

Background: The EGF receptor exhibits negative cooperativity in ligand binding.
Results: Mutation of the most membrane-proximal portion of the intracellular domain abrogates negative cooperativity.
Conclusion: Residues 645–665 of the EGF receptor are involved in the genesis of negative cooperativity.
Significance: These data demonstrate how alterations of the intracellular domain of the EGF receptor can lead to changes in ligand binding by the extracellular domain.
Keywords: Allosteric Regulation, Cooperativity, Epidermal Growth Factor (EGF), Epidermal Growth Factor Receptor (EGFR), Ligand-binding Protein, Tyrosine Protein Kinase (Tyrosine Kinase)
Abstract
The binding of EGF induces dimerization of its receptor, leading to the stimulation of its intracellular tyrosine kinase activity. Kinase activation occurs within the context of an asymmetric dimer in which one kinase domain serves as the activator for the other kinase domain but is not itself activated. How ligand binding is related to the formation and dynamics of this asymmetric dimer is not known. The binding of EGF to its receptor is negatively cooperative—that is, EGF binds with lower affinity to the second site on the dimer than to the first site on the dimer. In this study, we analyzed the binding of 125I-EGF to a series of EGF receptor mutants in the intracellular juxtamembrane domain and demonstrate that the most membrane-proximal portion of this region plays a significant role in the genesis of negative cooperativity in the EGF receptor. The data are consistent with a model in which the binding of EGF to the first site on the dimer induces the formation of one asymmetric kinase dimer. The binding of EGF to the second site is required to disrupt the initial asymmetric dimer and allow the formation of the reciprocal asymmetric dimer. Thus, some of the energy of binding to the second site is used to reorient the first asymmetric dimer, leading to a lower binding affinity and the observed negative cooperativity.
Introduction
The EGF receptor is a classical receptor tyrosine kinase with an extracellular ligand-binding domain and an intracellular kinase domain connected by a single transmembrane α-helical segment (1). Binding of an agonist ligand leads to the activation of the intracellular tyrosine kinase domain and autophosphorylation of the EGF receptor on its C-terminal tail. The phosphorylated tyrosines then serve as docking sites for the binding of SH2- and PTB domain-containing proteins that mediate the intracellular effects of the growth factor (2–4).
X-ray crystallography has shown that the unliganded EGF receptor exists as a monomer held in a closed conformation via an intramolecular tether (5). Binding of ligand releases this tether and allows the receptor to adopt an open or extended conformation. This open form of the receptor interacts with another open receptor monomer to form a back-to-back receptor dimer (6, 7). Inactive receptor “predimers” that may arise from interactions between the intracellular kinase domains have also been observed (8–18).
Ligand-induced dimerization of the extracellular domains of two EGF receptors induces the activation of their intracellular kinase domains. Zhang et al. (19) have shown that the EGF receptor kinase is activated by the formation of an asymmetric kinase dimer. In this dimer, the C-lobe of the activator kinase interacts with the N-lobe of the receiver kinase. This results in the activation of the receiver kinase, which then phosphorylates the C-terminal tail of the activator kinase.
Crystal structures of this asymmetric dimer show that residues ∼665–682 of the cytoplasmic domain, just N-terminal to the kinase domain, contribute significantly to the dimer interface. In the receiver kinase, this juxtamembrane segment forms a cradle around the C-lobe of the activator kinase (20), a structure that has been referred to as the juxtamembrane latch (18). Mutations in this juxtamembrane segment, such as the L680N mutation, result in a nearly complete loss of EGF-stimulated kinase activity in the mutant receptor (19). Thus, this juxtamembrane latch appears to be crucial for activation of the kinase domain.
The most membrane-proximal residues of the intracellular juxtamembrane domain (∼residues 645–665) form an α-helix in the available crystal structure (20). Jura et al. (18) used a combination of mutational analysis and NMR structural determination of soluble peptides corresponding to this region to suggest that this portion of the juxtamembrane domain forms an anti-parallel helical dimer, referred to as the juxtamembrane clasp. Mutation of residues in this region led to a decrease in the ability of EGF to activate the tyrosine kinase, suggesting that, like the juxtamembrane latch, this helical segment may contribute to the stabilization of the asymmetric dimer (18).
We have previously probed the dimerization of the EGF receptor using 125I-EGF radioligand binding studies (21–23). We have shown that the binding of EGF to the dimeric form of the receptor is negatively cooperative (22). EGF binds with high affinity to the first site on the dimer but with substantially lower affinity to the second site on the dimer. Binding studies on nested C-terminal truncations of the EGF receptor suggested that the intracellular juxtamembrane domain, but not the kinase domain, was necessary for the maintenance of this negative cooperativity (23).
In this report, we use site-directed mutagenesis to further examine the role of the intracellular juxtamembrane domain in the genesis of negative cooperativity in the EGF receptor. Our data suggest that interactions mediated by residues in and immediately adjacent to the membrane-proximal helical region of this domain are the main contributors to the establishment of negative cooperativity in the EGF receptor. They suggest a model of EGF receptor kinase activation in which the binding of EGF to the first subunit of the receptor dimer leads to the formation of an asymmetric dimer and the activation of the first kinase domain. This involves stabilizing interactions mediated by both the juxtamembrane latch and the juxtamembrane clasp. Ligand binding to the second site disrupts the juxtamembrane clasp, allowing the formation of the reciprocal asymmetric dimer and activation of the second kinase domain. Because a fraction of the energy derived from ligand binding to the second site on the dimer is used to reorient the asymmetric dimer, binding affinity is reduced compared with the first site on the dimer, giving rise to the observed negative cooperativity.
