

Background: CARD-containing proteins participate in immune responses, inflammation, and apoptosis.
Results: CARD-derived polypeptides displayed selective inhibition of apoptosome activation of procaspase-9 and/or of inflammasome activation of procaspase-1.
Conclusion: Peptides from the CARDs of CARD-containing proteins are useful tools to analyze the role of the CARD in signaling.
Significance: CARD-CARD interactions represent targets for inhibition to understand cellular function and pathology.
Keywords: Apoptosis, Apoptosome, Caspase, Caspase-1, Caspase-9, Inflammation, Innate Immunity, Peptide Chemical Synthesis, Peptides, Peptide Interactions, Protein Domains, Apaf-1, CARD, Inflammasome, Inhibitors, NLRP-1, NOD, ASC, Procaspase, Procaspase-1, Procaspase-9, Protein-Protein
Abstract
The caspase recruitment domain (CARD) is present in a large number of proteins. Initially, the CARD was recognized as part of the caspase activation machinery. CARD-CARD interactions play a role in apoptosis and are responsible for the Apaf-1-mediated activation of procaspase-9 in the apoptosome. CARD-containing proteins mediate the inflammasome-dependent activation of proinflammatory caspase-1. More recently, new roles for CARD-containing proteins have been reported in signaling pathways associated with immune responses. The functional role of CARD-containing proteins and CARDs in coordinating apoptosis and inflammatory and immune responses is not completely understood. We have explored the putative cross-talk between apoptosis and inflammation by analyzing the modulatory activity on both the Apaf-1/procaspase-9 interaction and the inflammasome-mediated procaspase-1 activation of CARD-derived polypeptides. To this end, we analyzed the activity of individual recombinant CARDs, rationally designed CARD-derived peptides, and peptides derived from phage display.
Introduction
Protein-protein interactions and the formation of multiprotein complexes in signaling pathways are involved in most physiological processes. Proteins often use complex interactions to produce a sophisticated signaling network that will ensure the most appropriate cellular response to environmental changes or pathogen invasion attempts. Signaling network understanding will facilitate the analysis of cellular pathways and their intricate cross-connectivity. The unregulated induction of signaling complexes and its cellular consequences is now an active area of research, given its pharmaceutical relevance. However, it is still unclear how multiple signaling pathways interact and how this communication is regulated; the postulated cross-talk between cell death, innate immunity, and inflammatory response is of particular interest (1–6).
The formation of apoptotic and inflammatory multiprotein complexes together with defined signaling episodes in innate immunity heavily relies on members of the death domain family and particularly on the family subclass of the caspase recruitment domain (CARD).6 The interaction between the CARD of Apaf-1 (apoptotic protease-activating factor) and the CARD of procaspase-9 (PC9) in the mitochondria-mediated apoptotic intrinsic pathway is essential for the recruitment of PC9 into the apoptosome and its subsequent activation (7). On the other hand, proteins like those of the NOD-like receptor (NLR) family (in particular NOD-1, NOD-2, and NLRP-1) act as intracellular scrutiny devices and signaling initiators to face microbial aggressions (8). The NLR proteins utilize the CARD for binding to downstream signaling molecules through CARD-CARD interactions in order to ultimately initiate the innate immune and inflammatory responses (9, 10). As a general organization, the CARD module is characterized by a Greek key of six antiparallel amphipathic closely packed α-helices (11–13). However, primary sequence alignments reveal a very low sequence identity (<20%), emphasizing that some critical conserved residues could mediate initial CARD-CARD electrostatic interactions and that the surrounding residues in this region would define the specificity of CARD-CARD interactions (8, 12). Such interactions therefore represent challenging targets for inhibition, not only to understand their function but also in pathology. The main issue ahead is to achieve a high degree of specificity within such a complex and large number of CARD proteins and to target protein-protein interactions that are not yet well characterized. Accordingly, if the CARD-CARD interaction was suppressed pharmacologically, the downstream signaling would be interrupted and allow a chance to control the progression of the process. In this sense, small molecule inhibitors of the apoptosome formation have been described to inhibit mitochondria-mediated apoptosis (14–17). Less information has been released on the pharmacological modulation of CARD-CARD-mediated protein-protein interactions that mediate innate immunity and inflammation, probably due to the intrinsic complexity of the protein complexes. Hence, it is difficult to define a chemical biology-based approach to explore a putative cross-connection between the biological processes implying CARD-CARD interactions. Proteins involved in signal transduction utilize adaptor domains to modulate protein-protein interactions during the formation of multiprotein signaling complexes. When these domains are overexpressed in defined cells, they behave like dominant negative inhibitors of protein-protein interactions (18–21). This strategy together with others that produce a complete loss-of-function or a null phenotype, such as those based on siRNA technologies, help to understand the function of defined proteins in specific signaling pathways (22–24). However, quantitative evaluation is difficult with such approaches. Protein domains produced as recombinant proteins or synthetic peptides derived from such domains could define useful analysis tools when analyzed in the appropriate in vitro assays. Therefore, the still unexplored design and evaluation of polypeptides defined as short protein domains and synthetic peptides targeting CARD-CARD interactions could provide a first level of ground information in this direction. We have therefore adopted an integrated strategy to define polypeptide modulators of a representative CARD-CARD-mediated protein interaction by first producing individual CARDs in bacteria; second, devising a rational approach based on the design of peptide mimics of the interacting proteins (25); and third, using phage display technology (26). In particular, we initially paid attention to the Apaf-1/PC9 interaction and to the different in vitro formats described to analyze this interaction. An initial attempt to explore the putative cross-talk between apoptosis and inflammation was also made by analyzing the inflammasome-mediated procaspase-1 (PC1) activation in cell extracts. The results show that the CARDs of Apaf-1, PC9, and NLRP-1 and defined CARD-derived peptides from both apoptosome-related (Apaf-1 and PC9) and -unrelated (NOD-1, NLRP-1, and ASC) CARD-containing proteins displayed inhibitory activity on the Apaf-1-mitochondria-dependent intrinsic pathway of apoptosis. However, only the CARD of NLRP-1 and selected peptides derived from the CARDs of Apaf-1, NOD-1, NLRP-1, and ASC showed inhibitory activity in PC1 activation.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
Peptides were prepared by Fmoc (N-(9-fluorenyl)methoxycarbonyl)-based solid phase synthesis in a 433A Applied Biosystems peptide synthesizer as reported previously (27). Purification was performed in a C18 preparative RP-HPLC system up to 95% of peptide purity as determined by analytical RP-HPLC. Identity was confirmed by MALDI-TOF mass spectroscopy. A Tyr residue followed by a Gly-Gly spacer was incorporated into the N terminus of those peptides without aromatic amino acid in the designed sequence to facilitate peptide concentration determination by UV spectroscopy. To obtain the cyclic peptides, the Cys-containing linear peptides were dissolved in HOAc/DMSO/H2O (1:3:16) at 0.5 mg/ml. A neutral pH was achieved after treatment with ammonium carbonate. The solution was then stirred at room temperature for 24 h to allow air oxidation (28).
Circular dichroism (CD) Measurements
CD spectra were recorded at 25 °C on a Jasco J-810 spectropolarimeter in quartz cells of 0.1-cm path length. Peptides were dissolved at 10 μm in phosphate buffer (50 mm, pH 7.0), and their ability to adopt a secondary conformation was analyzed with a 2,2,2-trifluoroethanol (TFE) titration. Each CD spectrum was the average of 20 scans performed at 1-nm intervals. CD spectra were interpreted with the K2D software provided by Dichroweb (available on the World Wide Web). The results are expressed as mean molar residue ellipticities (degrees cm2 dmol−1). The Apaf-1, PC9, and NLRP1 CARDs were evaluated under similar conditions at 4 μm.
