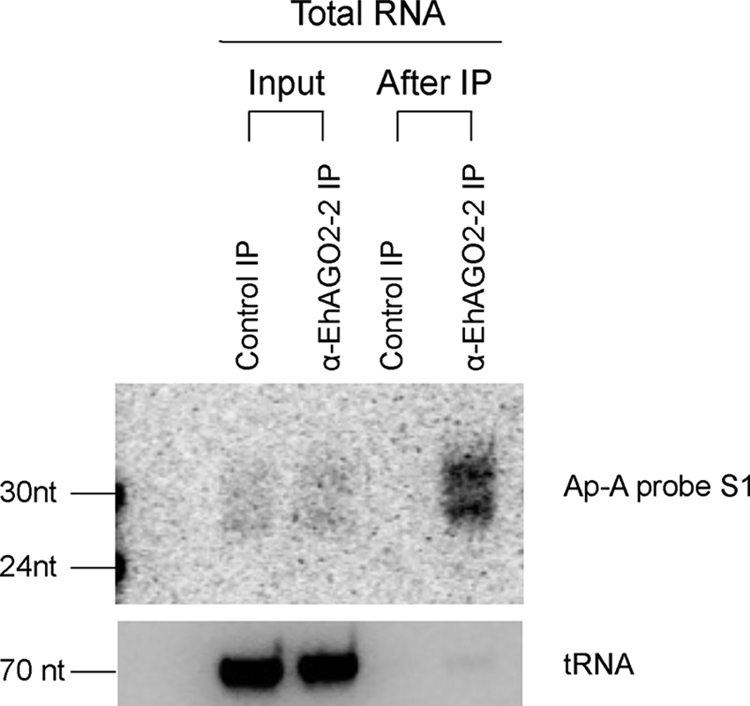
Antisense sRNAs are in direct association with EhAGO2-2. Total RNA was extracted from input ( input volume), α-EhAGO2-2 IP, and control IP (beads-only) samples. Northern blot analysis detected enriched antisense sRNA signal in the α-EhAGO2-2 IP lane but not the control, using ap-a sense probe S1. As a loading control, tRNA was probed on the same blot, which was reduced more than 3000-fold after IP.
EhAGO2-2 Is Enriched at Silenced Gene Loci by ChIP-PCR Analysis
In order to more clearly define the mechanism of sRNA-mediated gene silencing in the G3 parasites, we performed ChIP analysis using antibodies against EhAGO2-2 and histone 3. Comparisons were made between highly expressed and silenced genes and also between the G3 and control parasites, HM-1:IMSS. In G3 parasites, ChIP-PCR revealed a significant enrichment for the silenced ap-a gene for both antibodies, but no enrichment was noted for the highly expressed genes EhROM1 and EhRpl21 (EhRpl21 was used as a reference gene) (Fig. 9). Similarly, a significant enrichment for the silenced genes, ap-b and SAPLIP 1, was observed in G3 parasites compared with the HM-1:IMSS control. The nonspecific binding was determined by using a non-related rabbit anti-Myc antibody, which showed a very faint band for the ChIP conditions used in this study (data not shown). We extended the similar analysis to G3:ROM(KD) and G3:5′STIRP parasites and in all cases found that both antibodies (anti-EhAGO2-2 and histone 3) were enriched in the silenced gene loci compared with the expressed gene loci (supplemental Fig. S5). Our results have therefore not only confirmed the previous finding that histone H3 is enriched for the silenced ap-a gene in the G3 strain (27) but also provided the direct evidence that the EhAGO2-2 is in close contact with the silenced gene loci in G3 parasites. In other systems, DNA methylation, histone modification, and DNA elimination have been noted with siRNA-mediated TGS (43). Thus, our data indicate a new function of 5′-polyphosphate antisense sRNA in regulating TGS and additionally extend the RNAi-mediated TGS mechanism from model systems to a single-celled protist.