EXPERIMENTAL PROCEDURES
DNA Constructs and Cell Lines
The EGF receptor mutants (R656G,R657G-EGFR, E661A,E663A,E666A-EGFR, T654A-EGFR, T669A-EGFR, and T669D,S671D-EGFR) were generated in the pcDNA5/FRT vector using QuikChange mutagenesis. Once the mutations were confirmed by sequencing, the EGF receptor was transferred to the pBI-Tet vector using the NheI and EcoRV sites. The V665M-EGF receptor and the Δ648–662-EGF receptor were the generous gift of Dr. Graham Carpenter (Vanderbilt University) and were moved into the pBI-Tet vector. The EGF receptor mutants in the pBI-Tet vector were cotransfected with pTK-Hyg (Clontech) into Tet-on CHO-K1 cells (Clontech) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Stable clones were selected by growth in 500 μg/ml hygromycin.
The T669D, T669E, and T669R point mutations were generated in pcDNA 3.1 and moved into pcDNA5/FRT. The constructs were transfected into Flp-in CHO cells (Invitrogen), and stable lines were selected by growth in 500 μg/ml ZeocinTM (Invitrogen).
125I-EGF Radioligand Binding Assays
EGF was purchased from Biomedical Technologies (Stoughton, MA). 125I-EGF was synthesized using the ICl method of Doran and Spar (24). The cells were plated into 6-well dishes 48 h prior to assay and grown in DMEM supplemented with 10% Fetalplex (Gemini Bio-Products, West Sacramento, CA), penicillin/streptomycin, and the desired concentration of doxycycline. For assay, the cells were incubated overnight at 4 °C in Ham's F-12 medium containing 25 mm HEPES, pH 7.2, 5 mg/ml bovine serum albumin, 40 pm 125I-EGF, and increasing concentrations of unlabeled EGF. At the end of the incubation, the cells were washed three times in ice-cold phosphate-buffered saline, and the monolayers were dissolved in 1 m NaOH and counted in a Beckman gamma counter. All of the assays were done in triplicate.
The data were analyzed using GraphPad Prism 4.0 as described previously (22). Nonspecific binding was determined by fitting the data to the Prism equation for competition binding and was subtracted from all data points. Data from all the binding isotherms from a single mutant were globally fit to the following equation,
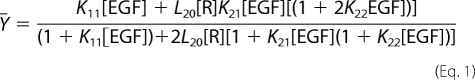 |
where Ȳ= is the fractional saturation of receptor with ligand, and [R] is the concentration of unoccupied EGF receptors (25). This can be calculated from the following equation,
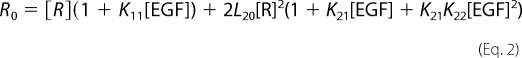 |
where R0 = total concentration of EGF receptors as derived by Wyman and Gill (26).
Receptor Kinase Assays
The cells were plated into 6-well dishes 48 h prior to use and grown in DMEM supplemented with 10% Fetalplex and penicillin/streptomycin. For assay, the cells were transferred to Ham's F-12 medium containing 25 mm HEPES, pH 7.2, and 1 mg/ml bovine serum albumin and incubated at 37 °C. The indicated concentration of EGF was then added for 5 min. At the end of the incubation, the medium was aspirated, and the cells were washed with ice-cold phosphate-buffered saline. Radioimmune precipitation assay buffer was added to each well, and lysates were prepared and centrifuged. Equal amounts of protein were analyzed by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride. After blocking with powdered milk, the membranes were probed with antibodies to phosphotyrosine (BD Biosciences), phospho-Thr-654 (Santa Cruz Biotechnology), phospho-Thr-669 (Santa Cruz Biotechnology), or the EGF receptor (Cell Signaling Technology). Antibody binding was detected by chemiluminescence.
Phorbol 12-Myristate 13-Acetate Treatments
For binding assays, the cells were chilled to 4 °C and treated with 100 nm PMA3 (Sigma) for 30 min prior to switching the cells to standard binding medium containing 125I-EGF and 100 nm PMA in a final concentration of 0.01% Me2SO. Controls contained 0.01% Me2SO. Binding assays were incubated overnight at 4 °C and processed as usual. For kinase assays, the cells were treated overnight at 4 °C with 100 nm PMA and stimulated with the indicated concentrations of EGF for 5 min at 4 °C.
RESULTS
EGF exhibits a heterogeneous binding affinity for its receptor as evidenced by curvilinear Scatchard plots (27–30). By carrying out 125I-EGF radioligand binding assays in cells expressing increasing levels of EGF receptors, we have shown that this heterogeneity is due to the existence of negative cooperativity in EGF receptor dimers (22). Fig. 1A shows our model for the binding of EGF in a dimerizing system.
FIGURE 1.

