

Abstract
p-Hydroxyphenylacetate (HPA) 3-hydroxylase is a two-component flavoprotein monooxygenase that catalyzes the hydroxylation of p-hydroxyphenylacetate to form 3,4-dihydroxyphenylacetate. Based on structures of the oxygenase component (C2), both His-120 and Ser-146 are located ∼2.8 Å from the hydroxyl group of HPA. The variants H120N, H120Q, H120Y, H120D, and H120E can form C4a-hydroperoxy-FMN (a reactive intermediate necessary for hydroxylation) but cannot hydroxylate HPA. The impairment of H120N is not due to substrate binding because the variant can still bind HPA. In contrast, the H120K variant catalyzes hydroxylation with efficiency comparable with that of the wild-type enzyme; the hydroxylation rate constant for H120K is 5.7 ± 0.6 s−1, and the product conversion ratio is 75%, compared with values of 16 s−1 and 90% for the wild-type enzyme. H120R can also catalyze hydroxylation, suggesting that a positive charge on residue 120 can substitute for the hydroxylation function of His-120. Because the hydroxylation reaction of wild-type C2 is pH-independent between pH 6 and 10, the protonation status of key components required for hydroxylation likely remains unchanged in this pH range. His-120 may be positively charged for selective binding to the phenolate form of HPA, i.e. to form the Hisδ+·HPAδ− complex, which in turn promotes oxygen atom transfer via an electrophilic aromatic substitution mechanism. Analysis of Ser-146 variants revealed that this residue is necessary for but not directly engaged in hydroxylation. Product formation in S146A is pH-independent and constant at ∼70% over a pH range of 6–10, whereas product formation for S146C decreased from ∼65% at pH 6.0 to 27% at pH 10.0. These data indicate that the ionization of Cys-146 in the S146C variant has an adverse effect on hydroxylation, possibly by perturbing formation of the Hisδ+·HPAδ− complex needed for hydroxylation.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, Flavin, Hydroxylase, C4a-hydroperoxyflavin, FMN, Oxygenase, Transient Kinetics
Introduction
Incorporation of single oxygen atoms into organic compounds (monooxygenation) by hydroxylation or epoxidation is an important biological process for aerobic organisms. The monooxygenation reaction is generally catalyzed by cytochrome P450, metalloenzymes, pterin-dependent and flavin-dependent monooxygenases (1). Flavin-dependent monooxygenases are involved in a wide variety of biological processes (2, 3). These enzymes catalyze the monooxygenation of many aromatic and aliphatic compounds (3).
Based on the number of proteins required for catalysis, flavin-dependent monooxygenases can be classified into single-protein component and two-protein component types (2–5). Besides an organic substrate, both types of enzymes require NAD(P)H and oxygen as co-substrates. The initial part of the reaction is the reduction of an enzyme-bound flavin by NAD(P)H to generate reduced flavin followed by the reaction of reduced flavin with oxygen to form the C4a-(hydro)peroxy flavin, a key intermediate that is required for oxygenation of an organic substrate. Oxygenation occurs via an oxygen atom transfer from C4a-hydroperoxy flavin to an organic substrate, resulting in the formation of a C4a-hydroxy flavin intermediate and an oxygenated product. The C4a-hydroxy flavin dehydrates at the last step of the reaction to form the final species, oxidized flavin (2–4, 6). All steps for the reactions of single-protein component flavin-dependent monooxygenases occur within the same polypeptide, whereas for the two-protein component type the flavin reduction occurs on a reductase component and the oxygenation occurs on an oxygenase component (4–8). The mechanism by which the reduced flavin is transferred involves simple diffusion or protein-protein contacts (5, 8). Although single-component flavin-dependent monooxygenases have been studied for more than 40 years, two-component flavin-dependent monooxygenases have only received significant attention during the past decade after recent discoveries of their involvement in a wide variety of reactions (2, 5).
The mechanism by which the oxygen atom transfer occurs in flavin-dependent monooxygenases is well understood for only a few enzymatic systems. The best understood oxygenation reaction is the hydroxylation of aromatic compounds catalyzed by a single-component flavoenzyme, p-hydroxybenzoate (pOHB)5 hydroxylase (PHBH) (4, 6, 9). Studies of PHBH and 2-methyl-3-hydroxypyridine-5-carboxylic acid (MHPC) monooxygenase (MHPCO) reactions showed that hydroxylation occurs via an electrophilic aromatic hydroxylation mechanism in which an aromatic substrate acts as a nucleophile and a terminal -OH group of C4a-hydroperoxy flavin acts as an electrophile (9–12). For the PHBH reaction, the hydroxylation rate constant increases upon an increase in pH with a pKa of 7.1 (13). This pKa value corresponds to the pKa of His-72, which is located on the protein surface and controls the H-bond network that facilitates removal of the phenolic proton of pOHB (9, 13, 14). For two-component flavin-dependent monooxygenases, the mode of oxygen atom transfer in hydroxylation of aromatic compounds has never been explored. Recently, pH-dependent studies of the oxygenase component (C2) of p-hydroxyphenylacetate (HPA) hydroxylase (HPAH) have shown that, unlike the PHBH reaction, the hydroxylation rate constant and the efficiency of product formation by C2 is constant in a pH range of 6–10 (15). These data indicate that the factors facilitating the hydroxylation reaction are different in C2 and PHBH.
HPAH is one of the prototype enzymes for investigations of the kinetics and mechanisms of two-component flavin-dependent monooxygenases that catalyze the hydroxylation of aromatic compounds. The enzyme from Acinetobacter baumannii is composed of a reductase component (C1) and an oxygenase component (C2), and it catalyzes the hydroxylation of HPA to 3,4-dihydroxyphenylacetate (DHPA) (16, 17). Kinetic studies of HPAHs from Pseudomonas putida (18), Pseudomonas aeruginosa (19), Escherichia coli W (20), and A. baumannii (8, 15, 21, 22) have been reported. X-ray structures of the oxygenase components from A. baumannii at 2.3–2.8 Å resolution (23) and Thermus thermophilus HB8 at 1.3–2.0 Å resolution (24, 25) and the reductase component from Sulfolobus tokodaii (26) at 1.7–2.3 Å resolution have been solved. Recent site-directed mutagenesis studies of the HPAH from A. baumannii have shown that Ser-171 is a key residue for stabilization of the reactive intermediate, C4a-hydroperoxy flavin, whereas His-396 facilitates intermediate formation (27). Although the ability of C2 to prolong the lifetime of C4a-hydroperoxy flavin is necessary for substrate hydroxylation (27), the enzyme must have another fine-tuned mechanism for efficient oxygen transfer because between pH 6.0 and 10.0 the product conversion percentage is ∼90% without wasteful H2O2 production, and the rate constants for hydroxylation remain constant at 15–17 s−1 (15). The pH independence of the C2 reaction implies that the protonation status of key components required for oxygen atom transfer is unchanged in this pH range. Based on the x-ray structure of C2 (23), the hydroxyl group of HPA is ∼2.8 Å away from the Nϵ atom of His-120 and the Oγ atom of Ser-146, a distance that should allow the formation of H-bonding interactions (Fig. 1). However, previous studies could not distinguish whether HPA was bound in the phenolic or phenolate form, and the protonation status of His-120 remains unknown.
FIGURE 1.

HPA binding in the active site of C2. HPA and FMNH− are shown as labled in purple and yellow, respectfully (PDB code 2JBT). Residues His-120 and Ser-146 are labeled. HPA is located ∼2.8 Å from His-120 and Ser-146.
Therefore, in this work we investigated the protonation status of His-120 and HPA bound at the C2 active site and the functional roles of His-120 and Ser-146 using site-directed mutagenesis, transient kinetics (stopped-flow and rapid-quench techniques), and product analysis at various pH values. The residue His-120 was mutated to Asn (H120N), Gln (H120Q), Tyr (H120Y), Asp (H120D), Glu (H120E), Lys (H120K), and Arg (H120R) to explore if the active form of His-120 is required to be neutral, negatively, or positively charged. The residue Ser-146 was mutated to Ala (S146A) and Cys (S146C) to explore whether the H-bonding ability or a suitable van der Waals contact of Ser-146 is necessary for the hydroxylation reaction.