Protein and Protein Domain Production
Overexpression and purification of His-tagged Apaf 1–591 and PC9 were developed as previously reported (29, 30). Recombinant Apaf-1 XL was obtained from a baculovirus expression system as described previously (14).
The CARDs from Apaf-1, PC9, NLRP-1, NOD-1, and ASC were cloned into pET15b plasmid and expressed in Bl21(DE3)pLysS codon+. Briefly, cells were grown at 37 °C, and protein expression was induced at A600 0.6 with 0.5 mm of IPTG overnight at 30 °C. Bacterial pellets were resuspended in lysis buffer (5 mm Tris-HCl, pH 8, 300 mm NaCl, 10% glycerol) and sonicated. After centrifugation, proteins were placed in a BD Talon affinity column and recovered in elution buffer (25 mm Tris-HCl, pH 8, 300 mm NaCl, 10% glycerol, 250 mm imidazole). The Apaf-1 CARD protein was dialyzed in a molecular weight 6000–8000 membrane against the maintenance buffer (20 mm Hepes, pH 7.5, 100 mm KCl). NLRP-1 CARD and PC9 CARD preparations were fractionated by anion exchange chromatography (HiTrap Q Sepharose) and maintained in the same buffer at pH 11, where they remained soluble. NOD-1 CARD and ASC CARD were insoluble proteins under our experimental conditions.
Caspase-9 Activation Assays (LEHD Activity)
Activation of PC9 by Apaf-1 1–591 was done as described elsewhere (29) with minor modifications. The protein concentrations in the assay were 0.5 μm for Apaf-1 1–591 and 100 nm for PC9, and the reaction was performed in the presence of 10 μm ATP. Peptides were preincubated with Apaf-1 1–591 for 25 min at room temperature. Then 100 μm Ac-LEHD-afc was added, and fluorescent afc release was continuously monitored using a Victor Wallace 1420 workstation spectrofluorimeter at 30 °C (λex = 390 nm; λem = 510 nm).
In the sodium citrate-dependent PC9 activation, 5 μm PC9 was preactivated for 20 min in SC buffer (50 mm Na2HPO4, 150 mm NaCl, 1.5% sucrose, 0.05% CHAPS, 10 mm DTT, 0.7 m sodium citrate, pH 7.4) at room temperature. Once preactivated, the complete reaction was diluted to 100 nm PC9 in assay buffer without sodium citrate (S buffer) and incubated for 25 min at room temperature in the presence or absence of peptides. Ac-LEHD-afc substrate was added at 40 μm, and afc release was monitored at 25 °C as described above.
Cell Lines and Cultures
The human embryonic kidney (HEK) 293, human cervix adenocarcinoma (HeLa), and human acute monocytic leukemia (THP-1) cell lines were obtained from the American Type Culture Collection (Manassas, VA). HEK 293 and HeLa were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS). THP-1 was cultured in RPMI 1640 supplemented with 10% FCS, 1 mm sodium pyruvate, and 50 μm β-mercaptoethanol. All of the cell lines were maintained at 37 °C in an atmosphere of 5% carbon dioxide. LipofectamineTM 2000 (Invitrogen) was used according to the manufacturer's instructions.
Cell-free Caspase Activation Assays (DEVDase Activity)
Cytosolic extracts from HEK293 were depleted of Apaf-1 as described previously (31). rApaf-1 (20 nm) was incubated with either CARDs or peptides for 20 min at 37 °C, in 20 mm Hepes-KOH, pH 7.5, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 0.1 mm PMSF buffer. After incubation, cytosolic extract and dATP were added, and the mixture was incubated at 37 °C for 30 min. Ac-DEVD-afc substrate (20 μm) was used to measure caspase 3 activity.
Cell-based Caspase Activation Assay
HeLa cells (2 × 105 cells seeded in 6-well plates) were treated with a Lipofectamine/peptide (100 μm) mixture for 4 h followed by administration of 20 μm cis-diammineplatinum(II) dichloride (cisplatin, CDDP). Cells were harvested after 24 h, and cytosolic extracts were obtained as described previously (14). The cell extract was protein-quantified, and 50-μg protein equivalents of cell extract were supplemented with caspase assay buffer containing 20 μm Ac-DEVD-afc substrate for caspase-3 activity evaluation.
Cell-free Inflammasome-dependent Procaspase-1-activating Activity
Extracts from THP-1 cells were prepared as described previously (32). Peptides were incubated with 25 μg of THP-1 extract for 25 min at 4 °C. Extract activation was performed at 37 °C and continuously monitored in the presence of 100 μm of the fluorogenic caspase-1 substrate Ac-WEDH-afc.
Phage Display Libraries and Screening
The peptide libraries (12-mer, 7-mer, and 7-mer cysteine-constrained) were purchased from New England BioLabs Inc. (Beverly, MA). All of the libraries have complexities on the order of 109 independent clones; the random peptides are fused to a minor coat protein (pIII) of the M13 phage and expressed at its N terminus separated by a Gly-Gly-Gly-Ser spacer. The His6-tagged Apaf-1 CARD was used as the target protein. The selection procedure was developed as described previously (33) with some adjustments. Briefly, for each library, a single well of Ni2+-NTA HisSorb plates (Qiagen) was coated with the purified protein at 150 μg/ml (20 mm Hepes, pH 7.5, 100 mm KCl) in the first round and at 75 and 37.5 μg/ml for the second and third rounds, respectively. The plate was kept overnight at 4 °C with gentle shaking. The unbound protein was discarded, and wells were washed 6 times with PBS-T (PBS plus 0.1% Tween 20) and blocked with PBS plus 1% BSA for 1 h at 4 °C. After five washing steps, 10 μl of the corresponding library (2 × 1010 pfu) diluted in PBST up to 100 μl/well were added, and the plate was incubated for 1 h at room temperature while shaking gently to allow phages to bind. The unbound phages were discarded, and wells were washed 10 times with PBS-T at room temperature. The bound phages were eluted by stirring with 100 μl of 0.1 n HCl-glycine, pH 2.2, for 10 min. The eluted solution was immediately neutralized using 2 m Tris-HCl, pH 9.1. The eluted phages were amplified by infecting the Escherichia coli strain ER2738. The amplified phages were precipitated, purified, and titered for the next round of panning using a phage number of 2 × 1011 pfu. During the third round, the stringency of the wash was increased by using 0.5% Tween 20 in the wash buffer. At the end of the third panning, the eluted phages were used to infect E. coli ER2738 to isolate individual clones. For each clone, ssDNA was purified and sequenced.