Ligand binding to the EGF receptor. A, model for the binding of EGF to its receptor in a dimerizing system. The circles represent EGF receptor subunits. E represents a molecule of EGF. L20, K11, K21, and K22 refer to association constants for the indicated equilibria. Units for the three binding constants (K11, K21, and K22) are m−1. The unit for L20 is D−1 (mol/dm2), which represents the surface density of the receptor (22). B, 125I-EGF binding to the wild type EGF receptor. CHO cells expressing increasing levels of wild type EGF receptor were subjected to 125I-EGF radioligand binding as described under “Experimental Procedures.” The data were globally fit to the equation for binding in a dimerizing system as described. The fitted parameters are given in the inset.
In this model, there is a pre-existing equilibrium between unoccupied monomers and unoccupied dimers. EGF can thus bind to three different states of the receptor: the monomer, the first site on the dimer, or the second site on the dimer. The position of the monomer-dimer equilibrium is dependent on the number of EGF receptors expressed in a cell. At low levels of receptor, the monomeric form will be favored, whereas at high levels of receptor expression, the dimer will be favored. As a result, if the affinity of EGF for the monomer is different from its affinity for the dimer, then the position of the saturation binding isotherm for EGF will shift as the concentration of cell surface receptors increases. An example of this is shown in Fig. 1B for the binding of 125I-EGF to CHO cells expressing wild type EGF receptors.
As can be seen from the figure, the saturation binding isotherms shift from left to right with increasing levels of EGF receptor expression. Global modeling of all the binding curves to the equation describing binding in this dimerizing system (see “Experimental Procedures”) yields fitted values for the equilibrium association constants. As can be seen from the values reported in the inset in Fig. 1B, EGF binds with higher affinity to the first site on the dimer (K21 = 1.5 × 109, corresponding to a KD of 660 pm) than to the second site on the dimer (K22 = 3 × 108, corresponding to a KD of 3.3 nm). This is classic negative cooperativity.
Mutations in the Intracellular Juxtamembrane Domain
We have previously shown that the intracellular juxtamembrane domain is required for negative cooperativity (23). This part of the EGF receptor has been roughly divided into two regions termed JM-A, which includes residues 645–664, and JM-B, which includes residues 665–682 (18). In the crystal structure of the asymmetric dimer (20), the JM-A segment appears as a helix oriented away from the kinase domains (Fig. 2A). However, NMR studies and mutational analyses suggest that this region may form an anti-parallel helical dimer that functions as a clasp to stabilize the asymmetric dimer (18). To assess the contribution of the JM-A domain to negative cooperativity in the EGF receptor, we made an internal deletion of this region (Δ648–662) as well as several point mutations as outlined in Fig. 2B. The effect of these mutations on the kinase and binding activity of the EGF receptor is shown in Fig. 3.
FIGURE 2.

The juxtamembrane domain of the EGF receptor. A, structure of the asymmetric EGF receptor kinase dimer (Protein Data Bank number 3GOP) from Brewer et al. (20). The positions of key residues mutated in this work are indicated. The arrowhead indicates the position of Leu-680. B, mutations in the JM-A and JM-B domains used in these studies.
FIGURE 3.

Kinase and 125I-EGF binding in JM-A domain mutants. A, autophosphorylation of wild type, Δ648–662-EGFR, E661,663,666A-EGFR, and R656,657G-EGFR. CHO cells expressing the indicated mutant were incubated with the indicated concentration of EGF for 5 min. Receptor autophosphorylation was determined following SDS-polyacrylamide gel electrophoresis and Western blotting with antibodies against phosphotyrosine (pTyr) and the EGF receptor. B–D, 125I-EGF binding to CHO cells expressing Δ648–662-EGF receptor (B), E661,663,666A-EGF receptor (C), or the R656,657G-EGF receptor (D). Binding studies and data analysis were done as described under “Experimental Procedures.” The fitted parameters are given in the insets.
As reported previously (31), deletion of essentially the entire JM-A domain, residues 648–662, resulted in the complete loss of EGF-stimulated receptor autophosphorylation (Fig. 3A). The effect of this mutation on ligand binding was similarly stark (Fig. 3B). Despite a greater than 100-fold change in the level of Δ648–662-EGF receptor expression, the binding isotherms did not change position and were best fit to the equation for binding to a single class of sites. This result indicates that deletion of the JM-A region abrogates negative cooperativity in the EGF receptor.
In the proposed JM-A anti-parallel helical dimer (18), a number of salt bridges between acidic and basic residues can form that would stabilize this helical clasp. To determine whether such interactions might play a role in negative cooperativity in the EGF receptor, a triple point mutant was generated that replaced all three glutamic acid residues in this region with alanines. This mutant is the E661A, E663A, E666A-EGF receptor.
The E661A,E663A,E666A-EGF receptor was expressed in CHO cells and assayed for receptor autophosphorylation. As shown in Fig. 3A, EGF-stimulated receptor autophosphorylation was severely compromised in this mutant. The level of receptor phosphorylation was only ∼20% of that observed in the wild type receptor. The E661A,E663A,E666A-EGF receptor also showed major changes in its ligand binding properties (Fig. 3C). Although the saturation binding isotherms shifted with increasing levels of receptor expression, the shift was from right to left, the opposite direction from that seen for the wild type receptor. Global fitting of the binding curves yielded fitted values for the equilibrium constants showing that the affinity of EGF for the second site on the dimer was increased compared with the wild type receptor, and in fact, K22 was slightly greater than K21 in this receptor mutant. Thus, this triple point mutation abolishes negative cooperativity in the EGF receptor, making it easier for EGF to bind to the second site on the dimer.