FIGURE 9.

EhAGO2-2 and histone 3 are enriched at the silenced gene loci in G3 parasites by ChIP-PCR analysis. Shown is ChIP analysis of HM-1:IMSS and G3 parasites using anti-histone 3 and anti-EhAGO2-2. The enrichment for each gene primer pair was calculated relative to the control gene primer pairs in whole cell extract (WCE) and is depicted below each lane (see “Experimental Procedures”).
DISCUSSION
Our findings have linked nuclear enriched 5′-polyphosphorylated antisense sRNAs to the silenced gene loci in E. histolytica G3 parasites, indicating that these sRNA molecules mediate gene silencing in this strain. We have previously demonstrated that EhAGO2-2 is bound to endogenous 5′-polyphosphorylated antisense sRNAs in Entamoeba (20). We now show that EhAGO2-2 is in direct association with exogenously triggered 5′-polyphosphorylated antisense sRNAs in the G3 strain, that the complex is predominantly nuclear, and that it is in direct contact with the silenced gene loci. Previous publications suggested that the mechanism of gene silencing in the G3 strain was a non-RNAi-mediated process and instead was related to chromatin remodeling (27). However, we have now shown that 5′-polyphosphorylated antisense sRNAs are key mediators for the gene silencing in these parasites, probably acting through the previously noted histone modifications. Overall, the combined data implicate a core-silencing complex, which contains nuclear EhAGO2-2 and 5′-polyphosphorylated antisense siRNAs targeting the silenced genes and mediating gene silencing via histone modifications. These data justify the conclusion that the siRNA pathway is the underlying mechanism for TGS in the G3 parasite strain.
Links between the RNAi pathway and DNA and chromatin modification proteins, such as DNA methytransferases, histone deacetylases, and histone methyltransferases, have been reported in S. pombe, A. thaliana, D. melanogaster, and mammalian cells (5). In E. histolytica, 5-methylcytosine modifications have been confirmed, an active DNA methyltransferase has been reported (44), and an E. histolytica methylated LINE binding protein that binds preferentially to methylated DNA has been characterized (45). In addition, histone acetylation and deacetylation are operational in E. histolytica (46). It was noted that gene silencing in G3 requires both a T-rich region and sequences at the 5′-end of the SINE element (24). The fact that this particular SINE1 is actively transcribed indicates that retrotransposon activity may somehow be related to the induction of gene silencing in G3 parasites (24). To date, there has been no direct evidence of DNA methylation in the region of the SINE1 or the ap-a gene in G3 parasites; nor have inhibitors such as trichostatin A (histone deacetylase) or 5-azacytidine (an inhibitor of DNA methylation) been shown to reverse the ap-a silencing in G3 parasites (24). However, studies on histone methylation of H3 Lys-4 in the coding region of the silenced genes showed significantly reduced methylation levels, supporting the idea that histone modifications occur at the chromatin level at silenced loci in G3 parasites (27). Given our data with EhAGO2-2 and the nuclear sRNAs to the silenced loci and their presence in the nucleus, it appears that the TGS pathway in E. histolytica G3 strain involves the interplay between the RNAi pathway, transposon elements, and chromatin modification.