RESULTS
Recombinant CARDs as Modulators of Apaf-1-dependent PC9 Activation
We initially approached the analysis of CARD-mediated protein-protein interactions using the recombinant CARDs of Apaf-1 (Apaf-1 CARD), procaspase-9 (PC9 CARD), NOD-1 (NOD-1 CARD), ASC (ASC CARD), and NLRP-1 (NLRP-1 CARD). NOD-1 CARD and ASC CARD precipitated out from the solution in the required experimental conditions. The secondary structure of the recombinant CARDs was investigated by CD spectroscopy. All three (Apaf-1 CARD, PC9 CARD, and NLRP-1 CARD) domains showed the double minima at 207 and 220 nm that are characteristic of α-helical conformation (Fig. 1). The in vitro activity of the CARDs modulating the CARD-CARD-mediated Apaf-1/PC9 interaction was investigated in an assay based on the reconstitution of a functional apoptosome from the recombinant proteins. The apoptosome is a multiprotein complex formed upon the release of cytochrome c from mitochondria when cells activated the intrinsic pathway of apoptosis (34, 35). Cytochrome c binds to Apaf-1 and, in the presence of ATP/dATP, the complex recruits PC9 to proceed with caspase activation. Apaf-1 consists of an N-terminal CARD, a central nucleotide-binding domain, and multiple C-terminal WD40 repeats employed to bind to cytochrome c. The last repeats are dispensable in apoptosome in vitro reconstitution assays (35, 36). Recombinant WD40 repeat-depleted Apaf-1 (Apaf-1 1–591) and recombinant PC9 were produced to set up the apoptosome in vitro reconstitution assay (29). Apaf-1 1–591 is able to promote the processing and activation of PC9 as followed by the release of the fluorophore afc from caspase-9-specific peptide substrate Ac-LEHD-afc (37). We tested the ability of the recombinant CARDs to inhibit the Apaf-1 1–591-mediated PC9 activation. PC9 was incubated in the presence or absence of the CARDs at different concentrations for 10 min before the addition of Apaf-1 1–591. After another 10-min period, the caspase-9 substrate was added, and PC9 activity was measured as a function of time. The inhibition percentages of caspase-9 activity were calculated and used to estimate the relative IC50 values (defined as the recombinant CARD concentration required inhibiting 50% of Apaf-1 1–591-mediated PC9 activation). The recombinant CARDs of Apaf-1, PC9, and NLRP-1 exhibited a similar inhibitory activity (Table 1). The inhibitory activity of the CARDs was also evaluated in a cell extract-based assay (see below), which confirmed that the CARDs are capable of inhibiting the apoptosome-mediated activation of the apoptotic pathway (Table 1).
FIGURE 1.

Recombinant CARDs adopt α-helical conformation. Circular dichroism spectra for the Apaf-1 CARD (♢), PC9 CARD (−), and NLRP-1 CARD (○) domains are shown. CD spectra were recorded at 4 μm in 50 mm phosphate, pH 7.0, and the average of 20 scans is represented. The percentages of α-helical structure (inset) were evaluated by the K2D software available in Dichroweb.
TABLE 1.
CARDs inhibit Apaf-1-dependent PC9 activation
IC50 values are defined as the recombinant CARD concentration required for inhibiting 50% of PC9 activation. ND, not determined.
| CARD | IC50a | IC50b |
|---|---|---|
| μm | μm | |
| Apaf-1 | 1.7 ± 0.2 | 5.6+ 0.4 |
| PC9 | 0.8 ± 0.1 | 0.8 ± 0.1 |
| NLRP-1 | 0.9 ± 0.1 | 1.6 ± 0.1 |
| NOD-1 | ND | ND |
| ASC | ND | ND |
a Apaf-1 1–591-mediated PC9 activation in an in vitro reconstituted apoptosome.
b Apaf-1XL cell extract-based assay.
In order to gain further knowledge of the cross-interactions and the nature of the CARD-CARD pathway connections, we evaluated the ability of the different CARDs to mediate PC9 activation. The isolated recombinant Apaf-1 CARD has been reported to be able to activate PC9 (37, 38). PC9 was preincubated with the different CARDs at increasing concentrations. After 10 min of preincubation, Ac-LEHD-afc was added to the medium, and PC9 activity was measured as a function of time. Recombinant PC9 showed a basal activity (38) as evidenced by the cleavage of the fluorogenic substrate and increased fluorescence. This activity was notably enhanced in the presence of Apaf-1 CARD but not in the presence of PC9 CARD or NLRP-1 CARD (Fig. 2).
FIGURE 2.
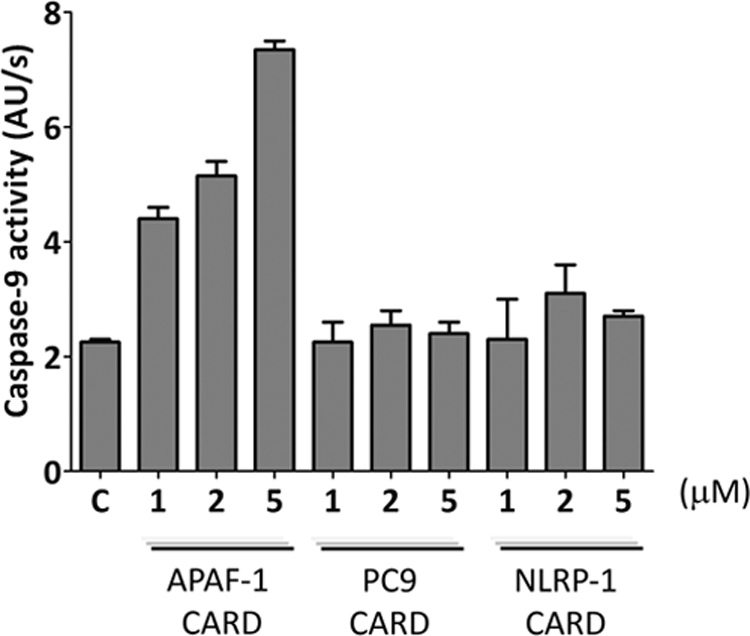
CARD-mediated activation of procaspase-9. Apaf-1 CARD, PC9 CARD, and NLRP-1 CARD were incubated at 1, 2, and 5 μm with recombinant non-active PC-9 in S buffer. Caspase-9 activity was measured in the presence of the fluorogenic Ac-LEHD-afc substrate. Caspase-9 activity is represented as increase in fluorescence arbitrary units/s (AU/s). Control (C) refers to the basal activity of PC-9. Data correspond to the mean ± S.D. (error bars) of three independent experiments.
CARD-derived Peptides as Modulators of Apaf-1-dependent Activation of PC9
Peptides generated as copies of protein fragments, in particular those sequences that participate in protein-protein interactions, have been shown to be effective modulators of signaling events (39–42). In order to design CARD-derived peptides that could modulate CARD-CARD-mediated protein-protein interactions, we analyzed the primary sequence of the CARDs of Apaf-1, PC9, NOD-1, ASC, and NLRP-1 (PDB accession numbers: 3YGS for Apaf-1 and PC9 and 2NZ7, 2KN6, and 3KAT for NOD-1, ASC, and NLRP-1, respectively). Previous studies have evidenced the common six-helix bundle structural organization of CARDs (8, 12), especially those selected for this study (Fig. 3A). The multiple-sequence alignment (Fig. 3B) reveals the strongly conserved amino acids among the five selected domains. The CARD contains a basic and acidic patch on opposite sides, including critical residues that mediate the postulated initial electrostatic interaction (13, 43). In Apaf-1 CARD, residues Asp-27, Glu-39, Glu-40, and Glu-41 are localized within the acidic region and could be the equivalents of residues Asp-42, Asp-48, Glu-53, Asp-54, and Glu-56 in NOD-1 CARD as well as of residues Asp-1401, Glu-1411, and Glu-1414 in NLRP-1. On the opposite side, the basic and concave region of PC9 includes residues Arg-6, Arg-7, Arg-10, Arg-13, Arg-52, and Arg-56 (8, 13). To accomplish the design, we analyzed the CARD-CARD complex described for the target Apaf-1/PC9 interaction (13). Then the peptides derived from helices 2 and 3 of Apaf-1 CARD were selected (peptides A2.Y24G33 and A3.F34V43; see Fig. 3B and Table 2). These peptides included some of the above mentioned critical residues, such as Asp-27 from helix 2 and Glu-39, Glu-40, and Glu-41 from helix 3. From counterpart protein PC9, the peptides derived from helices 1 and 4, which included residues Arg-10 and Arg-13 and residues Arg-52 and Arg-56, respectively, were then selected (peptides C1.L8E17 and C4.R52I60; see Fig. 3B and Table 2). In an attempt to further analyze peptides derived from the apoptosis-unrelated CARD-containing proteins, a similar exercise was applied to NOD-1, NLRP-1, and ASC. Although the CARD-CARD-mediated Apaf-1·PC9 structure has been solved (13), less is known about the CARD-CARD interaction that participates in inflammasome formation. Then we explored peptides derived from both the acidic and basic amino acid patches of these CARDs. From the acidic patch, the peptides designed were N2.T37D48 (including residues Asp-42 and Asp-48) and N3.Y49A60 (containing residues Glu-53 and Asp-54) from NOD-1 CARD helices 2 and 3, respectively; peptide Np2.V1398Q1406 containing the residue Asp-1401 from the NLRP-1 CARD helix 2; peptide As2.N128L136 containing residue Asp-134 (helix 2); and As3.D143A151 containing residues Asp-143 and Glu-144 (helix 3) from the ASC CARD. From the basic patch, we synthesized the following peptides: N4.Q64V75 (helix 4 NOD-1 CARD), Np1.L1379R1392 (helix 1 NLRP-1 CARD), Np4.R1422W1436 (helix 4 NLRP-1 CARD), As1.D116R125 (helix 1 ASC CARD), and As4.N155T166 (helix 4 ASC CARD).