The binding isotherms for the E661A,E663A,E666A-EGF receptor shift from right to left because of the continued presence of a limited amount of positive linkage in this mutant. Positive linkage refers to the situation in which the affinity for the dimeric form is higher than the affinity for the monomeric form. Under these conditions, the binding of ligand induces the assembly of receptor dimers. We have previously shown that the kinase-dead, K721A-EGF receptor exhibits positive linkage and, correspondingly, a shift of its binding isotherms from right to left with increasing receptor levels (23). Positive linkage is almost certainly present in the wild type receptor but is masked by changes in receptor affinity caused by phosphorylation of the receptor (23). The kinase-dead K721A-EGF receptor and the highly kinase-impaired L680N-EGF receptor both retain negative cooperativity in ligand binding (22, 23). Thus, the decreased kinase activity observed in the Δ648–662- and E661A,E663A,E666A-EGF receptors is not responsible for their loss of negative cooperativity.
Jura et al. (18) reported that a mutation in the JM-A domain, R656G,R657G, led to a significant loss in EGF-stimulated kinase activity. They speculated that this was due to the weakening of the juxtamembrane helix and hence the helical dimer that helps to stabilize the asymmetric dimer. As shown in Fig. 3A, receptor autophosphorylation was only slightly reduced in this mutant, reaching approximately two-thirds of the level of phosphorylation seen in the wild type receptor. Consistent with this limited change in kinase activity, the R656G,R657G-EGF receptor showed only modest differences from the wild type receptor in terms of ligand binding properties (Fig. 3D). The EGF binding isotherms shifted from left to right as the level of EGF receptor expression increased from 15,000 receptors/cell to ∼1 million receptors/cell. The fitted parameters for all four equilibrium constants were fairly similar to those observed in cells expressing wild type receptors: in particular, K21 > K22, indicating the continued presence of negative cooperativity in this mutant. Thus, this mutation led to only modest effects on binding and kinase activity, suggesting that these residues play a relatively minor role in determining negative cooperativity.
Brewer et al. (20) recently showed that the V665M mutation of the EGF receptor, at the junction of JM-A and JM-B domains, led to ligand-independent phosphorylation of the EGF receptor and enhanced its transforming activity. Based on the crystal structure of the asymmetric kinase dimer, they proposed that this mutation stabilized the asymmetric dimer because the larger side chain of methionine packed more effectively into a hydrophobic pocket on the C-lobe of the activator kinase than did the smaller side chain of valine.
To determine the effect of stabilization of the asymmetric dimer on EGF binding, the V665M-EGF receptor was expressed in CHO cells, and analyses of its kinase and radioligand binding properties were performed. The results are shown in Fig. 4. As observed by Brewer et al. (20), the V665M mutation resulted in significant ligand-independent kinase activity of the EGF receptor that was further enhanced by the addition of EGF (Fig. 4A). In 125I-EGF binding experiments (Fig. 4B), the saturation binding isotherms showed the expected shift from left to right with increasing levels of EGF receptor. However, global fitting of the data from all of the curves revealed that there was a striking decrease of ∼2 orders of magnitude in the value of K22. Thus, stabilizing the asymmetric dimer with this mutation enhances negative cooperativity, making it more difficult for EGF to bind to the second site on the dimer.
FIGURE 4.

Kinase and 125I-EGF binding in the V665M-EGF receptor. A, CHO cells expressing wild type or V665M-EGF receptors were stimulated with the indicated concentration of EGF for 5 min. Receptor autophosphorylation was assessed by Western blotting with antibodies against phosphotyrosine (pTyr) and the EGF receptor. B, 125I-EGF binding to CHO cells expressing increasing levels of the V665M-EGF receptor. Binding studies and data analysis were done as described under “Experimental Procedures.” The fitted parameters are given in the inset.
Mutation of the Sites of Threonine Phosphorylation in the Juxtamembrane Domain
Thr-654 in the JM-A segment is the site of phosphorylation of the EGF receptor by protein kinase C (32). The activity of protein kinase C can be stimulated by treatment of cells with PMA (33). Fig. 5A shows the effect of PMA pretreatment on the binding and kinase activity of cells expressing the wild type EGF receptor. In untreated cells, the 125I-EGF binding isotherm exhibited 50% saturation at ∼0.9 nm EGF. After pretreatment with PMA, the binding curve was significantly shifted to the right (p < 0.001), with 50% saturation occurring at ∼1.5 nm EGF. As shown in the inset of the figure, under normal conditions, the EGF receptor is not phosphorylated on Thr-654. However, treatment of the cells with PMA led to the constitutive phosphorylation of the EGF receptor at this site. This phosphorylation was associated with a significant decrease in the ability of EGF to stimulate the autophosphorylation of its receptor but no change in EGF receptor levels. Thus, as has been reported previously, phosphorylation of the EGF receptor on Thr-654 results in a decrease in ligand binding affinity and reduced EGF-stimulated receptor autophosphorylation (28, 30).