EXPERIMENTAL PROCEDURES
Reagents
All chemicals and reagents used were analytical grade and purchased from commercial sources. High purity FMN was produced by converting FAD to FMN using snake venom from Crotalus adamanteus (28) according to the protocol described in Sucharitakul et al. (22). C1, wild-type C2, and C2variants were expressed and purified as previously described (15–17). The concentrations of the following compounds were determined using the known extinction coefficients at pH 7.0: NADH, ϵ340 = 6.22 mm−1cm−1; HPA, ϵ277 = 1.55 mm−1cm−1 (16); FMN, ϵ446 = 12.2 mm−1cm−1; DHPA, ϵ281 = 2.74 mm−1cm−1; C1, ϵ458 = 12.8 mm−1cm−1 (22); wild-type C2 and all C2variants, ϵ280 = 56.7 mm−1cm−1.
Spectroscopic Studies and Site-directed Mutagenesis
Details are provided in the supplemental Procedures.
Rapid Kinetics Measurement
Experimental protocols for maintaining anaerobic conditions are similar to those described in Refs. 15 and 21). Details are provided in the supplemental Procedures.
For the reactions of the His-120 and Ser-146 variants at pH 7.0, solutions of enzymes and oxidized FMN in the absence or presence of HPA in 50 mm sodium phosphate buffer (pH 7.0), were equilibrated in the anaerobic glove box. The anaerobic enzyme solution was reduced with an equal reducing equivalent of sodium dithionite (a solution of 5 mg/ml in 100 mm potassium phosphate buffer pH 7.0). Spectra of FMN were monitored with a diode-array spectrophotometer inside the glove box to ensure complete reaction. Then, the reduced enzyme solution was transferred into a glass tonometer. Solutions of various concentrations of oxygen were prepared by equilibrating buffers with air and certified oxygen-nitrogen gas mixtures (29).
Rapid Quench-flow Experiments
All experiments used acid as the quenching solution and were performed at 4 °C using a TgK Scientific Model RQF-63, DimentionTM D1 rapid quench-flow system in an anaerobic glove box (Belle Technology). The rapid quench-flow system employed three syringes. An anaerobic solution of a C2variant (H120K, S146A, or S146C), FMN, and HPA in 50 mm sodium phosphate buffer (pH 7.0), was reduced with a solution of sodium dithionite (5 mg/ml in 100 mm potassium phosphate buffer, pH 7.0) in the anaerobic glove box. The solution of reduced enzyme·substrate complex from one syringe was mixed with the air-saturated buffer plus HPA from the second syringe. The reaction mixture was incubated for various time periods before quenching with a 0.15 m HCl solution from a third syringe. The reaction mixture after mixing and before quenching contained H120K, S146A, or S146C (100 μm), FMNH− (50 μm), and HPA (2 mm for S146A, 20 mm for H120K and S146C). The quenched samples were collected from the sample loop, and the enzyme was removed by ultrafiltration with a Microcon unit (Amicon YM-10). The filtrates were analyzed for the amount of DHPA produced from the reaction using an HPLC method, as previously described (16, 17).
Product Analysis; Measurement of DHPA
Anaerobic solutions of His-120 and Ser-146 variants (100 μm), FMNH− (50 μm), and HPA (2 mm or 20 mm) in 10 mm sodium phosphate buffer (pH 7.0), at 25 °C were reduced with a solution of sodium dithionite in an anaerobic glove box. An anaerobic solution of reduced enzyme·substrate complex was added in closed vessels containing air-saturated buffers at various pH values in the anaerobic glove box. The air-saturated buffers used for maintaining the various pH values were 100 mm sodium phosphate buffer (pH 6.0–7.5), 100 mm Tris-HCl buffer (pH 8.0–8.5), and 100 mm glycine buffer adjusted with NaOH (pH 9.0–10.0). After the reaction was complete (5 min), 0.15 m HCl was immediately added to the mixture to a final concentration of 0.08 m. Denatured protein was removed from the quenched solution using a Centricon YM-10 concentrator. Samples were analyzed for the amount of DHPA formed using the HPLC method previously described (16, 17). Measurements of H2O2 formed in C2 reactions were carried out as described in the supplemental Procedures.
Data Analysis
Kinetic data were analyzed by fitting the data using exponential functions in Program A, which was developed by Rong-yen Chu, Joel Dinverno, and D. P. Ballou at the University of Michigan (Ann Arbor, MI) to obtain the observed rate constants (kobs). Determinations of rate constants were calculated from plots of kobs versus substrate concentration using Levenber-Marquardt algorithms that were included in the KaleidaGraph software (Synergy Software).
RESULTS
Reactions of H120N·FMNH−, H120Q·FMNH−, H120Y·FMNH−, H120D·FMNH−, H120E·FMNH−, and H120K·FMNH− with Oxygen in Absence of HPA
A solution of His-120 variant (40 μm) and FMN (16 μm) in 50 mm sodium phosphate buffer (pH 7.0) was made anaerobic and reduced with a solution of sodium dithionite. The resulting enzyme-FMNH− complex was mixed with the same buffer containing the following oxygen concentrations: 0.13, 0.31, 0.61, and 1.03 mm (concentrations after mixing). The reactions were monitored by the changes in absorbance at 390 and 446 nm. For the H120K·FMNH− variant, the reaction exhibited biphasic kinetics (Fig. 2A). The first phase (reaction time ∼ 0.002–0.1 s) was an increase in absorbance at 390 nm without any significant change at 446 nm. The rate constants during this phase were oxygen-dependent, consistent with a bimolecular rate constant of 6.3 ± 0.2 × 105 m−1s−1 (k1) (Table 1). The second phase (0.1–20 s) was a small decrease in absorbance at 390 nm and a large increase in absorbance at 446 nm. The rate constants during this phase (k2) were independent of oxygen concentration and measured 0.52 ± 0.01 s−1. The reaction at oxygen concentration of 1.03 mm was measured with wavelengths of detection ranging from 310–550 nm, and the absorbances at a reaction time of 0.1 s were plotted (filled circle spectrum in the inset in Fig. 2A) to obtain an intermediate spectrum after the first phase. The spectrum in the inset in Fig. 2A had a maximum absorption peak at 390 nm, similar to that of a C4a-hydroperoxy-FMN intermediate in the reactions of wild-type C2 and other variants (15, 21, 27). Therefore, the first phase shown in Fig. 2A was the formation of C4a-hydroperoxy-FMN, and the second phase was the elimination of H2O2 from the intermediate to generate oxidized FMN (Fig. 3). For other His-120 variants, the kinetics of the reactions are shown in supplemental Figs. 1–5. All His-120 variants show similar kinetic patterns as those of H120K and wild-type enzymes (15), and the reactions are depicted in Fig. 3. Rate constants for the formation and decay of C4a-hydroperoxy flavin are summarized in Table 1. The rate constants for C4a-hydroperoxy flavin formation in these variants are similar to that of wild-type C2, whereas those for H2O2 elimination are similar for H120Q and H120Y, ∼7–10-fold higher for H120N, H120D, and H120E and ∼170-fold higher for H120K.
FIGURE 2.

Kinetics of the reaction of H120K·FMNH− with oxygen in the absence and presence of HPA. A, reaction of H120K·FMNH− with oxygen in the absence of HPA. A solution of H120K (40 μm) and FMNH− (16 μm) in 50 mm sodium phosphate buffer, pH 7.0, at 4 °C was mixed with the same buffer containing the following concentrations of oxygen: 0.13, 0.31, 0.61, and 1.03 mm (concentrations after mixing). The traces at 390 nm are shown as the lower to upper traces from low to high oxygen concentrations, whereas all traces monitored at 446 nm are superimposed. The inset in A shows the spectra of the species involved in the reaction of H120K·FMNH− with oxygen. The filled circle spectrum represents the C4a-hydroperoxy-FMN intermediates accumulated at a reaction time of 0.1 s. The empty square spectrum represents the spectrum of the starting species (the H120K·FMNH− complex), and the solid line represents that of the oxidized FMN. B, kinetics of the reaction of the H120K·FMNH−·HPA complex with oxygen at pH 7.0. A solution of H120K (40 μm), FMNH− (16 μm), and HPA (20 mm) in 50 mm sodium phosphate, pH 7.0, at 4 °C was mixed with the same buffer containing various oxygen concentrations plus 20 mm HPA (concentrations after mixing). The kinetic traces were obtained by monitoring the changes at 390 and 446 nm. The traces at 390 nm are shown as the lower to upper traces from low to high oxygen concentrations, whereas all traces monitored at 446 nm are superimposed. The filled circle kinetic trace is a plot of DHPA analyzed from the rapid quench experiment of the reaction of H120K·FMNH−·HPA with oxygen at pH 7.0, 4 °C (details are provided under “Results”). The plot was fitted with a single exponential curve with a rate constant of 7.1 ± 0.8 s−1. The percentage of DHPA formed at a reaction time of 5.03 min was 80 ± 4%.