FIGURE 3.

Multiple-protein alignment of CARDs. A, structural overlapping of CARDs. The coordinates were obtained from the Protein Data Bank (accession number 3YGS for Apaf-1 and PC9 CARDs, in black and yellow respectively; 2NZ7 for NOD1 CARD in green; 3KAT for NLRP-1 CARD in blue; and 2KN6 for ASC CARD in gray). Structural modeling was performed with MISTRAL (the Multiple Protein Structure Alignment Server), and PyMOL (Schrodinger LLC) was used to obtain the graphical representation. B, multiple-sequence alignment (ClustalW2). Underlined fragments represent the sequences considered for the synthetic peptides. Conserved amino acids are indicated in red.
TABLE 2.
CD characterization and biological activity of CARD-derived peptides
| Peptide | Patch | Sequence | Percentage of α-helixa |
IC50b | IC50c | |
|---|---|---|---|---|---|---|
| 0% TFE | 50% TFE | |||||
| % | % | μm | μm | |||
| A2.Y24G33 | Acidic | YIMDHMISDG | 0 | 28 | 92 ± 12 | 63 ± 1 |
| A3.F34V43 | Acidic | YGG-FLTISEEEKV | 7 | 28 | 102 ± 7 | 105 ± 10 |
| C1.L8E17 | Basic | YGG-LRRCRLRLVE | 15 | 28 | 68 ± 7 | 113 ± 10 |
| C4.R52I60 | Basic | YGG-RDQARQLII | 25 | 28 | 81 ± 9 | NId |
| N2.T37D48 | Acidic | YGG-TQCLVDNLLKND | 8 | 32 | NI | NI |
| N3.Y49A60 | Acidic | YFSAEDAEIVCA | 27 | 47 | NI | NI |
| N4.Q64V75 | Basic | YGG-QPDKRVRKILDLV | 7 | 85 | 4 ± 1 | 75 ± 10 |
| Np1.L1379R1392 | Basic | LHFVDQYREQLIAR | 8 | 87 | 21 ± 3 | 62 ± 8 |
| Np2.V1398Q1406 | Acidic | YGG-VVLDKLHGQ | 7 | 39 | NI | NI |
| Np4.R1422W1436 | Basic | RPSQMRKLFSLSQSW | 29 | 31 | NI | NI |
| As1.D116R125 | Basic | YGG-DQHRAALIAR | 31 | 86 | 83 ± 10 | 136 ± 10 |
| As2.N128L136 | Acidic | NVEWLLDAL | 28 | 100 | NI | NI |
| As3.D143A151 | Acidic | DEQYQAVRA | 39 | 74 | NI | NI |
| As4.N155T166 | Basic | NPSKMRKLFSFT | 29 | 100 | 27 ± 7 | 69 ± 5 |
a Percentages of α-helical secondary structure obtained from the CD data interpreted with the K2D program of Dichroweb (available on the World Wide Web).
b Activity was analyzed in the in vitro reconstituted apoptosome.
c Activity was analyzed in a cell extract-based assay.
d NI, no inhibition found at the peptide concentration range analyzed.
CD spectroscopy revealed that all of the designed peptides displayed low helicity in solution and that they thus predominantly exist as a random coil (Table 2). However, in the presence of the α-helix stabilizer 2,2,2-trifluoroethanol (44), peptides demonstrated helical stabilization (Table 2). Therefore, the full CARD-derived peptide set displayed environment-dependent conformational adaptability and was seen to be capable of populating the CARD native helical conformation beyond the tertiary context of the protein domain.
The in vitro biological activity of the peptides was investigated in the functional apoptosome assay. Peptides A2.Y24G33 and A3.F34V43 and peptides C1.L8E17 and C4.R52I60, derived from the CARDs of Apaf-1 and PC9, respectively, exerted a moderate inhibitory effect on the enzymatic activity of the apoptosome, unlike those peptides derived from the acidic patch of the CARDs of NOD-1, NLRP-1, and ASC, which did not (Table 2). However, all of the peptides derived from the basic patch from the CARDs of NOD-1, NLRP-1, and ASC except for Np4.R1422W1436, which derived from helix 4 of NLRP-1 CARD, inhibited apoptosome activity (Table 2).
The in vitro results suggested that the peptides derived from the CARD-CARD interacting surface of Apaf-1·PC9 and those derived from the basic patch of the CARDs of NOD-1, NLRP-1, and ASC could be potential hits as selective inhibitors of CARD/CARD-mediated protein-protein interactions in the apoptosome. However, in a cellular milieu, there are a large number of proteins and cellular metabolites that could compromise the activity of peptides and their accessibility to the molecular target. To further explore the efficacy of the peptide hits in a cellular environment, their activity was initially evaluated in mammalian cellular extracts. Cytosolic extracts from human embryonic kidney 293 cells (HEK 293) were depleted from endogenous Apaf-1 by chromatography (FT fraction; see “Experimental Procedures”). Cellular extracts were activated to enter the apoptotic pathway by the addition of baculovirus-produced recombinant full-length Apaf-1XL and incubation with ATP and cytochrome c, which induced caspase-3 activity (14, 31). All of the peptides that inhibited the in vitro activity of recombinant apoptosome, except C4.R52I60, inhibited the apoptosome-mediated caspase-3 activity in cellular extracts (Table 2).
As explained above, CARD-derived peptides were designed and inspired by the reported high resolution structure of the CARD-CARD-mediated Apaf-1/PC9 interaction (13, 43). Peptides A2.Y24G33 and A3.F34V43 contained those proposed as key residues for the interaction and also covered most of the Apaf-1 CARD surface defined as the Apaf-1 interactive surface with PC9 CARD. Similar premises were used for the design of PC9 CARD-derived peptides C1.L8E17 and C4.R52I60. Then we wondered whether the active peptides would show an additive or a synergic effect as inhibitors of apoptosome activity. To this end, we evaluated the activity of peptide combinations at sub-IC50 concentrations (Fig. 4). Only the Apaf-1 CARD-derived peptides had a synergic effect, in fact a combination of peptide A2.Y24G33 and peptide A3.F34V43 at 50 μm each came close to a 100% inhibition of the apoptosome-mediated caspase-3 activity in the cell extract assay (Fig. 4A; the expected percentage of inhibition for a simple additive effect came close to 25%). Furthermore, the assay output confirmed the increased activity of peptide A2.Y24G33 in the overall inhibitory activity. Actually, when peptides A2.Y24G33 and A3.F34V43 were combined at ratios of 1:2 or 2:1, the later combination displayed increased activity. These results agree with previously reported observations from the Apaf-1 CARD-PC9 CARD interaction analysis (13), which highlighted the predominant role of Apaf-1 CARD helix 2 over the other helices (helices 3 and 5) encompassing the binding surface that Apaf-1 CARD offers to PC9 CARD for a productive interaction to occur. Peptide A2.Y24G33 upon binding to PC9 CARD would induce minor conformational changes that facilitated the accessibility of other inhibitory peptides. In contrast, the PC9 CARD-derived peptides (C1.L8E17 and C4.R52I60) did not show a synergic inhibitory effect (Fig. 4B).