FIGURE 5.

Effect of phosphorylation of Thr-654 on ligand binding affinity and kinase activity. A, CHO cells expressing wild type EGF receptors were treated without or with 100 nm PMA prior to and during 125I-EGF binding assays. The assays were processed, and the data were analyzed as outlined under “Experimental Procedures.” Inset, CHO cells expressing wild type EGF receptors were treated without or with 100 nm PMA overnight at 4 °C and then stimulated with the indicated concentrations of EGF for 5 min at 4 °C. The lysates were prepared and analyzed by Western blotting with antibodies against phosphotyrosine, phosphothreonine 654, and the EGF receptor. B, same as A except that the studies were performed on cells expressing T654A-EGF receptors.
These effects of PMA on the binding and kinase activity of the EGF receptor were abolished in the nonphosphorylatable T654A-EGF receptor. Treatment of cells expressing the T654A-EGF receptor with PMA failed to alter the position of the 125I-EGF binding isotherm (Fig. 5B). As expected, there was no detectable phosphorylation at Thr-654, and the ability of EGF to stimulate receptor autophosphorylation was similar in control and PMA-treated cells (Fig. 5B, inset). Thus, the effects of PMA on the EGF receptor appear to be consequences of the phosphorylation of Thr-654.
Although the T654A mutation completely ablated the effects of PMA on the EGF receptor, it did not affect the overall binding and kinase properties of the EGF receptor. As shown in Fig. 6A, the binding isotherms of cells expressing the T654A-EGF receptor shifted from left to right with increasing levels of EGF receptor expression. Global fitting of the data yielded parameters very similar to those seen for the wild type receptor, with the continued presence of negative cooperativity. Likewise, the ability of EGF to stimulate receptor autophosphorylation was not different in cells expressing wild type or T654A-EGF receptors (Fig. 6B). A low level of ligand-independent receptor autophosphorylation was consistently observed in the T654A-EGF receptor, suggesting that phosphorylation at this site may provide some suppression of basal receptor kinase activity. However, the lack of phosphorylation at this site in the absence of PMA treatment (Fig. 5A) indicates that under the conditions of our assays, phosphorylation at this site is limited.
FIGURE 6.

Role of Thr-654 in the regulation of ligand binding. A, 125I-EGF binding to cells expressing increasing levels of the T654A-EGF receptor. Binding assays and data analyses were done as described under “Experimental Procedures.” The fitted parameters are given in the inset. B, cells expressing either wild type or T654A-EGF receptors were treated with the indicated concentrations of EGF for 5 min. Receptor autophosphorylation was assessed by Western blotting with antibodies against phosphotyrosine (pTyr) and the EGF receptor. C, 125I-EGF binding to cells expressing wild type EGF receptors treated concomitantly with 100 nm PMA to induce phosphorylation of Thr-654. Binding studies and data analyses were done as described under “Experimental Procedures.” The fitted parameters are given in the inset.
In contrast to the results obtained with the nonphosphorylatable T654A-EGF receptor, constitutive phosphorylation of Thr-654 by pretreatment of the cells with PMA led to marked changes in the 125I-EGF saturation binding isotherms. As shown in Fig. 6C, in cells expressing wild type EGF receptors treated with PMA, all of the binding isotherms collapsed to a single position regardless of how many EGF receptors were present in the cells. The data were best fit by the equation for binding to a single class of sites with a shared KA = 6 × 108 m−1 (KD ≈ 1.6 nm). These data demonstrate that phosphorylation of the EGF receptor at Thr-654 abolishes both linkage and cooperativity in the EGF receptor and strongly implicate the JM-A region in the genesis of negative cooperativity.
Thr-669 is present in the JM-B domain that comprises the juxtamembrane latch and is the site of phosphorylation of the EGF receptor by MAP kinase (34). This phosphorylation is associated with desensitization of the EGF receptor (35). This is likely due to phosphorylation-induced destabilization of the juxtamembrane latch (20). To assess the role of this residue in regulating EGF receptor binding and kinase activity, several different single and double point mutations were made at this position and assayed for their effect on EGF-stimulated receptor autophosphorylation. The results are shown in Fig. 7.
FIGURE 7.

Role of Thr-669 in the regulation of kinase activity. A, CHO cells expressing wild type, T669A-, T669R-, T669D-, or T669E-EGF receptors were stimulated with the indicated doses of EGF for 5 min. Receptor autophosphorylation, phosphorylation at Thr-669, and EGF receptor level were assessed by Western blotting with the appropriate antibodies. B, CHO cells expressing wild type or the T669D,S671D-EGFR double point mutant were stimulated with the indicated doses of EGF for 5 min. Receptor autophosphorylation and EGF receptor level were assessed by Western blotting.