We previously reported that the endogenous sRNA repertoire in E. histolytica is composed of multiple sRNA populations, including an abundant 27-nt sRNA population, which specifically associates with EhAGO2-2 and has an uncommon 5′-polyphosphate structure (20). This structural feature is reminiscent of secondary siRNAs in C. elegans (13), where these 5′-polyphosphate siRNAs function in silencing of transposons and repetitive elements as well as chromosome segregation (47, 48). Two recent papers have further shown that 5′-polyphosphate siRNAs associate with Argonaute-like protein NRDE-3, which upon binding of these siRNAs redistributes the complex to the nucleus, a process that is essential for silencing nucleus- localized RNAs (42). E. histolytica 5′-polyphosphate sRNAs are also implicated in silencing functions with inverse correlation between gene and antisense sRNA expression levels (20). The IFA and RNA FISH data demonstrate that both EhAGO2-2 and the 5′-polyphosphate sRNAs are enriched in the nucleus, and ChIP analysis further showed that both histone 3 and EhAGO2-2 are enriched at the silenced gene loci. Whether binding of 5′-polyphosphate siRNA is a prerequisite condition for EhAGO2-2 to be localized in the nucleus needs to be further determined. Because 5′-polyphosphate antisense sRNA generation probably involves RNA-dependent RNA polymerase processing, and in E. histolytica the full-length RNA-dependent RNA polymerase protein is localized in the cytoplasm,3 we speculate that a similar translocation may happen in Entamoeba; upon a trigger transcript being recognized by the RNAi machinery, RNA-dependent RNA polymerase starts to generate 5′-polyphosphate antisense sRNAs, which are then bound to EhAGO2-2 and redistributed to the nucleus, where they find their targeted genomic loci. Further investigations in this area are needed.
The mechanisms by which the RNAi pathways function in Entamoeba are still largely unknown. By utilizing two constructs to silence EhSTIRP, we had an opportunity to probe for sRNAs in different regions of the gene. To our surprise, transfectants from the psAP-2/3′STIRP construct had antisense sRNAs not only in the trigger region but also in the 5′-end of the EhSTIRP gene. In addition, probes outside the trigger regions detected antisense sRNAs in both the G3:5′STIRP and G3:3′STIRP cell lines. We speculate that there are two pools (rounds) of antisense sRNA generation. The first pool (round) of antisense sRNAs corresponds to and is restricted to the trigger transcript. The second pool (round) of antisense sRNAs is generated at the site of the endogenous target gene transcript. Upon recognition of the first pool of antisense sRNAs to the targeted endogenous gene, RNAi/siRNA machinery starts to degrade the endogenous transcript, which results in more amplified antisense sRNA signal that is predominantly toward the 5′-end of gene (Fig. 10). Both pools of sRNAs can be loaded into EhAGO2-2, and sRNA leads the complex to the nucleus and targets the genomic locus for chromatin modifications, which mark it for permanent gene silencing.
FIGURE 10.

A proposed model for antisense sRNA-mediated gene silencing in G3. Upon trigger recognition as an aberrant transcript by the cell, the initial pool of antisense sRNAs is generated, which corresponds to and is restricted to the trigger transcript. An additional pool of antisense sRNAs is further formed at the site of the endogenous target gene transcript upon recognition of the first pool of antisense sRNAs to the targeted endogenous gene. RNAi/siRNA machinery further induces the chromatin modifications for permanent silencing at the targeted genomic loci in the nucleus.
In conclusion, we have shown that 5′-polyphosphorylated antisense sRNAs are linked to TGS in E. histolytica G3 parasites. These antisense sRNAs not only respond to the trigger transcript but also to the endogenous target genes. EhAGO2-2 associates with 5′-polyphosphorylated antisense sRNAs and is localized predominantly in the nucleus, where antisense sRNAs act as a guide for target recognition and induce the chromatin modifications required for permanent gene silencing. Our finding of 5′-polyphosphorylated antisense sRNAs mediating TGS in Entamoeba is the first to show that RNAi-mediated TGS can be mediated by this type of sRNA species and is the first to demonstrate that this pathway can be functional in a single-celled protist.
Supplementary Material
Acknowledgments
We gratefully acknowledge Gretchen Ehrenkaufer for critical reading of the manuscript and all members of the Singh laboratory for helpful comments and suggestions. The G3 parasites were a kind gift of David Mirelman.
This work was supported, in whole or in part, by National Institutes of Health Grants AI-053724 and AI-085178 (to U. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S-I to S-V and Figs. S1–S5.