FIGURE 4.

Synergistic inhibitory effect of Apaf-1-CARD-derived peptides in cellular extracts. A, peptides A2.Y24G33 and A3.F34V43 were incubated at different ratios in the micromolar range in the reconstituted apoptosome assay in cell-free extracts. Caspase-3 activity was determined in the presence of the fluorogenic Ac-DEVD-afc substrate. B, similar procedures were used for the PC9 CARD-derived peptides C1.L8E17 and C4.R52I60. Data represent the inhibition percentages taking as a control a sample incubated in the absence of peptide. Data correspond to the mean ± S.D. (error bars) of three independent experiments.
According to the in vitro and cell extract results, peptides A2.Y24G33, A3.F34V43, C1.L8E17, C4.R52I60, N4.Q64V75, Np1.L1379R1392, As1.D116R125, and As4.N155T166 inhibited the CARD-dependent activation of PC9 in the apoptosome. The peptides showed a dose-dependent activity, which allowed the determination of IC50 values (Table 2). It is noteworthy that the experimental procedures required for an unequivocal determination of the type of inhibition exerted were not possible, given the complexity of the apoptosome as a macromolecular enzyme. Therefore, in order to corroborate the mechanism of action of the peptides, we turned out our attention to a CARD-independent PC9 activation procedure. Recombinant PC9 has been described as being activated by high concentrations of Hofmeister effect-inducing salts (45, 46). In particular, a 0.7 m concentration of sodium citrate activates both full-length recombinant PC9 and CARD-depleted recombinant PC9 (45, 46). Peptides A2.Y24G33, A3.F34V43, C1.L8E17, C4.R52I60, As1.D116R125, and As4.N155T166 did not inhibit the sodium citrate-dependent activation of PC9 (Fig. 5), and peptides Np1.L1379R1392 and N4.Q64V75 displayed partial inhibitory activity in this assay. This result suggests that six of the eight peptides identified as apoptosome inhibitors are specific to exclusively inhibit the CARD-dependent apoptosome activation of PC9.
FIGURE 5.
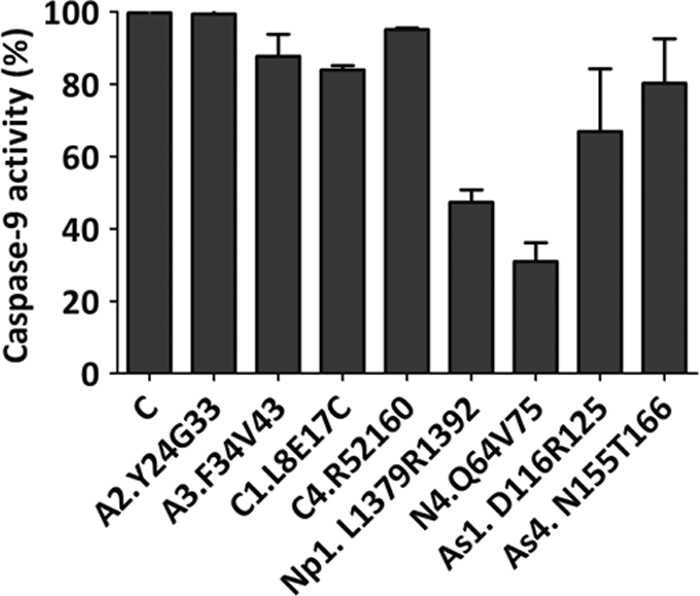
Apaf-1, PC9, and ASC CARD-derived peptides do not inhibit caspase-9 enzymatic activity. Caspase-9 preactivated in SC buffer was incubated with CARD-derived peptides (70 μm). Caspase-9 activity was determined in the presence of the Ac-LEHD-afc substrate. Data represent the inhibition percentages with reference to a control sample incubated in the absence of peptides. Data correspond to the mean ± S.D. (error bars) of three independent experiments.
Phage Display Screening to Identify Apaf-1 CARD-binding Peptides
We also evaluated phage display libraries to identify selective binding peptide ligands of the CARDs. We initially used the Apaf-1 CARD as a target and high diversity random libraries (12-mer, 7-mer, and 7-mer constrained) of around 2 × 109 different peptide sequences fused to a minor coat protein III from phage M13. The selection supposed an affinity interaction between the target and the peptides through the biopanning process. The recombinant Apaf-1 CARD was fixed to Ni2+ plates in order to use the His tag as an anchoring platform, which in turn, allowed a setting with a uniformly oriented protein. The most enriched selection was obtained from the 12-mer library. However, some interesting particularities were observed between peptides derived from the 7-mer and 7-mer constrained libraries. In a first global sequence analysis, we were able to identify a total of 37 different peptide sequences from the three independent screenings after three rounds of phage population enrichment. The peptide contingent was clustered according to sequence characteristics supported by analysis with the RELIC (receptor ligand contacts) bioinformatics server (47); specifically, the peptide motif identification programs (Motif 1 and Motif 2) and information associated with the observation of a peptide (Info program), which estimates the likelihood of random occurrence of sequence, were used. In order to test specific peptide binding to the Apaf-1 CARD, we performed a phage ELISA assay (48) using amplified individual phage clones. The phage-peptide binding to the Apaf-1 CARD was evaluated through anti-M13 antibodies. From the contingent phage-peptide population, only a few sequences interacted with the target with different affinities (results not shown), and a series of peptides were selected for chemical synthesis. This series included the most reacting linear sequences (WPTPPYA, ASLRTLTSLLPA, NFMESLPRLGMH, and AHLELRSNNMYF) and the constrained sequences cyclo-CSWFEASYC and cyclo-CLPTLHLLC. The peptides' biological activity was investigated in both the in vitro functional apoptosome assay and the mammalian cellular extract-based assay. Only the constrained peptide cyclo-CSWFEASYC exhibited moderate inhibitory activity in the two assays, whereas the linear sequence ASLRTLTSLLPA showed inhibitory activity in the in vitro assay but not in the cell extract-based assay (Table 3). The peptide cyclo-CSWFEASYC did not inhibit the sodium citrate-dependent activation of PC9 (results not shown).
TABLE 3.
Biological activity of synthetic peptides derived from Apaf-1 CARD phage display
| Peptide | Sequence | IC50a | IC50b |
|---|---|---|---|
| μm | μm | ||
| Phg1 | WPTPPYA | NIc | NI |
| Phg2 | ASLRTLTSLLPA | 92 ± 10 | NI |
| Phg3 | NFMESLPRLGMH | NI | NI |
| Phg4d | Cyclo-CSWFEASYC | 65 ± 7 | 90 ± 10 |
| Phg5d | Cyclo-CLPTLHLLC | NI | NI |
| Phg6 | AHLELRSNNMYF | NI | NI |
a Activity was analyzed in the in vitro reconstituted apoptosome.
b Activity was analyzed in a cell extract-based assay.
c NI, no inhibition found at the peptide concentration range analyzed.
d Peptides Phg4 and Phg5, which contain two Cys residues in the N and the C terminus, were cycled (see “Experimental Procedures”).