As shown in Fig. 7A, in the wild type EGF receptor, EGF stimulated receptor autophosphorylation, as well as phosphorylation on Thr-669. Replacement of Thr-669 with an alanine residue led to an increase in EGF receptor autophosphorylation. This is the expected result because this substitution precludes phosphorylation at Thr-669 and blocks desensitization of the receptor. This leads to enhanced kinase activity. If EGF-induced phosphorylation at Thr-669 alters the ligand binding properties of the receptor, then substitution of the threonine with the nonphosphorylatable alanine should result in a change in the characteristics of EGF binding. However, as shown in Fig. 8A, the T669A mutation did not significantly alter the binding properties of the receptor compared with those of the wild type receptor. The curves shifted from left to right with increasing receptor levels, and the fitted equilibrium constants were similar to those seen in the wild type receptor. In particular, the receptor exhibited normal levels of negative cooperativity (K21 > K22). This suggests that phosphorylation at Thr-669 does not modulate EGF binding.
FIGURE 8.

Role of Thr-669 phosphorylation on ligand binding by the EGF receptor. A, 125I-EGF binding to CHO cells expressing increasing levels of the T669A-EGF receptor. B, 125I-EGF binding to CHO cells expressing increasing levels of the T669D,S671D-EGF receptor. Binding studies and data analysis were done as described under “Experimental Procedures.” The fitted parameters are given in the insets.
To further examine the role of Thr-669 phosphorylation in the regulation of ligand binding properties, the threonine was substituted with potentially phosphomimetic aspartic acid or glutamic acid. As shown in Fig. 7A, these substitutions actually increased receptor autophosphorylation as did replacement of the threonine with the positively charged arginine residue. Thus, single point mutations of Thr-669 appear to enhance kinase activity and hence fail to reproduce the desensitizing effect of phosphorylation at this site.
A phosphate group carries significantly more charge than a carboxyl group. Thus, substitution of a single threonine with an acidic amino acid may not adequately recapitulate the effects of phosphorylation. We therefore turned to a double point mutation to mimic phosphorylation of the EGF receptor at this site. Heisermann and Gill (36) have reported that Thr-669, as well as Ser-671, is a site of phosphorylation of the EGF receptor. Therefore, we generated and stably expressed the double point mutant, T669D,S671D-EGF receptor and examined both its kinase activity and ligand binding properties.
As shown in Fig. 7B, maximal receptor autophosphorylation was reduced by approximately one-third in the T669D,S671D-EGF receptor as compared with the wild type EGF receptor. Thus, unlike the single point mutants, the double point mutation does recapitulate the desensitizing effect of phosphorylation of Thr-669 on EGF receptor kinase activity. Despite the significant change in EGF-stimulated kinase activity in this double point mutant, the binding of EGF was not significantly altered (Fig. 8B). This suggests that phosphorylation of this site is unlikely to be associated with changes in the ligand binding properties of the EGF receptor.
DISCUSSION
We have previously shown that the binding of EGF to its receptor is negatively cooperative (22) and that the intracellular juxtamembrane domain is required for this allosteric regulation of ligand binding (23). The studies reported here were designed to more precisely define the region of the juxtamembrane domain that contributes to negative cooperativity in EGF binding.
Internal deletion of the JM-A region (residues 645–664) resulted in the complete loss of all cooperativity in EGF binding, suggesting a role for this portion of the juxtamembrane domain in determining the ligand binding properties of the receptor. NMR studies of a peptide corresponding to two tandem copies of this region suggest that it can form an anti-parallel helical dimer (18). In this dimer, Glu-663 and Glu-666 are predicted to be involved in interhelical salt bridges that would stabilize the helical dimer and lock its C-terminal end against the C-lobe of the activator kinase. Our finding that negative cooperativity is abrogated when these ionic interactions are removed (the E661A,E663A,E666A-EGFR) suggests that the proposed helical dimer could contribute to negative cooperativity in the EGF receptor. Contrary to the findings of Jura et al. (18), we noted only a modest effect of the R656G,R657G mutation on kinase activity. The difference between our results and theirs may be due to the fact that our studies were done using stable transfectants, whereas Jura et al. (18) used transient transfectants. The lack of effect of the R656G,R657G mutation on the ligand binding properties of the EGF receptor is consistent with its limited effect on kinase activity and suggests that these residues are not particularly important to the function of the juxtamembrane domain.
It has long been recognized that phosphorylation of the EGF receptor on Thr-654 leads to a decrease in the affinity of EGF and a loss of EGF-stimulated receptor autophosphorylation (28, 30, 37, 38). Substitution of Thr-654 with alanine did not alter the ligand binding properties of the EGF receptor. This may be due to the fact that under the conditions of our assay, the EGF receptor is not phosphorylated on this residue. Thus, the mutation would not substantially change the properties of the helix. By contrast, the PMA-induced phosphorylation of this site led to the complete loss of cooperativity as assessed in our binding assays. The fact that a physiologically relevant site of regulatory phosphorylation of the EGF receptor is located in the JM-A domain and is associated with changes in ligand binding affinity supports the conclusion that the JM-A domain represents an important structural component underlying negative cooperativity in the EGF receptor. Our previous observation that substitution of residues 647 and 650 with cysteines to allow palmitoylation of these sites also abolishes negative cooperativity (23, 39) further supports a role for the JM-A in determining negative cooperativity.