TABLE 1.
Summary of the rate constants for the reactions of H120N·FMNH−, H120Q·FMNH−, H120Y·FMNH−, H120D·FMNH−, H120E·FMNH−, H120K·FMNH−, S146A·FMNH−, and S146C·FMNH− with oxygen in the absence and presence of HPA
Rate constants are as described in Figs. 3 and 4 and from stopped-flow and rapid-quench data in Figs. 2 and 6 and supplemental Figs. 1–5 and 8–9.
| Enzymes | Rate constant of C4a-hydroperoxy-FMN formation (k1) | Rate constant of H2O2 elimination from C4a-hydroperoxy-FMN (k2) | Rate constant of C4a-hydroperoxy-FMN·HPA formation (k3) | Rate constant of hydroxylation (k4) | Rate constant of C4a-hydroxy-FMN dehydration (k5) |
|---|---|---|---|---|---|
| m−1s−1 | s−1 | m−1s−1 | s−1 | s−1 | |
| C2 wild type | 1.1 × 106a | 3 × 10−3a | 4.8 × 104a | 16 a | 0.35 a |
| H120 variants | |||||
| H120N | 6.2 ± 0.3 × 105 | 1.9 ± 0.2 × 10−2 | |||
| 6.7 ± 0.2 × 105b | 1.2 ± 0.1 × 10−2b | ||||
| H120Q | 7.6 ± 0.2 × 105 | 7.0 ± 0.1 × 10−3 | |||
| H120Y | 5.8 ± 0.1 × 105 | 4.0 ± 0.1 × 10−3 | |||
| H120D | 8.4 ± 0.3 × 105 | 3.3 ± 0.6 × 10−2 | |||
| 8.4 ± 0.3 × 105b | 3.4 ± 0.2 × 10−2b | ||||
| H120E | 5.6 ± 0.1 × 105 | 2.1 ± 0.1 × 10−2 | |||
| H120K | 6.3 ± 0.2 × 105 | 5.2 ± 0.1 × 10−1 | 4.6 ± 0.2 × 104c | 5.7 ± 0.6 | 0.75 ± 0.01 |
| S146 variants | |||||
| S146A | 3.1 ± 0.1 × 105 | 2.0 ± 0.3 × 10−3 | 2.2 ± 0.1 × 104 | 2.6 ± 0.1 | 0.031 ± 0.001 |
| S146C | 1.1 ± 0.1 × 106 | 8.0 ± 0.2 × 10−3 | 6.2 ± 0.3 × 105 | 0.35 ± 0.05 | >0.35 |
a Rate constants of the wild-type enzyme at pH 7.0, 4 °C from (15).
b Rate constants of the reactions of H120N·FMNH− and H120D·FMNH− with oxygen in the presence of HPA. No hydroxylation occurs in these reactions.
c Rate constant of C4a-hydroperoxy-FMN formation in the reaction of H120K in the presence of HPA was taken from the reaction with 0.13 mm oxygen (supplemental Fig. 6).
FIGURE 3.

The reaction of C2·FMNH−, His-120 variants·FMNH−, or Ser-146 variants·FMNH− with oxygen in the absence of HPA.
Formation of DHPA from the Reactions of the His-120 Variants
To determine whether the His-120 variants (H120N, H120Q, H120Y, H120D, H120E, H120K, and H120R) could catalyze hydroxylation of HPA, we measured the amount of DHPA obtained from single-turnover reactions. Solutions of His-120 variants (200 μm), FMNH− (100 μm), and HPA (20 mm) in 10 mm sodium phosphate buffer (pH 7.0), was mixed with air-saturated buffers containing 20 mm HPA at 4 °C (pH 6.0–10.0) inside an anaerobic glove box (“Experimental Procedures”). Reaction samples (100 μl) were added to 0.15 m HCl (100 μl) to make a final concentration of 0.08 m HCl in the quenched solution. DHPA in the sample was measured using HPLC (“Experimental Procedures”), which can detect DHPA with a lower detection limit of 5%, i.e. 1.3 μm DHPA. No DHPA was detected for the reactions of H120N, H120Q, H120Y, and H120E. However, DHPA (75 ± 6%) was formed in the H120K reactions in the pH range of 6.0–10.0 (Table 2). For H120R, ∼10–15% DHPA formed in the same pH range (Table 2). These data indicate that H120K and H120R are active and can perform hydroxylation, whereas the neutral or negatively charged variants are inactive and cannot catalyze the hydroxylation of HPA.
TABLE 2.
Formation of the hydroxylated product (DHPA) from single-turnover reactions of His-120 and Ser-146 variants
DHPA product was not detected for the reactions of H120N·FMNH−, H120Q·FMNH−, H120Y·FMNH−, H120D·FMNH−, and H120E·FMNH− with oxygen in the presence of HPA (2 or 20 mm) using the HPLC method. The limit of detection was 5% (1.3 μm).
| pH | %DHPA |
|||
|---|---|---|---|---|
| H120K | H120R | S146A | S146C | |
| 6.0 | 76 ± 4 | 9 ± 1 | 72 ± 4 | 65 ± 6 |
| 6.5 | 80 ± 5 | 74 ± 1 | 59 ± 1 | |
| 7.0 | 81 ± 6 | 16 ± 1 | 73 ± 1 | 65 ± 2 |
| 7.5 | 78 ± 10 | 75 ± 8 | 65 ± 5 | |
| 8.0 | 80 ± 5 | 11 ± 1 | 76 ± 7 | 65 ± 4 |
| 8.5 | 84 ± 11 | 76 ± 3 | 64 ± 1 | |
| 9.0 | 72 ± 3 | 11 ± 1 | 76 ± 3 | 66 ± 1 |
| 9.5 | 71 ± 4 | 79 ± 8 | 47 ± 1 | |
| 10.0 | 70 ± 5 | 7 ± 1 | 78 ± 7 | 27 ± 4 |
To confirm the results of the DHPA detection, an analysis of H2O2 formation by H120N and H120D was also performed (supplemental Table 2). The results indicated that 90–100% of H2O2 was detected in the reaction of H120N and H120D in a pH range of 6–10. These data agree with the HPLC analysis; both H120N and H120D are inactive and cannot convert HPA to DHPA.
Reactions of H120K·FMNH−, H120N·FMNH−, and H120D·FMNH− with Oxygen in the Presence of HPA
A solution of H120K (40 μm), FMNH− (16 μm), and HPA (20 mm) in 50 mm sodium phosphate buffer, pH 7.0, was mixed with the same buffer containing 20 mm HPA and various concentrations of oxygen at 4 °C. The kinetic traces of the H120K reaction monitored at 390 and 446 nm showed three phases (Fig. 2B). The first phase (0.002–0.02 s) was an increase in absorbance at 390 nm (∼0.01 AU) without a significant change in absorbance at 446 nm. The observed rate constants of this phase were linearly dependent on oxygen concentration with a bimolecular rate constant of ∼6.3 ± 0.2 × 105 m−1s−1. This phase was likely C4a-hydroperoxy-FMN formation in the H120K·FMNH− complex without HPA bound (∼15% of the total enzyme) as shown in Fig. 2A. The second phase (0.02–0.8 s) was an increase in absorbance at 390 nm (∼0.05 AU) without any significant change in absorbance at 446 nm. Based on the absorption characteristics, this phase should be related to the formation of a C4a-adduct intermediate. The observed rate constants of this phase were dependent on oxygen concentration in a nonlinear fashion (supplemental Fig. 6). Based on the results from rapid-quench analysis (described later), the second phase should result from the combined signal changes from the formation of C4a-hydroperoxy-FMN·HPA and C4a-hydroxy-FMN (Fig. 4) because this phase was synchronized with the hydroxylation step. The inability to differentiate between the formation of C4a-hydroperoxy-FMN·HPA and C4a-hydroxy-FMN (Fig. 4) may be due to similarities in the absorption characteristics and rate constants of the formation of the two intermediates. The final, third phase of the reaction exhibited a large change in absorbance at 446 nm. The observed rate constants of this phase were independent of the oxygen concentration and consistent with a value of 0.75 ± 0.01 s−1 (k5). This phase represents the decay of a C4a-adduct intermediate (C4a-hydroperoxy-FMN or C4a-hydroxy-FMN) to yield oxidized FMN.