Biological Activity of CARD-derived Peptides in Cell-based Assays
Apaf-1-dependent apoptosome formation is required to induce apoptosis in response to the DNA-damaging agent cisplatin (CDDP) (15). We therefore studied the response of HeLa cells to CDDP in the presence of not only the CARD-derived apoptosome peptide inhibitors that were active in the two in vitro assays shown in Table 2 but also the most active peptide derived from the phage display approach (Table 3). In general terms, synthetic peptides have low cell permeability, and different strategies have been developed for the cytosolic delivery of peptides. Here we have used the lipid Lipofectamine as a vehicle for peptide delivery. After 24 h of treatment, caspase-3 activity was monitored by evaluating the enzymatic activity of the cell extract on the caspase-3 fluorogenic substrate Ac-DEVD-afc. The peptides (in particular C1.L8E17, As1.D116R125, As4.N155T166, and cyclo-CSWFEASYC) relevantly induced inhibition of CDDP apoptosome-mediated caspase-3 activity (Fig. 6).
FIGURE 6.
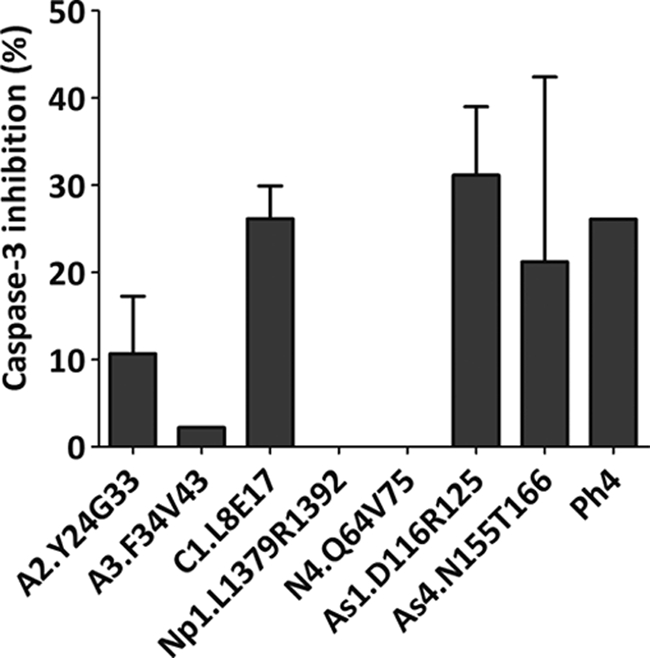
Inhibition of cisplatin-dependent apoptosis in HeLa cells by CARD-derived peptides. HeLa cells were treated with a Lipofectamine/peptide (100 μm) mixture for 4 h, followed by administration of 20 μm CDDP. Cells were harvested after 24 h, and cytosolic extracts were supplemented with caspase assay buffer containing 20 μm Ac-DEVD-afc substrate for caspase-3 activity evaluation. Data correspond to the mean ± S.D. (error bars) of three independent experiments and represent the inhibition percentages obtained with reference to a control sample incubated in the absence of peptides.
CARD-derived Peptides as Modulators of Procaspase-1-activating Inflammasome Complexes
In order to initially explore the influence of the CARD-related peptides on the inflammasome-mediated PC1 activation, we used a mammalian cellular extract-based assay (32). Cytosolic extracts from the human acute monocytic leukemia cell line (THP-1) were prepared, and inflammasome-dependent PC1-activating activity was induced by incubation at 37 °C in the presence of the different CARDs and CARD-derived peptides. NLRP-1 CARD and peptides A2.Y24G33, N3.Y49A60, Np1.L1379R1392, and As2.N128L136 inhibited more than 25% of inflammasome-mediated PC1 activation (Fig. 7).
FIGURE 7.

Inhibition of CARDs and CARD-derived peptides over procaspase-1-activating inflammasome complexes. CARD domains (5 μm) and CARD-derived peptides (100 μm) were incubated with THP-1 cell extracts. Caspase-1 activity was determined in the presence of the Ac-WEDH-afc substrate. Data represent the inhibition percentages with reference to a control sample incubated in the absence of peptides. Data correspond to the mean ± S.D. (error bars) of three independent experiments.
DISCUSSION
In this report, we initiated the in vitro evaluation of the role of polypeptides derived from CARD-containing proteins in the modulation of CARD-CARD-mediated protein-protein interactions. From the different CARD proteins, for the present study, we selected Apaf-1 and PC9, with a well characterized signaling role in the mitochondria-mediated pathway (intrinsic) of apoptosis (7, 49, 50) and NLRP-1 (NLR family, pyrin domain-containing 1; also named NALP-1) and NOD-1 (nucleotide-binding oligomerization domain-containing 1) as representative members of the inflammasome constituent NLR family of proteins (51). Protein ASC (apoptosis-associated speck-like protein containing a CARD), described as an adapter protein in assembly of different inflammasome complexes (52), was also included in this study. NLRP-1 and NOD-1 contain an N terminus and a C terminus CARD, respectively (53). ASC contains two structurally independent pyrin and CARDs (54). The NLR family of proteins is central for the regulation of immunity (51), and different studies have proposed cross-signaling interactions among immunity, inflammation, and apoptosis (4, 51, 55). The three recombinant CARDs selected for the study (Apaf-1 CARD, PC9 CARD, and NLRP-1 CARD; the recombinant CARDs from NOD-1 and ASC turned out to be insoluble under the experimental conditions required for our experimental settings) inhibited the Apaf-1-dependent activation of PC9 at low micromolar concentrations (Table 1), but only Apaf-1 CARD was able to in vitro activate PC9 in the absence of the Apaf-1 protein (Fig. 2). In contrast, only NLRP-1 CARD showed inhibitory activity against inflammasome-mediated PC1 activation (Fig. 7). These results suggest that mutual recognition of CARD-mediated inhibitory events can occur among the different CARD-containing proteins. However, the CARD-CARD interaction should induce additional conformational events among the protein partners to trigger their specific gain of function signaling purpose. The apoptosome is an efficient and selective multiprotein complex for the recruitment and processing of PC9. It has been proposed that the intracellular concentration of PC9 is a key determinant in activity regulation, given the apoptosome's greater affinity to bind, through CARD-CARD-dependent interactions, to PC9 than to processed caspase-9 (56). Furthermore, Stephanou et al. (57) have reported an antiapoptotic activity of the PC9 CARD when released from caspase-3-processed PC9. Such activity was initially thought to be based on a putative CARD-CARD-mediated interaction with the adaptor protein RICK and activation of NF-κB. Here we show that the antiapoptotic activity of PC9 CARD may also have a direct effect component because of its inhibitory activity on the apoptosome. When the intrinsic apoptosis pathway reaches a threshold of apoptosome- and caspase-9-dependent active caspase-3, this enzyme cleaves PC9, releasing the CARD, which would participate in the signaling process to slow down apoptosome activity when it is required. An early study has reported that NLRP-1 influenced the activity of the apoptosome when analyzed in cell extract-based experiments (58). It was found that full-length NLRP-1 enhanced the activity of the apoptosome, whereas the CARD of NLRP-1 had the opposite effect. Here we demonstrate the inhibitory effect of NLRP-1 CARD on an in vitro reconstituted apoptosome with an IC50 value close to those of Apaf-1 CARD and PC9 CARD. In contrast, we found that only Apaf-1 CARD, but not NLRP-1 CARD and PC9 CARD, were able to in vitro activate PC9 (Fig. 2). These results suggest a putative CARD-mediated or CARD-related regulation at the cross-talk point between cell death and inflammation. In defined cell types, it is possible that early episodes of cell death could be turned down to favor an inflammatory response. This could be initiated by an overexpression of inflammasome constituent proteins with a 2-fold aim: 1) conformation of the inflammasome platform to activate the maturation of proinflammatory cytokines (53) and 2) CARD-dependent inhibition of the apoptosome, followed by inhibition of cell death.