The V665M mutation in the EGF receptor lies immediately C-terminal to the JM-A helix. This mutation results in a receptor that exhibits ligand-independent kinase activity (20). Analysis of the ligand binding properties of this receptor demonstrated that although the binding affinity of EGF for the monomer and the first site on the dimer did not differ significantly from that seen in the wild type receptor, the affinity of EGF for the second site on the dimer was ∼2 orders of magnitude lower than that seen in the wild type receptor. Thus, this mutation, which is thought to stabilize the asymmetric dimer, makes it more difficult for ligand to bind to the second site on the dimer.
Interestingly, a completely different mutation, C571A,C593A, in the tethering arm of the extracellular domain also results in a receptor with ligand-independent kinase activity and a severely decreased affinity of EGF for the second site on the dimer (21). Thus, these two phenomena may be functionally related. In particular, binding of EGF to the second site on the dimer may be associated with the dissociation or inactivation of the asymmetric dimer. Because JM-A mutations, such as E661A,E663A,E666A, that would destabilize the asymmetric dimer abolish negative cooperativity and the V665M mutation that stabilizes the asymmetric dimer enhances negative cooperativity, we speculate that the negative cooperativity associated with the binding of EGF to the second site on the dimer arises at least in part because some of the binding energy is used to dissociate or reorient the asymmetric dimer.
The JM-B domain (residues 665–682) comprises the juxtamembrane latch that is the major interface in the asymmetric kinase dimer. The L680N mutation in the distal portion of the JM-B domain destabilizes this interface and inhibits kinase activation (19). However, we have shown previously that it does not abolish negative cooperativity (22). This suggests that although the JM-B segment is important for formation of the asymmetric dimer, it is structurally less important than the JM-A segment in supporting negative cooperativity. The results of the binding studies on the T669A-EGF receptor are consistent with this conclusion. Despite the fact that the EGF receptor is phosphorylated on Thr-669 following the binding of EGF, blocking this phosphorylation event in the T669A-EGF receptor did not alter the ligand binding properties of the receptor. Indeed, phosphorylation of Thr-669 by MAP kinase (34, 40) has never been shown to alter ligand binding affinity, although it clearly leads to desensitization of receptor tyrosine kinase activity (20, 35). Thus, the central region of the JM-B domain containing Thr-669 also appears to be relatively unimportant in determining negative cooperativity in the EGF receptor.
Using purified intracellular kinase domains in vitro, Brewer et al. (20) showed that replacement of Thr-669 with an aspartic acid led to the inhibition of kinase activity, consistent with the notion that phosphorylation of this site destabilizes the asymmetric dimer and leads to desensitization. However, in our experiments, which used full-length receptor in intact cells, replacement of Thr-669 with either Glu or Asp led to an enhancement of EGF-stimulated receptor autophosphorylation. This discrepancy may arise because in cells, the EGF receptor is quickly phosphorylated on Thr-669 following the binding of EGF. Thus, in intact cells, the comparison being made is actually between the effect of a phosphorylated threonine residue at this position and a Glu or Asp at this position. Because a doubly charged phosphate group is likely to be more destabilizing than a singly charged Glu or Asp residue, kinase activity is enhanced by the Thr → Glu or Thr → Asp substitutions because they preclude phosphorylation. Consistent with this hypothesis, we found that kinase activity was compromised in the double point mutant, the T669D,S671D-EGF receptor. However, the ligand binding properties of this mutant were not significantly different from those of the wild type EGF receptor. This supports the conclusion that phosphorylation in this region of the receptor is not associated with changes in the negative cooperativity seen in EGF binding.
Together, our data suggest that the JM-A region and the most proximal residues of the JM-B region of the intracellular juxtamembrane domain are intimately involved in the genesis of negative cooperativity within the EGF receptor dimer. The JM-A residues may form an anti-parallel helical dimer that stabilizes the asymmetric dimer, whereas the proximal JM-B residues may lock the JM-A helix tightly against the C-lobe of the activator kinase. Although necessary for kinase activation, the central and distal parts of the JM-B domain appear to play a lesser role in supporting negative cooperativity.
Based on our data, we propose a model for EGF receptor activation in which the binding of EGF to the first site on a receptor dimer leads to the formation of the asymmetric kinase dimer and activation of one kinase subunit. This involves intersubunit interactions mediated by residues 645–665, possibly involving the formation of an anti-parallel helical dimer (18). Binding of EGF to the second site on the dimer breaks these JM-A domain-mediated interactions, facilitating the formation of the reciprocal asymmetric dimer and the activation of the other kinase domain. Negative cooperativity arises because some of the energy derived from ligand binding is used to reorganize the asymmetric dimer, leading to a reduced affinity of EGF for the second site on the dimer.
This work was supported, in whole or in part, by National Institutes of Health Grant GM064491 (to L. J. P.).