FIGURE 4.

Kinetic mechanism proposed for the reaction of H120K·FMNH−, S146A·FMNH−, and S146C·FMNH− with oxygen in the presence of HPA. The reduced enzyme·HPA complex (H120K·FMNH−·HPA, S146A·FMNH−·HPA, or S146C·FMNH−·HPA) reacts with molecular oxygen to form C4a-hydroperoxy-FMN·HPA (k3). HPA is then hydroxylated to yield DHPA and C4a-hydroxy-FMN (k4). The C4a-hydroxy-FMN dehydrates to generate oxidized FMN (k5). The elimination of H2O2 from the C4a-hydroperoxy-FMN can occur as an uncoupling pathway (k6).
Reactions of neutral and negatively charged variants (H120N and H120D) that cannot hydroxylate HPA were also investigated to detect whether HPA can bind and influence kinetics of these variants. A solution of H120N or H120D (40 μm), FMNH− (16 μm), and HPA (2 mm) in 50 mm sodium phosphate buffer, pH 7.0, was mixed with the same buffer containing 2 mm HPA and various concentrations of oxygen at 4 °C. The kinetic traces of H120N·FMNH− in the presence of HPA (supplemental Fig. 1B) at 390 and 446 nm exhibited two phases similar to the reaction without HPA (supplemental Fig. 1A). The rate constant of the first phase (6.7 ± 0.2 × 105 m−1s−1) was similar to that of the reaction in the absence of HPA (6.2 ± 0.3 × 105 m−1s−1, Table 1, inset in supplemental Fig. 1B). The rate constants of the second phase were independent of the oxygen concentration and equivalent to 1.2 ± 0.1 × 10−2 s−1, which is in the same range as the value in the absence of HPA (1.9 ± 0.2 × 10−2 s−1, Table 1). Likewise, the reaction of H120D in the presence of HPA (supplemental Fig. 4B) was similar to the reaction in the absence of HPA (Table 1 and supplemental Fig. 4A).
The slight difference in the kinetics of the second phase of H120N reactions, i.e. C4a-hydroperoxy-FMN oxidized with a rate constant of 1.9 ± 0.2 × 10−2 s−1 in the absence of HPA versus 1.2 ± 0.1 × 10−2 s−1 in the presence of 2 mm HPA (supplemental Fig. 1, A and B) was investigated to assess whether it was due to the binding of HPA to the enzyme. An experiment similar to that in supplemental Fig. 1B was performed by mixing a solution of H120N·FMNH− with air-saturated buffer containing various concentrations of HPA (Fig. 5). The rate constant for formation of C4a-hydroperoxy-FMN was constant at ∼6.7 ± 0.2 × 105 m−1s−1, whereas that of H2O2 elimination varied from 1.1 × 10−2 s−1 (0.16 mm HPA) to 7.0 × 10−3 s−1 (8 mm HPA) (supplemental Fig. 7). For wild-type C2, elimination of H2O from C4a-hydroxyflavin to generate oxidized flavin (k5, Fig. 4) is inhibited by the binding of excess HPA (21). In this case, although no hydroxylation was detected for the reaction of H120N·FMNH− (Table 1), high concentrations of HPA decreased the rate constant for H2O2 elimination from 1.9 × 10−2 s−1 (no HPA) to 7.0 × 10−3 s−1 (8 mm HPA). The data were analyzed according to a model in which HPA binds to the C4a-hydroperoxy-FMN intermediate and inhibits H2O2 elimination (supplemental Fig. 7). The analysis yielded a Kd value for binding of 5.0 ± 1.1 mm.
FIGURE 5.

Binding of HPA to H120N. A solution of H120N (40 μm)·FMNH− (16 μm) was mixed with air-saturated sodium phosphate buffer (pH 7.0) containing various concentrations of HPA: 0, 0.16, 0.4, 1, 2, 4, and 8 mm (concentrations after mixing). The kinetics was monitored at 390 and 446 nm. The traces at 390 and 446 nm from low to high HPA concentrations are indicated by the vertical and horizontal arrows, respectively. The inset is a plot of the absorbance change at 390 nm at a reaction time of 2 s versus the concentration of HPA. The data analysis in the inset indicates that HPA bound to C4a-hydroperoxy-FMN with a Kd value of 2.2 ± 0.7 mm.
The kinetic traces in Fig. 5 at a reaction time of ∼0.2–10 s also showed an absorbance decrease at 390 nm when the HPA concentration was increased. These changes may also be due to the binding of HPA to C4a-hydroperoxy-FMN. When the absorbance change at a reaction time of 2 s was plotted as a function of HPA concentration, a Kd value of 2.2 ± 0.7 mm was obtained (inset in Fig. 5).
All of these data indicate that HPA can bind to the H120N:C4a-hydroperoxy-FMN, although with a much weaker affinity than to the wild-type enzyme; the Kd value for H120N is ∼2.2–5.0 versus 0.35 mm for the wild-type enzyme (21). Under the experimental conditions employed (20 mm HPA), some HPA should bind to H120N, but it cannot be hydroxylated (Table 2). These data emphasized the important role of His-120 for the hydroxylation activity of C2. Neutral and negatively charged variants failed to catalyze hydroxylation; only a positive charge at residue 120 (H120K and H120R) could compensate for His-120 and thus may be required for the hydroxylation reaction.
Measurement of the Hydroxylation Rate Constant of H120K
Because the hydroxylation rate constant could not be determined based on the difference in the absorption characteristics of C4a-hydroperoxy flavin and C4a-hydroxy flavin formation (Fig. 2B), rapid-quench techniques were used to measure the hydroxylation rate constant of H120K. A solution of the reduced enzyme·substrate complex (H120K·FMNH−·HPA) in 50 mm sodium phosphate buffer, pH 7.0, at 4 °C from one syringe was mixed with an air-saturated sodium phosphate buffer (50 mm, pH 7.0) containing 20 mm HPA from a second syringe. The reaction mixture was incubated for various time periods between 0.03 and 5.03 s before the solution was quenched by mixing with 0.15 m HCl from a third syringe. The amount of DHPA formed at various time points was analyzed using HPLC. A plot of DHPA formed versus time (Fig. 2B) showed a single-exponential curve that was consistent with a rate constant of 7.1 ± 0.8 s−1. Therefore, the overall reaction of H120K·FMNH− with oxygen in the presence of HPA can be summarized as shown in Fig. 4. The reaction of the reduced enzyme·HPA complex with oxygen results in the initial formation of C4a-hydroperoxy-FMN·HPA (k3, Fig. 4) followed by formation of C4a-hydroxy-FMN and DHPA (k4, Fig. 4). At the last step of the hydroxylation pathway, C4a-hydroxy-FMN dehydrates to generate oxidized FMN and water (k5, Fig. 4). The product analysis data also imply that the reaction of H120K is bifurcated; in addition to the regular hydroxylation pathway, a fraction of C4a-hydroperoxy-FMN (∼20%) generates H2O2 and oxidized FMN (k6, Fig. 4). Nevertheless, the rapid-quench and product analyses of H120K suggest that the enzyme is capable of performing hydroxylation. The results using H120K and H120R indicate that a positive charge at residue 120 can substitute for His-120 in hydroxylation. This implies that His-120 may possess a positive charge in the wild-type enzyme, as in the case in H120K or H120R.