V. Tran, H. Zhang, and U. Singh, unpublished data.
- nt
- nucleotide(s)
- miRNA
- microRNA
- piRNA
- PIWI-associated small RNA
- TGS
- transcriptional gene silencing
- sRNA
- small RNA
- IFA
- immunofluorescence analysis
- IP
- immunoprecipitation.
REFERENCES
- 1. Cerutti H., Casas-Mollano J. A. (2006) Curr. Genet. 50, 81–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nowotny M., Yang W. (2009) Curr. Opin. Struct. Biol. 19, 286–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chapman E. J., Carrington J. C. (2007) Nat. Rev. Genet. 8, 884–896 [DOI] [PubMed] [Google Scholar]
- 4. Krol J., Loedige I., Filipowicz W. (2010) Nat. Rev. Genet. 11, 597–610 [DOI] [PubMed] [Google Scholar]
- 5. Moazed D. (2009) Nature 457, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Djupedal I., Ekwall K. (2009) Cell Res. 19, 282–295 [DOI] [PubMed] [Google Scholar]
- 7. Verdel A., Moazed D. (2005) FEBS Lett. 579, 5872–5878 [DOI] [PubMed] [Google Scholar]
- 8. Pikaard C. S., Haag J. R., Ream T., Wierzbicki A. T. (2008) Trends Plant Sci. 13, 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin H., Lin H. (2007) Nature 450, 304–308 [DOI] [PubMed] [Google Scholar]
- 10. Grishok A., Sinskey J. L., Sharp P. A. (2005) Genes Dev. 19, 683–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hawkins P. G., Santoso S., Adams C., Anest V., Morris K. V. (2009) Nucleic Acids Res. 37, 2984–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki K., Kelleher A. D. (2009) Curr. Top. Med. Chem. 9, 1079–1087 [DOI] [PubMed] [Google Scholar]
- 13. Pak J., Fire A. (2007) Science 315, 241–244 [DOI] [PubMed] [Google Scholar]
- 14. Gent J. I., Lamm A. T., Pavelec D. M., Maniar J. M., Parameswaran P., Tao L., Kennedy S., Fire A. Z. (2010) Mol. Cell 37, 679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maniar J. M., Fire A. Z. (2011) Curr. Biol. 21, 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen X. S., Collins L. J., Biggs P. J., Penny D. (2009) Genome Biol. Evol. 1, 165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ullu E., Tschudi C., Chakraborty T. (2004) Cell Microbiol. 6, 509–519 [DOI] [PubMed] [Google Scholar]
- 18. Zhang H., Pompey J. M., Singh U. (2011) Future Microbiol. 6, 103–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atayde V. D., Tschudi C., Ullu E. (2011) Trends Parasitol. 27, 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang H., Ehrenkaufer G. M., Pompey J. M., Hackney J. A., Singh U. (2008) PLoS Pathog. 4, e1000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. World Health Organization (1997) Bull. World Health Organ. 75, 291–2949277015 [Google Scholar]
- 22. Bracha R., Nuchamowitz Y., Mirelman D. (2003) Eukaryotic Cell 2, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bracha R., Nuchamowitz Y., Anbar M., Mirelman D. (2006) PLoS Pathog. 2, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anbar M., Bracha R., Nuchamowitz Y., Li Y., Florentin A., Mirelman D. (2005) Eukaryotic Cell 4, 1775–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bracha R., Nuchamowitz Y., Wender N., Mirelman D. (2007) Eukaryotic Cell 6, 1758–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baxt L. A., Rastew E., Bracha R., Mirelman D., Singh U. (2010) Eukaryotic Cell 9, 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huguenin M., Bracha R., Chookajorn T., Mirelman D. (2010) Parasitology 137, 619–627 [DOI] [PubMed] [Google Scholar]