Defined synthetic peptides derived from the CARDs inhibited the PC9 Apaf-1 interaction. The apoptosome-related peptides A2.Y24G33, A3.F34V43 (from Apaf-1 CARD), C1.L8E17, and C4.R52I60 (from PC9 CARD), together with peptides derived from the respective basic patch of their CARDs (N4.Q64V75 (from NOD-1 CARD), Np1.L1379R1392 (from NLRP-1 CARD), and As1.D116R125 and As4.N155T166 (from ASC CARD)), displayed inhibitory activity. In contrast, those peptides derived from the acidic patch of the inflammasome-related protein CARDs and peptide Np4.R1422W1436 from the basic domain of NLRP-1 CARD did not inhibit the apoptosome activity (Table 2). Active peptides inhibited apoptosome activity in not only an in vitro apoptosome assay based on recombinant proteins but also in cell extract-based apoptosome assays (see Table 2 and Fig. 4). Furthermore, a subset of peptides exhibited biological activity in the cell-based assays as inhibitors of the CDDP-induced apoptosis (Fig. 6). The full set of peptides was also evaluated in a cellular extract-based inflammasome-mediated PC1 activation assay, where peptides A2.Y24G33, N3.Y49A60, Np1.L1379R1392, and As2.N128L136 showed inhibitory activity (Fig. 7). Although it would not be void of some uncertainties due to the inherent complexities associated with this first pass study on CARD-derived synthetic peptides, an attempt to perform an activity-based classification of the set of peptides analyzed would highlight that only peptide A2.Y24G33 (from Apaf-1 CARD) inhibits the apoptosome-mediated PC9 activation in all three apoptosome-related assays (in vitro, cell extract-based, and cell-based; see Table 2 and Fig. 6) as well as the inflammasome-mediated activation of PC1 (Fig. 7). Likewise, peptide Np1.L1379R1392 (from NLRP-1 CARD) was seen to be similarly active by inhibiting apoptosome activity both in vitro and in the cell extract-based assays (Table 2) and inflammasome activity (Fig. 7). At present, we cannot rule out the possibility that lack of inhibitory activity in the apoptosome cell-based assay can be ascribed to the low cell permeability of this particular peptide. Peptides N3.Y49A60 (from NOD-1 CARD) and As2.N128L136 (from ASC CARD) inhibited the inflammasome but were inactive as apoptosome inhibitors. Besides their own activity, these peptides could be valuable tools to drive the design of more pharmacologically relevant inhibitors, such as peptidomimetics or small molecules, of CARD-CARD-dependent multiprotein signaling complexes.
We also approached the identification of CARD-modulating peptides by exploring the phage display approach. After three selection rounds, RELIC-aided analysis of peptide sequences, and a further phage-ELISA-based selection, we obtained only one peptide with a moderate ability to inhibit the apoptosome activity in the three apoptosome-related assays (in vitro, cell extract-based, and cell-based), cyclo-CSWFEASYC (Table 3 and Fig. 6). In a future study, we will analyze the interaction site of these peptides with the Apaf-1 CARD, which should expose a binding site of low relevance for the CARD-CARD interaction.
The peptides described herein, together with those described by Marasco et al. (59) derived from the CARD of BCL10, are pioneering tools to interrogate not only the initial systems that inspired the design (the apoptosome and BCL10-interacting proteins, respectively) but also the complex and still poorly understood role of the CARD as signaling device. CARD-containing proteins exert relevant functions as scaffolds to build up key multiprotein complexes that amplify signaling in apoptosis, inflammation, and immunity (53, 60). Although other protein domain-dependent signaling pathways have been studied more and have helped draw pictures with cross-connections in the intricate cell signaling process (61), the contribution of CARD-dependent signaling complexes to the important decisions that cells, and probably tissues, have to make to prioritize apoptosis or inflammation is still in its initial stages and poses important open questions. The development of research tools, such as synthetic peptides derived from CARDs and the set up of reliable in vitro assays, will probably facilitate future research in this field.
Acknowledgments
We thank Alicia García-Jareño, Rebeca Montava, and Eliana Sirvent for technical assistance.
This work has been supported by Spanish Ministry of Science and Innovation (MICINN) Grants BIO2007-60066 and SAF2010 15512, Laboratorios Salvat, SA, Generalitat Valenciana Prometeo 2010/005 (partially funded with the European Regional Development Fund), and Consolider-Ingenio 2010 (MICINN Grant CSD2008-00005C) (to E. P.-P).
- CARD
- caspase recruitment domain
- TFE
- 2,2,2-trifluoroethanol
- NLR
- NOD-like receptor
- CDDP
- cis-diammineplatinum(II) dichloride
- afc
- 7-amino-4-trifluoromethylcoumarin.
REFERENCES
- 1. Seth R. B., Sun L., Ea C. K., Chen Z. J. (2005) Cell 122, 669-682 [DOI] [PubMed] [Google Scholar]
- 2. Hiscott J., Lin R., Nakhaei P., Paz S. (2006) Trends Mol. Med. 12, 53–56 [DOI] [PubMed] [Google Scholar]
- 3. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. (2009) Nature 458, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Inohara N., Koseki T., del Peso L., Hu Y., Yee C., Chen S., Carrio R., Merino J., Liu D., Ni J., Núñez G. (1999) J. Biol. Chem. 274, 14560–14567 [DOI] [PubMed] [Google Scholar]
- 5. Mahoney D. J., Cheung H. H., Mrad R. L., Plenchette S., Simard C., Enwere E., Arora V., Mak T. W., Lacasse E. C., Waring J., Korneluk R. G. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., Vucic D. (2007) Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 7. Acehan D., Jiang X., Morgan D. G., Heuser J. E., Wang X., Akey C. W. (2002) Mol. Cell 9, 423–432 [DOI] [PubMed] [Google Scholar]
- 8. Proell M., Riedl S. J., Fritz J. H., Rojas A. M., Schwarzenbacher R. (2008) PLoS One 3, e2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inohara N., Nuñez G. (2003) Nat. Rev. Immunol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 10. Park H. H., Lo Y. C., Lin S. C., Wang L., Yang J. K., Wu H. (2007) Annu. Rev. Immunol. 25, 561–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coussens N. P., Mowers J. C., McDonald C., Nuñez G., Ramaswamy S. (2007) Biochem. Biophys. Res. Commun. 353, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manon F., Favier A., Núñez G., Simorre J. P., Cusack S. (2007) J. Mol. Biol. 365, 160–174 [DOI] [PubMed] [Google Scholar]
- 13. Zhou P., Chou J., Olea R. S., Yuan J., Wagner G. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 11265–11270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malet G., Martín A. G., Orzáez M., Vicent M. J., Masip I., Sanclimens G., Ferrer-Montiel A., Mingarro I., Messeguer A., Fearnhead H. O., Pérez-Payá E. (2006) Cell Death Differ. 13, 1523–1532 [DOI] [PubMed] [Google Scholar]