Reactions of the S146A·FMNH− and S146C·FMNH− Variants with Oxygen in the Absence of HPA
A solution of 40 μm S146A or S146C and 16 μm FMNH− in 50 mm sodium phosphate buffer (pH 7.0) was mixed with the same buffer containing various oxygen concentrations in the stopped-flow spectrophotometer at 4 °C. The kinetics of S146A·FMNH− displayed two phases corresponding to the formation and decay of C4a-hydroperoxy-FMN, as observed when using the His-120 variants and wild-type enzymes (supplemental Fig. 8, Fig. 3, Table 1). The bimolecular rate constant for C4a-hydroperoxy-FMN formation was 3.1 ± 0.1 × 105 m−1s−1, whereas that for H2O2 elimination from the intermediate was 2.0 ± 0.3 × 10−3 s−1. For S146C, C4a-hydroperoxy-FMN formed with a bimolecular rate constant of 1.1 ± 0.1 × 106 m−1s−1 and eliminated H2O2 with a rate constant of 8.0 ± 0.2 × 10−3 s−1 (supplemental Fig. 9, Fig. 3, Table 1). All these rate constants are similar to those of wild-type C2 (Table 1). The C4a-hydroperoxy-FMN intermediates in both reactions have absorbance maxima at 390 nm (inset in supplemental Figs. 8 and 9), similar to those of wild-type C2 and other variants (15, 21, 27).
Kinetics of the Reactions of the S146A·FMNH− and S146C·FMNH− Variants with Oxygen in Presence of HPA
The reaction of S146A·FMNH− in the presence of HPA was investigated by mixing an anaerobic buffer of S146A (40 μm), FMNH− (16 μm), and HPA (2 mm) in 50 mm sodium phosphate buffer, pH 7.0, with the same buffer containing various concentrations of oxygen plus 2 mm HPA. All concentrations are after mixing. The kinetic traces in Fig. 6A indicate that the reaction kinetics contained four phases. The first phase (0.002–0.01 s) showed an increase in absorbance at 390 nm (∼0.025 AU) with no significant absorbance change at 446 nm, and the observed rate constants of this phase were dependent on the oxygen concentration, with a bimolecular rate constant of 3.1 ± 0.1 × 105 m−1s−1 (Table 1). This value is similar to that of the S146A·FMNH− complex with oxygen in the absence of HPA (supplemental Fig. 8). Therefore, this phase was likely the formation of C4a-hydroperoxy-FMN without HPA binding. The second phase (0.01–0.2 s) exhibited an increase in absorbance at 390 nm (∼0.085 AU) and no significant absorbance at 446 nm (∼0.035 AU). The observed rate constants of this phase were dependent on the oxygen concentration with a bimolecular rate constant of 2.2 ± 0.1 × 104 m−1s−1 (inset in Fig. 6A). The second phase was likely the formation of C4a-hydroperoxy-FMN·HPA (Fig. 4). The third phase (0.2–1.5 s) had no significant change in absorbance at 390 nm and an increase in absorbance at 446 nm (∼0.06 AU) that corresponded with an observed rate constant of 4.3 ± 0.3 s−1. The fourth phase (1.5–200 s) showed a decrease in absorbance at 390 nm (∼0.023 AU) and a large increase in absorbance at 446 nm (∼0.12 AU), consistent with a rate constant of 0.031 ± 0.001 s−1. The observed rate constants of the third and fourth phase were independent of oxygen concentration. These data suggest that, after forming S146A·C4a-hydroperoxy-FMN·HPA, the enzyme proceeds through bifurcated pathways in which one pathway generates DHPA and the other pathway generates H2O2 without hydroxylation (Fig. 4).
FIGURE 6.

Kinetics of the reactions of S146A·FMNH− and S146C·FMNH− with oxygen in the presence of HPA. A, A solution of S146A (40 μm)·FMNH− (16 μm)·HPA (2 mm) in 50 mm sodium phosphate (pH 7.0), was mixed with the same buffer containing the following oxygen concentrations, 0.13, 0.31, 0.61, and 1.03 mm, plus 2 mm HPA (concentrations after mixing). The kinetics of the reaction of S146A·FMNH−·HPA with oxygen at pH 7.0 was monitored by the change in absorbance at 390 and 446 nm. Traces at 390 nm are shown as the lower to the upper traces from low to high oxygen concentrations. The inset in A shows a plot of kobs of the second phase versus oxygen concentration with a bimolecular rate constant of 2.2 ± 0.1 × 104 m−1s−1. The filled circles represent the DHPA formed, which was measured from rapid-quench experiments of the reactions of S146A·FMNH−·HPA with oxygen at pH 7.0, 4 °C. The plot is consistent with a single exponential curve with a rate constant of 3.9 ± 0.8 s−1. The percentage of DHPA accumulated at a reaction time of 8.03 min was 66 ± 2%. B, kinetic traces for the reaction of S146C·FMNH−·HPA with oxygen are shown. The experimental conditions used were similar to the reaction of S146A except that 20 mm HPA was used. The lower to upper traces at 390 nm are shown from low to high oxygen concentrations, whereas all traces at 446 nm were superimposed. The inset in B is a plot of kobs of the first phase versus oxygen concentration. The plot shows a linear relationship with a bimolecular rate constant of 6.2 ± 0.3 × 105 m−1s−1. The filled circled trace is a plot of DHPA formed from the reaction of S146C versus reaction time in the rapid-quench instrument. The plot shows a single exponential curve with a rate constant of 0.52 ± 0.05 s−1. At a reaction time of 8.30 min, DHPA (68 ± 3%) was formed.
For S146C, a solution of S146C (40 μm), FMNH− (16 μm), and HPA (20 mm) in 50 mm sodium phosphate, pH 7.0, was mixed with the same buffer containing 20 mm HPA and various oxygen concentrations at 4 °C. Reaction kinetics showed three phases (Fig. 6B). The first phase (0.002–0.08 s) was an increase in absorbance at 390 nm without any significant change at 446 nm, and the observed rate constants of this phase were consistent with a bimolecular rate constant of 6.2 ± 0.3 × 105 m−1s−1 (inset in Fig. 6B). This value was significantly different from the rate constant of C4a-hydroperoxy-FMN formation in the absence of HPA (supplemental Fig. 9); thus, this phase was likely the formation of S146C·C4a-hydroperoxy-FMN·HPA. The second phase (0.08–0.4 s) showed a small increase in absorbance at 390 nm (∼0.005 AU) that was independent of oxygen concentration and consistent with a rate constant of 2.7 ± 0.5 s−1. This phase may result from a conformational change of C4a-hydroperoxy-FMN·HPA. The third phase (0.4–20 s) displayed a large increase in absorbance at 446 nm (∼0.16 AU) that coincided with a decrease in absorbance at 390 nm (∼0.03 AU), indicating that this phase was involved in flavin oxidation. The rate constant for this phase (0.52 ± 0.01 s−1) was independent of oxygen concentration.
Given the results of the reactions of S146A and S146C in the presence of HPA, the reactions of both variants may occur through bifurcated pathways in which one pathway generates DHPA and the other generates H2O2 without hydroxylation (Fig. 4). Therefore, the hydroxylation rate constants and product formation of S146A and S146C were measured using rapid quench-flow techniques.
Measurement of the Hydroxylation Rate Constants and Product Formation of S146A and S146C
A solution of the S146A·FMNH−·HPA complex was mixed with an air-saturated buffer containing 2 mm HPA at 4 °C and quenched at various time points during 0.045–8.03 s using a quench-flow apparatus (“Experimental Procedures”). The amount of DHPA formed was analyzed using HPLC. A plot of DHPA versus the delayed time before the quench yielded a single exponential curve that was consistent with a rate constant of 3.9 ± 0.8 s−1. This rate constant value is in the same range as the observed rate constant (4.3 ± 0.3 s−1) found for the third phase of the stopped-flow data (Fig. 6A). At an age time of 8.03 s, the amount of DHPA formed was 66 ± 2%, which is similar to the amount of DHPA formed when the solution of the S146A·FMNH−·HPA complex was incubated with oxygen for 5 min (73 ± 1%, Table 2).
For S146C, a rapid-quench experiment similar to that for S146A was performed, except that 20 mm HPA was used. A plot of DHPA concentration versus age time yielded a single exponential curve with an observed rate constant of 0.52 ± 0.05 s−1 (Fig. 6B), which is similar to the observed rate constant of the third phase monitored by stopped-flow experiments (0.52 ± 0.01 s−1). When a solution of S146C·FMNH−·HPA was mixed with air-saturated buffer and left for 5 min, the reaction resulted in the formation of 65 ± 2% DHPA, in agreement with the extrapolated value from the rapid-quench plot (68 ± 3%, Fig. 6B).