- 28. Diamond L. S., Harlow D. R., Cunnick C. C. (1978) Trans. R. Soc. Trop. Med. Hyg. 72, 431–432 [DOI] [PubMed] [Google Scholar]
- 29. MacFarlane R. C., Singh U. (2006) Infect. Immun. 74, 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu J., Tsourkas A. (2009) Nucleic Acids Res. 37, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silahtaroglu A. N. (2010) Methods Mol. Biol. 659, 165–171 [DOI] [PubMed] [Google Scholar]
- 32. Pall G. S., Hamilton A. J. (2008) Nat. Protoc. 3, 1077–1084 [DOI] [PubMed] [Google Scholar]
- 33. Gilchrist C. A., Leo M., Line C. G., Mann B. J., Petri W. A., Jr. (2003) J. Biol. Chem. 278, 4646–4653 [DOI] [PubMed] [Google Scholar]
- 34. Chookajorn T., Dzikowski R., Frank M., Li F., Jiwani A. Z., Hartl D. L., Deitsch K. W. (2007) Proc. Natl. Acad. Sci. U. S. A. 104, 899–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ehrenkaufer G. M., Hackney J. A., Singh U. (2009) Cell. Microbiol. 11, 898–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacFarlane R. C., Singh U. (2007) Eukaryotic Cell 6, 2139–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hutvagner G., Simard M. J. (2008) Nat. Rev. Mol. Cell Biol. 9, 22–32 [DOI] [PubMed] [Google Scholar]
- 38. Ehrenkaufer G. M., Haque R., Hackney J. A., Eichinger D. J., Singh U. (2007) Cell Microbiol. 9, 1426–1444 [DOI] [PubMed] [Google Scholar]
- 39. Li C. F., Pontes O., El-Shami M., Henderson I. R., Bernatavichute Y. V., Chan S. W., Lagrange T., Pikaard C. S., Jacobsen S. E. (2006) Cell 126, 93–106 [DOI] [PubMed] [Google Scholar]
- 40. Brower-Toland B., Findley S. D., Jiang L., Liu L., Yin H., Dus M., Zhou P., Elgin S. C., Lin H. (2007) Genes Dev. 21, 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buker S. M., Iida T., Bühler M., Villén J., Gygi S. P., Nakayama J., Moazed D. (2007) Nat. Struct. Mol. Biol. 14, 200–207 [DOI] [PubMed] [Google Scholar]
- 42. Guang S., Bochner A. F., Pavelec D. M., Burkhart K. B., Harding S., Lachowiec J., Kennedy S. (2008) Science 321, 537–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verdel A., Vavasseur A., Le Gorrec M., Touat-Todeschini L. (2009) Int. J. Dev. Biol. 53, 245–257 [DOI] [PubMed] [Google Scholar]
- 44. Fisher O., Siman-Tov R., Ankri S. (2004) Nucleic Acids Res. 32, 287–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lavi T., Isakov E., Harony H., Fisher O., Siman-Tov R., Ankri S. (2006) Mol. Microbiol. 62, 1373–1386 [DOI] [PubMed] [Google Scholar]
- 46. Ramakrishnan G., Gilchrist C. A., Musa H., Torok M. S., Grant P. A., Mann B. J., Petri W. A., Jr. (2004) Mol. Biochem. Parasitol. 138, 205–216 [DOI] [PubMed] [Google Scholar]
- 47. Claycomb J. M., Batista P. J., Pang K. M., Gu W., Vasale J. J., van Wolfswinkel J. C., Chaves D. A., Shirayama M., Mitani S., Ketting R. F., Conte D., Jr., Mello C. C. (2009) Cell 139, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gu W., Shirayama M., Conte D., Jr., Vasale J., Batista P. J., Claycomb J. M., Moresco J. J., Youngman E. M., Keys J., Stoltz M. J., Chen C. C., Chaves D. A., Duan S., Kasschau K. D., Fahlgren N., Yates J. R., 3rd, Mitani S., Carrington J. C., Mello C. C. (2009) Mol. Cell 36, 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.