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Ullrich A., Coussens L., Hayflick J. S., Dull T. J., Gray A., Tam A. W., Lee J., Yarden Y., Libermann T. A., Schlessinger J., Downward J., Mayes E. L., Whittle N., Waterfield M. D., Seeburg P. H. (1984) Nature 309, 418–425 [DOI] [PubMed] [Google Scholar]
- 2. Burgess A. W. (2008) Growth Factors 26, 263–274 [DOI] [PubMed] [Google Scholar]
- 3. Hynes N. E., Lane H. A. (2005) Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 4. Lemmon M. A., Schlessinger J. (2010) Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H. S., Leahy D. J., Lemmon M. A. (2003) Mol. Cell 11, 507–517 [DOI] [PubMed] [Google Scholar]
- 6. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Zhu H. J., Walker F., Frenkel M. J., Hoyne P. A., Jorissen R. N., Nice E. C., Burgess A. W., Ward C. W. (2002) Cell 110, 763–773 [DOI] [PubMed] [Google Scholar]
- 7. Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J. H., Saito K., Sakamoto A., Inoue M., Shirouzu M., Yokoyama S. (2002) Cell 110, 775–787 [DOI] [PubMed] [Google Scholar]
- 8. Clayton A. H., Walker F., Orchard S. G., Henderson C., Fuchs D., Rothacker J., Nice E. C., Burgess A. W. (2005) J. Biol. Chem. 280, 30392–30399 [DOI] [PubMed] [Google Scholar]
- 9. Martin-Fernandez M., Clarke D. T., Tobin M. J., Jones S. V., Jones G. R. (2002) Biophys. J. 82, 2415–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sako Y., Minoguchi S., Yanagida T. (2000) Nat. Cell Biol. 2, 168–172 [DOI] [PubMed] [Google Scholar]
- 11. Tao R. H., Maruyama I. N. (2008) J. Cell Sci. 121, 3207–3217 [DOI] [PubMed] [Google Scholar]
- 12. Yu X., Sharma K. D., Takahashi T., Iwamoto R., Mekada E. (2002) Mol. Biol. Cell 13, 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arteaga C. L., Ramsey T. T., Shawver L. K., Guyer C. A. (1997) J. Biol. Chem. 272, 23247–23254 [DOI] [PubMed] [Google Scholar]
- 14. Bublil E. M., Pines G., Patel G., Fruhwirth G., Ng T., Yarden Y. (2010) FASEB J. 24, 4744–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chantry A. (1995) J. Biol. Chem. 270, 3068–3073 [PubMed] [Google Scholar]
- 16. Gan H. K., Walker F., Burgess A. W., Rigopoulos A., Scott A. M., Johns T. G. (2007) J. Biol. Chem. 282, 2840–2850 [DOI] [PubMed] [Google Scholar]
- 17. Lichtner R. B., Menrad A., Sommer A., Klar U., Schneider M. R. (2001) Cancer Res. 61, 5790–5795 [PubMed] [Google Scholar]
- 18. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., Kuriyan J. (2009) Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 20. Brewer M. R., Choi S. H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M. A., Carpenter G. (2009) Mol. Cell 34, 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adak S., DeAndrade D., Pike L. J. (2011) J. Biol. Chem. 286, 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macdonald J. L., Pike L. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Macdonald-Obermann J. L., Pike L. J. (2009) J. Biol. Chem. 284, 13570–13576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doran D. M., Spar I. L. (1980) J. Immunol. Methods 39, 155–163 [DOI] [PubMed] [Google Scholar]
- 25. Wong I., Lohman T. M. (1995) Methods Enzymol. 259, 95–127 [DOI] [PubMed] [Google Scholar]
- 26. Wyman J., Gill S. J. (1990) Binding and Linkage: Functional Chemistry of Biological Macromolecules, University Science Books, Mill Valley, CA [Google Scholar]
- 27. King A. C., Cuatrecasas P. (1982) J. Biol. Chem. 257, 3053–3060 [PubMed] [Google Scholar]
- 28. Magun B. E., Matrisian L. M., Bowden G. T. (1980) J. Biol. Chem. 255, 6373–6381 [PubMed] [Google Scholar]
- 29. Rees A. R., Gregoriou M., Johnson P., Garland P. B. (1984) EMBO J. 3, 1843–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shoyab M., De Larco J. E., Todaro G. J. (1979) Nature 279, 387–391 [DOI] [PubMed] [Google Scholar]
- 31. Thiel K. W., Carpenter G. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19238–19243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hunter T., Ling N., Cooper J. A. (1984) Nature 311, 480–483 [DOI] [PubMed] [Google Scholar]
- 33. Castagna M., Takai Y., Kaibuchi K., Sano K., Kikkawa U., Nishizuka Y. (1982) J. Biol. Chem. 257, 7847–7851 [PubMed] [Google Scholar]
- 34. Northwood I. C., Gonzalez F. A., Wartmann M., Raden D. L., Davis R. J. (1991) J. Biol. Chem. 266, 15266–15276 [PubMed] [Google Scholar]
- 35. Li X., Huang Y., Jiang J., Frank S. J. (2008) Cell Signal. 20, 2145–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heisermann G. J., Gill G. N. (1988) J. Biol. Chem. 263, 13152–13158 [PubMed] [Google Scholar]
- 37. Downward J., Waterfield M. D., Parker P. J. (1985) J. Biol. Chem. 260, 14538–14546 [PubMed] [Google Scholar]
- 38. Friedman B., Frackelton A. R., Jr., Ross A. H., Connors J. M., Fujiki H., Sugimura T., Rosner M. R. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 3034–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Macdonald-Obermann J. L., Pike L. J. (2009) Biochemistry 48, 2505–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takishima K., Griswold-Prenner I., Ingebritsen T., Rosner M. R. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 2520–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]