Kinetic Mechanism of the H120K, S146A, and S146C Reactions in the Presence of HPA
The results from Fig. 2B, Fig. 6, A and B, and Table 2 indicate that after formation of C4a-hydroperoxy-FMN·HPA, the enzymatic pathway is divided into two pathways in which one pathway generates the product and the other generates H2O2 without hydroxylation. Therefore, the observed rate constants measured from the rapid-quench experiments (7.1 ± 0.8 s−1 for H120K, 3.9 ± 0.8 s−1 for S146A, and 0.52 ± 0.05 s−1 for S146C) or from the second phase of the H120K reaction (6.2 ± 0.5 s−1) and the third phase of the Ser-146 variants (4.3 ± 0.3 s−1 for S146A and 0.52 ± 0.01 s−1 for S146C) from the stopped-flow data are combined values (kOH +kH2O2) from both pathways according to the nature of parallel reactions (see Equation 1 and Connors (30)). The percentage of DHPA formed (80 ± 4, 66 ± 2, and 68 ± 3% from the reactions of H120K, S146A, and S146C at pH 7.0, respectively) is equivalent to the ratio of kOH/(kOH + kH2O2) (see Equation 2).
In Equation 1, kobs is the observed rate constant for the parallel reactions of A → B and A → C. kB is the rate constant associated with the former reaction, whereas kC is that of the latter reaction (30). In Equation 2, kOH is the rate constant of HPA hydroxylation (k4, Fig. 4), and kH2O2 is the rate constant of H2O2 formation (k6, Fig. 4).
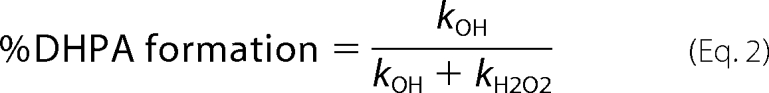 |
The results in Figs. 2B and 6, A and B, and Table 2 were analyzed according to Equations 1 and 2 to obtain the values of kOH and kH2O2 for both variants (Fig. 4 and Table 1). For H120K, the hydroxylation rate constant was 5.7 ± 0.6 s−1 (k4, Fig. 5), and the rate constant of H2O2 elimination due to the uncoupling pathway (k6, Fig. 5) was 1.4 ± 0.6 s−1. For the S146 variants, the rate constants of hydroxylation were 2.6 ± 0.1 s−1 for S146A and 0.35 ± 0.05 s−1 for S146C. The rate constants of H2O2 elimination from the uncoupling pathway (k6, Fig. 5) were 1.3 ± 0.1 s−1 for S146A and 0.17 ± 0.05 s−1 for S146C. This analysis also suggested that the fourth phase (k5 = 0.031 ± 0.001 s−1) of the S146A reaction was likely the hydration of C4a-hydroxy-FMN to form the oxidized flavin. However, for S146C, this dehydration process must be rapid and at least 10 times greater than 0.35 s−1 because the formation of oxidized FMN was synchronized with the hydroxylation step. These data suggest that the hydroxylation rate constant of H120K is about one-third of the wild-type value (∼16 s−1) and about 2- and 20-fold greater than the rate constants of S146A and S146C, respectively.
pH Effects on Product Formation by S146C and S146A
For the wild-type C2 reaction, HPA (∼90%) is hydroxylated to form DHPA in the pH range of 6.2–9.9 (15). Therefore, we investigated the effect of pH on DHPA formation in the Ser-146 variants (S146A and S146C). A solution of S146C (100 μm), FMNH− (50 μm), and HPA (20 mm) in 10 mm sodium phosphate buffer, pH 7.0, was mixed with air-saturated buffers of various pH values in vessels inside the anaerobic glove box. All concentrations represent those after mixing. After mixing for 5 min, the reactions were quenched with 0.15 m HCl, and the sample was analyzed using HPLC (“Experimental Procedures”). The analysis showed that 64 ± 3% of HPA was hydroxylated to form DHPA for the reactions of S146C·FMNH−·HPA in the pH range of 6.0–9.0. At pH 9.5 and 10.0, the hydroxylation percentage decreased to 47 ± 1 and 27 ± 4%, respectively (Table 2). This effect may be due to ionization of the cysteine residue of S146C at higher pH (more details are provided under “Discussion”).
For S146A, a solution of S146A·FMNH−·HPA (100 μm S146A, 50 μm FMNH−, 2 mm HPA) was mixed with air-saturated buffers in vessels inside the anaerobic glove box at various pH values, similar to the experiment performed with S146C. At all pH values in the range of 6.0–10.0, ∼70% DHPA formed per FMNH− used (Table 2). These results differed from those of S146C because pH had no effect on product formation by S146A. The Ala-146 side chain in S146A is not ionizable, suggesting that the decreased production of DHPA in S146C at pH > 9.0 must be due to ionization of the Cys-146 side chain.
DISCUSSION
Our results show that His-120 and Ser-146 are crucial for the hydroxylation activity of C2. His-120 is particularly important because mutation of this residue to a neutral or negative charge can abolish the hydroxylation, whereas a positive charge residue can substitute its function in hydroxylation. Ser-146 interactions with the substrate are not mandatory for hydroxylation but assist in positioning the enzyme-bound HPA in an optimum geometry for hydroxylation. According to x-ray structures of two-component monooxygenases that utilize phenolic compounds as substrates, these two residues are well conserved (24, 31, 32). Our findings imply that the conserved His/Ser pairs found in other two-component monooxygenases may have similar functional roles.
The analysis of the His-120 variants (H120N, H120Q, H120Y, H120D, H120E, H120K, and H120R) indicates that a positive charge at residue 120 enables the hydroxylation process. H120K and H120R can catalyze HPA hydroxylation, whereas the rest are inactive. H120K catalyzes hydroxylation of HPA with efficiency similar to that of the wild-type enzyme. The hydroxylation ratio of H120K was 75 ± 6% in a pH range of 6.0–10.0, and the hydroxylation rate constant was 5.7 ± 0.6 s−1, which is comparable with a value of 16 s−1 for the wild-type enzyme. H120R can also catalyze the hydroxylation of HPA to yield ∼10–15% DHPA at pH 6.0–10.0. These results imply that a positive charge at residue 120 supports HPA hydroxylation, possibly through the selective binding of the HPA phenolate. The pKa values of the side chains of Lys and Arg in free solution are generally ∼10.4 and ∼12, respectively (33). Although the pKa value of Lys can vary depending on the microenvironment of the protein active site, the pKa of Arg is generally constant (34, 35). Because both H120K and H120R can hydroxylate HPA, it is reasonable to assume that the lysine and arginine side chains in H120K and H120R have a similar protonation status, i.e. that they are positively charged at all pH values in a pH range of 6.0–10.0. Because no variation in the hydroxylation efficiency or rate constants due to the pH change was observed for wild-type C2 (15), the data imply that the protonation status of the key components required for hydroxylation remain constant in this pH range, i.e. His-120, may be positively charged.
The findings that H120N, H120Q, H120Y, H120D, and H120E variants are inactive also implies that a neutral or negative charge cannot replace the positive charge required at this residue. All variants can form C4a-hydroperoxy flavin with rate constants similar to that of the wild-type enzyme (supplemental Fig. 1–5, Table 1). The results in Fig. 5 indicate that the aberrant hydroxylation ability of H120N is not due to an abolishment of substrate binding. H120N can still bind HPA, as indicated by the change in absorbance at 390 nm at a reaction time of 2 s at various HPA concentrations and the influence of HPA on the rate constants of H2O2 elimination from C4a-hydroperoxy-FMN (Fig. 5, supplemental Fig. 7). Although the Kd for binding of HPA to H120N was ∼10-fold higher than the value for wild-type C2 (inset in Fig. 5), the 20 mm concentration of HPA used in the product determination experiment should result in the formation of at least 43 μm H120N·C4a-hydroperoxy-FMN·HPA complex. Although we cannot completely rule out the possibility that HPA binds to the H120N, H120Q, H120Y, H120D, and H120E variants with the wrong geometry for hydroxylation, the observed hydroxylation in the H120K, H120R, S146A, and S146C variants suggests that imperfect binding of HPA can result in hydroxylation if the mechanistic requirement is met. Therefore, the absence of hydroxylation activity in H120N is likely to be due to the lack of a positive charge at residue 120.