- 15. Mondragón L., Galluzzi L., Mouhamad S., Orzáez M., Vicencio J. M., Vitale I., Moure A., Messeguer A., Perez-Paya E., Kroemer G. (2009) Apoptosis 14, 182–190 [DOI] [PubMed] [Google Scholar]
- 16. Mondragón L., Orzáez M., Sanclimens G., Moure A., Armiñán A., Sepúlveda P., Messeguer A., Vicent M. J., Pérez-Payá E. (2008) J. Med. Chem. 51, 521–529 [DOI] [PubMed] [Google Scholar]
- 17. Vicent M. J., Pérez-Payá E. (2006) J. Med. Chem. 49, 3763–3765 [DOI] [PubMed] [Google Scholar]
- 18. Caratù G., Allegra D., Bimonte M., Schiattarella G. G., D'Ambrosio C., Scaloni A., Napolitano M., Russo T., Zambrano N. (2007) Mol. Cell Proteomics 6, 333–345 [DOI] [PubMed] [Google Scholar]
- 19. Fenske S. A., Yesilaltay A., Pal R., Daniels K., Rigotti A., Krieger M., Kocher O. (2008) J. Biol. Chem. 283, 22097–22104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlegel B. P., Starita L. M., Parvin J. D. (2003) Oncogene 22, 983–991 [DOI] [PubMed] [Google Scholar]
- 21. Várnai P., Bondeva T., Tamás P., Tóth B., Buday L., Hunyady L., Balla T. (2005) J. Cell Sci. 118, 4879–4888 [DOI] [PubMed] [Google Scholar]
- 22. Day T. W., Safa A. R. (2009) Mini Rev. Med. Chem. 9, 741–748 [DOI] [PubMed] [Google Scholar]
- 23. Paddison P. J., Hannon G. J. (2002) Cancer Cell 2, 17–23 [DOI] [PubMed] [Google Scholar]
- 24. Zender L., Kubicka S. (2004) Apoptosis 9, 51–54 [DOI] [PubMed] [Google Scholar]
- 25. Rubinstein M., Niv M. Y. (2009) Biopolymers 91, 505–513 [DOI] [PubMed] [Google Scholar]
- 26. Scott J. K., Smith G. P. (1990) Science 249, 386–390 [DOI] [PubMed] [Google Scholar]
- 27. Orzáez M., Mondragón L., Marzo I., Sanclimens G., Messeguer A., Pérez-Payá E., Vicent M. J. (2007) Peptides 28, 958–968 [DOI] [PubMed] [Google Scholar]
- 28. Mora P., Mas-Moruno C., Tamborero S., Cruz L. J., Pérez-Payá E., Albericio F. (2006) J. Pept. Sci. 12, 491–496 [DOI] [PubMed] [Google Scholar]
- 29. Riedl S. J., Li W., Chao Y., Schwarzenbacher R., Shi Y. (2005) Nature 434, 926–933 [DOI] [PubMed] [Google Scholar]
- 30. Stennicke H. R., Salvesen G. S. (1999) Methods 17, 313–319 [DOI] [PubMed] [Google Scholar]
- 31. Fearnhead H. O. (2001) Methods Cell Biol. 66, 167–185 [DOI] [PubMed] [Google Scholar]
- 32. Gong Y. N., Wang X., Wang J., Yang Z., Li S., Yang J., Liu L., Lei X., Shao F. (2010) Cell Res. 20, 1289–1305 [DOI] [PubMed] [Google Scholar]
- 33. Palacios-Rodríguez Y., Gazarian T., Rowley M., Majluf-Cruz A., Gazarian K. (2007) J. Microbiol. Methods 68, 225–235 [DOI] [PubMed] [Google Scholar]
- 34. Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., Wang X. (1997) Cell 91, 479–489 [DOI] [PubMed] [Google Scholar]
- 35. Zou H., Henzel W. J., Liu X., Lutschg A., Wang X. (1997) Cell 90, 405–413 [DOI] [PubMed] [Google Scholar]
- 36. Saleh A., Srinivasula S. M., Acharya S., Fishel R., Alnemri E. S. (1999) J. Biol. Chem. 274, 17941–17945 [DOI] [PubMed] [Google Scholar]
- 37. Pérez-Payá E., Orzáez M., Mondragón L., Wolan D., Wells J. A., Messeguer A., Vicent M. J. (2011) Med. Res. Rev. 31, 649–675 [DOI] [PubMed] [Google Scholar]
- 38. Shiozaki E. N., Chai J., Shi Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 4197–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agopian A., Gros E., Aldrian-Herrada G., Bosquet N., Clayette P., Divita G., Caputo G. A., Litvinov R. I., Li W., Bennett J. S., Degrado W. F., Yin H. (2009) J. Biol. Chem. 284, 254–264 [DOI] [PubMed] [Google Scholar]
- 40. Churchill E. N., Qvit N., Mochly-Rosen D. (2009) Trends Endocrinol. Metab. 20, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walensky L. D., Kung A. L., Escher I., Malia T. J., Barbuto S., Wright R. D., Wagner G., Verdine G. L., Korsmeyer S. J. (2004) Science 305, 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeon S. W., Jeon Y. J., Hwang E. M., Kim T. Y. (2007) Peptides 28, 838–844 [DOI] [PubMed] [Google Scholar]
- 43. Qin H., Srinivasula S. M., Wu G., Fernandes-Alnemri T., Alnemri E. S., Shi Y. (1999) Nature 399, 549–557 [DOI] [PubMed] [Google Scholar]
- 44. Pérez-Payá E., Houghten R. A., Blondelle S. E. (1994) Biochem. J. 299, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J. E., Edris W. A., Sutherlin D. P., Green D. R., Salvesen G. S. (2003) Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 46. Pop C., Timmer J., Sperandio S., Salvesen G. S. (2006) Mol. Cell 22, 269–275 [DOI] [PubMed] [Google Scholar]
- 47. Mandava S., Makowski L., Devarapalli S., Uzubell J., Rodi D. J. (2004) Proteomics 4, 1439–1460 [DOI] [PubMed] [Google Scholar]
- 48. Serasinghe M. N., Seneviratne A. M., Smrcka A. V., Yoon Y. (2010) J. Biol. Chem. 285, 620–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu Y., Benedict M. A., Ding L., Núñez G. (1999) EMBO J. 18, 3586–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu X., Acehan D., Ménétret J. F., Booth C. R., Ludtke S. J., Riedl S. J., Shi Y., Wang X., Akey C. W. (2005) Structure 13, 1725–1735 [DOI] [PubMed] [Google Scholar]
- 51. Ting J. P., Willingham S. B., Bergstralh D. T. (2008) Nat. Rev. Immunol. 8, 372–379 [DOI] [PubMed] [Google Scholar]
- 52. Ippagunta S. K., Malireddi R. K., Shaw P. J., Neale G. A., Walle L. V., Green D. R., Fukui Y., Lamkanfi M., Kanneganti T. D. (2011) Nat. Immunol. 12, 1010–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schroder K., Tschopp J. (2010) Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 54. de Alba E. (2009) J. Biol. Chem. 284, 32932–32941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bruey J. M., Bruey-Sedano N., Luciano F., Zhai D., Balpai R., Xu C., Kress C. L., Bailly-Maitre B., Li X., Osterman A., Matsuzawa S., Terskikh A. V., Faustin B., Reed J. C. (2007) Cell 129, 45–56 [DOI] [PubMed] [Google Scholar]
- 56. Malladi S., Challa-Malladi M., Fearnhead H. O., Bratton S. B. (2009) EMBO J. 28, 1916–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stephanou A., Scarabelli T. M., Knight R. A., Latchman D. S. (2002) J. Biol. Chem. 277, 13693–13699 [DOI] [PubMed] [Google Scholar]
- 58. Chu Z. L., Pio F., Xie Z., Welsh K., Krajewska M., Krajewski S., Godzik A., Reed J. C. (2001) J. Biol. Chem. 276, 9239–9245 [DOI] [PubMed] [Google Scholar]
- 59. Marasco D., Stilo R., Sandomenico A., Monti S. M., Tizzano B., de Capua A., Varricchio E., Liguoro D., Zotti T., Formisano S., Ruvo M., Vito P. (2009) Biochem. J. 422, 553–561 [DOI] [PubMed] [Google Scholar]
- 60. Bouchier-Hayes L., Martin S. J. (2002) EMBO Rep. 3, 616–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith F. D., Scott J. D. (2002) Curr. Biol. 12, R32–40 [DOI] [PubMed] [Google Scholar]