Based on the current data, we propose that His-120 interacts with HPA via formation of the His-120δ+·HPAδ− complex. His-120 and HPA share a common proton for which most of the positive charge characteristic remains at His-120 (Fig. 7). This setup is advantageous for facilitating the subsequent HPA hydroxylation process via an electrophilic aromatic substitution mechanism. The formation of the His-120δ+·HPAδ− complex may be a key requirement for efficient hydroxylation of C2 throughout the pH range of 6.0–10.0. The model in Fig. 7 implies an unusually low pKa value for the phenolic group of HPA and an unusually high pKa for His-120 upon enzyme·substrate complex formation. In free solution, the pKa of His is ∼6.4 (36). Unusually high pKa values for His have been reported for tyrosine phosphatase (pKa of ∼9.2) (37) and the complex of gallium-protoporphyrin (pKa of ∼9.7) (38). Nevertheless, we cannot rule out a model in which His-120 and HPA interact via a neutral hydrogen bond interaction and a partial charge pair complex, as in Fig. 7, exists transiently during the hydroxylation.
FIGURE 7.

Proposed mechanism for the hydroxylation of HPA catalyzed by C2. Hydroxylation occurs through an electrophilic aromatic substitution reaction. The formation of Hisδ+·HPAδ− increases substrate nucleophilicity and facilitates oxygen atom transfer from C4a-hydroperoxy-FMN as an electrophile in the subsequent step. Ser-146 is not directly involved in the reaction but facilitates efficient hydroxylation by C2.
The analysis of S146A and S146C suggests that the role of Ser-146 is the optimal alignment of the C2-bound HPA for hydroxylation and that this residue, unlike His-120, is not directly engaged in HPA hydroxylation. The importance of Ser-146 lies in its proper size and its ability to form a hydrogen bond with an oxygen atom of HPA (Fig. 1). S146A forms DHPA with a rate constant of 2.6 ± 0.1 s−1 (∼6-fold lower than the wild-type value) and a product coupling ratio of 66 ± 2% at pH 7.0. For S146C, the hydroxylation rate constant is 0.35 ± 0.05 s−1 (45-fold less than wild-type C2), and 68 ± 3% DHPA was produced. These results indicate that replacing the side chain of residue 146 with a thiol group (S146C), which is capable of making a hydrogen-bond interaction but has a much larger van der Waals radius, impaired the hydroxylation step more than replacing it with a comparably sized methyl side chain (S146A). Therefore, the role of Ser-146 is to align the C2-bound HPA so that the substrate can be efficiently hydroxylated by C4a-hydroperoxy-FMN. These results also imply that Ser-146 is not a key residue for committing the substrate for hydroxylation. In this respect, Ser-146 and His-120 have interesting, discrete functions in substrate binding, although the Oγ atom of Ser-146 and Nϵ atom of His-120 are located ∼2.8 Å from the phenolic moiety of HPA (23).
The decreased hydroxylation efficiency of S146C at pH values greater than 9.0 (Table 2) also supports the model in Fig. 7, which depicts the His-120δ+·HPAδ− complex as a required enzyme·substrate complex for productive hydroxylation. Because no pH effect was observed for product formation by S146A (Table 2), the pH effect on S146C must be due to an ionization of the Cys-146 side chain. In general, the pKa of cysteine in aqueous solution is 9.1 (36). The observation that adverse effects on product formation began at pH 9.5 suggests that hydroxylation is disfavored when the side chain of Cys-146 is deprotonated. This may be due to a repulsive interaction between the negatively charged thiolate and the negatively charged HPA that may subsequently disturb the formation of the His-120δ+·HPAδ− complex.
The requirement for formation of the His-120δ+·HPAδ− complex suggests that the hydroxylation likely occurs via an electrophilic aromatic substitution mechanism (Fig. 7). For single-component flavin-dependent monooxygenases, studies using a series of 8-substituted flavin analogs in the reactions of PHBH and MHPCO have shown that oxygen atom transfer occurs by an electrophilic aromatic substitution mechanism (10–12). Bronsted plots of the hydroxylation rate constants versus the pKa values of the C4a-hydroxy flavin, which represent the stability of the C4a-flavin-alkoxide (a leaving group in the second step in Fig. 7), yield a negative slope. These data indicate that a partial negative charge is formed at the flavin leaving group during the transition state of the reaction (9–12). Because the hydroxylation of an aromatic compound catalyzed by C2 is similar to the reactions of PHBH and MHPCO, it can be envisaged that the electrophilic aromatic substitution could operate in the C2 reaction. The formation of the His-120δ+·HPAδ− complex should set a convenient stage for the electrophilic aromatic substitution reaction to proceed because it increases the nucleophilicity of HPA due to a higher electron density in the negatively charged HPA that facilitates the subsequent electrophilic attack by the terminal oxygen of C4a-hydroperoxy flavin (Fig. 7).
The increase in substrate nucleophilicity is important for the ability of flavin-dependent aromatic monooxygenases to enable efficient oxygen atom transfer reactions. Different enzymes achieve this process through different mechanisms. Our results suggest that C2 may allow formation of the His-120δ+·HPAδ− complex throughout pH range of 6.0–10.0 such that the negatively charged HPA is prone to receive the electrophile (Fig. 7). Selective binding of a deprotonated hydroxyl group to facilitate the electrophilic aromatic substitution has been observed for the reaction of MHPCO (11, 12, 39). Although the substrate (MHPC) of MHPCO exists as a tautomeric mixture at pH 7.0, only the tripolar ionic form in which the hydroxyl group (equivalent to the phenolic group in HPA) is deprotonated binds to MHPCO (12, 39). For the PHBH reaction, both the phenolic and phenolate forms of the substrate (pOHB) can bind to the enzyme, depending on the pH of the solution. However, once bound to the enzyme, the pKa of pOHB decreases to 7.4, ∼1.9 units below the regular pKa of pOHB in solution (pKa = 9.3) (6, 9, 40). The deprotonation of pOHB is necessary for facilitating the hydroxylation process because the hydroxylation rate constant increases ∼6-fold with a rise in pH according to a pKa of 7.1 (9, 13). The pKa of pOHB bound in PHBH is lowered by a hydrogen-bonding network that links pOHB with Tyr-201, Tyr-385, molecular water, and the surface residue His-72 (6, 9, 13, 14, 40). Recently, a study of styrene monooxygenase, another two-component flavin-dependent enzyme that also employs C4a-hydroperoxy flavin as an oxygenation species, has shown that the oxygenation (epoxidation) reaction of styrene monooxygenase is favored at lower pH values, consistent with a pKa of 7.7 (41). The functional group associated with this pKa value has not been assigned and is thought to be associated with a pKa value in the active site. The above examples demonstrate that flavin-dependent monooxygenases employ different active site fine-tuning mechanisms to achieve oxygen atom transfer.
The roles of His-120 and Ser-146 in the C2 active site in providing specific interactions with HPA to achieve effective hydroxylation may also be present in other flavin-dependent monooxygenases. Homologues of these residues are found in the oxygenase component (HpaB) of HPAH from T. thermophilus HB8, in which the O atom of Thr-104 and the Nϵ of His-142 are 2.5 and 2.8 Å from the hydroxyl group of HPA, respectively (24), and in the oxygenase involved in the cholesterol catabolic pathway of Mycobacterium tuberculosis, in which His-92 and Ser-118 are about 2.4 Å from the hydroxyl group of the modeled 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione substrate (32). For the oxygenase (TftD) of chlorophenol 4-monooxygenase from Burkholderia cepacia AC1100, His-289 was proposed to interact with a hydroxyl group of 2,5-dichlorohydroquinone docked in the structure of TftD (31). Site-directed mutagenesis of His-289 to alanine in TftD abolished the hydroxylation activity of TftD when 2,5-dichlorohydroquinone was used as a substrate (31). Based on our current data, we speculate that the His-289 residue in TftD may have a similar function as His-120 in C2.
In conclusion, His-120 and Ser-146 are important catalytic residues in C2. Formation of the His-120δ+·HPAδ− complex may be a key factor that prompts the enzyme to readily catalyze the oxygenation reaction via an electrophilic aromatic substitution mechanism. Ser-146 is important for proper binding of HPA for efficient hydroxylation. The mechanism by which C2 facilitates oxygen atom transfer is different from that of single component enzymes such as PHBH. The findings here are useful for the future application of two-component enzymes as biocatalysts.
Supplementary Material
Acknowledgment
We thank Barrie Entsch, University of New England, New South Wales, Australia, for critical reading of the manuscript.
This work was supported in part by Thailand Research Fund Grant BRG5480001 and a grant from the Faculty of Science, Mahidol University (to P. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Procedures, Tables 1 and 2, and Figs. 1–9.
- pOHB
- p-hydroxybenzoate
- PHBH
- p-hydroxybenzoate hydroxylase
- HPA
- p-hydroxyphenylacetate
- HPAH
- p-hydroxyphenylacetate hydroxylase
- DHPA
- 3,4-dihydroxyphenylacetate
- C1
- the reductase component of HPAH from A. baumannii
- C2
- the oxygenase component of HPAH from A. baumannii
- MHPCO
- 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase
- TftD
- the oxygenase component of chlorophenol 4-monooxygenase from B. cepacia AC1100
- AU
- absorbance units.
REFERENCES
- 1. Torres Pazmiño D. E., Winkler M., Glieder A., Fraaije M. W. (2010) J. Biotechnol. 146, 9–24 [DOI] [PubMed] [Google Scholar]
- 2. van Berkel W. J., Kamerbeek N. M., Fraaije M. W. (2006) J. Biotechnol. 124, 670–689 [DOI] [PubMed] [Google Scholar]
- 3. Fagan R. L., Palfey B. A. (2010) Comprehensive Natural Products II Chemistry and Biology, Vol. 7, pp. 37–117, Elsevier Science Publishing Co., Inc., Oxford [Google Scholar]
- 4. Ballou D. P., Entsch B., Cole L. J. (2005) Biochem. Biophys. Res. Commun. 338, 590–598 [DOI] [PubMed] [Google Scholar]
- 5. Ellis H. R. (2010) Arch. Biochem. Biophys. 497, 1–12 [DOI] [PubMed] [Google Scholar]
- 6. Palfey B. A., McDonald C. A. (2010) Arch. Biochem. Biophys. 493, 26–36 [DOI] [PubMed] [Google Scholar]
- 7. Palfey B. A., Massey V. (1998) Comprehensive Biological Catalysis, Vol. 3, pp. 37–83, Chapman & Hall, Glasgow, UK [Google Scholar]
- 8. Sucharitakul J., Phongsak T., Entsch B., Svasti J., Chaiyen P., Ballou D. P. (2007) Biochemistry 46, 8611–8623 [DOI] [PubMed] [Google Scholar]
- 9. Entsch B., Cole L. J., Ballou D. P. (2005) Arch. Biochem. Biophys. 433, 297–311 [DOI] [PubMed] [Google Scholar]
- 10. Ortiz-Maldonado M., Ballou D. P., Massey V. (1999) Biochemistry 38, 8124–8137 [DOI] [PubMed] [Google Scholar]
- 11. Chaiyen P., Sucharitakul J., Svasti J., Entsch B., Massey V., Ballou D. P. (2004) Biochemistry 43, 3933–3943 [DOI] [PubMed] [Google Scholar]
- 12. Chaiyen P. (2010) Arch. Biochem. Biophys. 493, 62–70 [DOI] [PubMed] [Google Scholar]
- 13. Ortiz-Maldonado M., Entsch B., Ballou D. P. (2004) Biochemistry 43, 15246–15257 [DOI] [PubMed] [Google Scholar]
- 14. Palfey B. A., Moran G. R., Entsch B., Ballou D. P., Massey V. (1999) Biochemistry 38, 1153–1158 [DOI] [PubMed] [Google Scholar]
- 15. Ruangchan N., Tongsook C., Sucharitakul J., Chaiyen P. (2011) J. Biol. Chem. 286, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chaiyen P., Suadee C., Wilairat P. (2001) Eur. J. Biochem. 268, 5550–5561 [DOI] [PubMed] [Google Scholar]
- 17. Thotsaporn K., Sucharitakul J., Wongratana J., Suadee C., Chaiyen P. (2004) Biochim. Biophys. Acta 1680, 60–66 [DOI] [PubMed] [Google Scholar]
- 18. Arunachalam U., Massey V., Vaidyanathan C. S. (1992) J. Biol. Chem. 267, 25848–25855 [PubMed] [Google Scholar]
- 19. Chakraborty S., Ortiz-Maldonado M., Entsch B., Ballou D. P. (2010) Biochemistry 49, 372–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galán B., Díaz E., Prieto M. A., García J. L. (2000) J. Bacteriol. 182, 627–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sucharitakul J., Chaiyen P., Entsch B., Ballou D. P. (2006) J. Biol. Chem. 281, 17044–17053 [DOI] [PubMed] [Google Scholar]
- 22. Sucharitakul J., Chaiyen P., Entsch B., Ballou D. P. (2005) Biochemistry 44, 10434–10442 [DOI] [PubMed] [Google Scholar]
- 23. Alfieri A., Fersini F., Ruangchan N., Prongjit M., Chaiyen P., Mattevi A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1177–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim S. H., Hisano T., Takeda K., Iwasaki W., Ebihara A., Miki K. (2007) J. Biol. Chem. 282, 33107–33117 [DOI] [PubMed] [Google Scholar]
- 25. Kim S. H., Hisano T., Iwasaki W., Ebihara A., Miki K. (2008) Proteins 70, 718–730 [DOI] [PubMed] [Google Scholar]
- 26. Okai M., Kudo N., Lee W. C., Kamo M., Nagata K., Tanokura M. (2006) Biochemistry 45, 5103–5110 [DOI] [PubMed] [Google Scholar]
- 27. Thotsaporn K., Chenprakhon P., Sucharitakul J., Mattevi A., Chaiyen P. (2011) J. Biol. Chem. 286, 28170–28180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duch D. S., Laskowski M., Sr. (1971) Anal. Biochem. 44, 42–48 [DOI] [PubMed] [Google Scholar]
- 29. Sucharitakul J., Prongjit M., Haltrich D., Chaiyen P. (2008) Biochemistry 47, 8485–8490 [DOI] [PubMed] [Google Scholar]
- 30. Connors K. A. (1990) Chemical Kinetics: The Study of Reaction Rates in Solution, 1st Ed., pp. 62–66, Wiley-VCH, New York [Google Scholar]
- 31. Webb B. N., Ballinger J. W., Kim E., Belchik S. M., Lam K. S., Youn B., Nissen M. S., Xun L., Kang C. (2010) J. Biol. Chem. 285, 2014–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dresen C., Lin L. Y., D'Angelo I., Tocheva E. I., Strynadka N., Eltis L. D. (2010) J. Biol. Chem. 285, 22264–22275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nozaki Y., Tanford C. (1967) Methods Enzymol. 11, 715–734 [Google Scholar]
- 34. Riordan J. F., McElvany K. D., Borders C. L., Jr. (1977) Science 195, 884–886 [DOI] [PubMed] [Google Scholar]
- 35. André I., Linse S., Mulder F. A. (2007) J. Am. Chem. Soc. 129, 15805–15813 [DOI] [PubMed] [Google Scholar]
- 36. Fersht A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, pp. 169–173, W. H. Freeman and Co, New York [Google Scholar]
- 37. Tishmack P. A., Bashford D., Harms E., Van Etten R. L. (1997) Biochemistry 36, 11984–11994 [DOI] [PubMed] [Google Scholar]
- 38. Wolff N., Deniau C., Létoffé S., Simenel C., Kumar V., Stojiljkovic I., Wandersman C., Delepierre M., Lecroisey A. (2002) Protein Sci. 11, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chaiyen P., Brissette P., Ballou D. P., Massey V. (1997) Biochemistry 36, 13856–13864 [DOI] [PubMed] [Google Scholar]
- 40. Entsch B., Palfey B. A., Ballou D. P., Massey V. (1991) J. Biol. Chem. 266, 17341–17449 [PubMed] [Google Scholar]
- 41. Kantz A., Gassner G. T. (2011) Biochemistry 50, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.