

Abstract
Background
In a context of climate change, phenotypic plasticity provides long-lived species, such as trees, with the means to adapt to environmental variations occurring within a single generation. In eucalyptus plantations, water availability is a key factor limiting productivity. However, the molecular mechanisms underlying the adaptation of eucalyptus to water shortage remain unclear. In this study, we compared the molecular responses of two commercial eucalyptus hybrids during the dry season. Both hybrids differ in productivity when grown under water deficit.
Results
Pyrosequencing of RNA extracted from shoot apices provided extensive transcriptome coverage - a catalog of 129,993 unigenes (49,748 contigs and 80,245 singletons) was generated from 398 million base pairs, or 1.14 million reads. The pyrosequencing data enriched considerably existing Eucalyptus EST collections, adding 36,985 unigenes not previously represented. Digital analysis of read abundance in 14,460 contigs identified 1,280 that were differentially expressed between the two genotypes, 155 contigs showing differential expression between treatments (irrigated vs. non irrigated conditions during the dry season), and 274 contigs with significant genotype-by-treatment interaction. The more productive genotype displayed a larger set of genes responding to water stress. Moreover, stress signal transduction seemed to involve different pathways in the two genotypes, suggesting that water shortage induces distinct cellular stress cascades. Similarly, the response of functional proteins also varied widely between genotypes: the most productive genotype decreased expression of genes related to photosystem, transport and secondary metabolism, whereas genes related to primary metabolism and cell organisation were over-expressed.
Conclusions
For the most productive genotype, the ability to express a broader set of genes in response to water availability appears to be a key characteristic in the maintenance of biomass growth during the dry season. Its strategy may involve a decrease of photosynthetic activity during the dry season associated with resources reallocation through major changes in the expression of primary metabolism associated genes. Further efforts will be needed to assess the adaptive nature of the genes highlighted in this study.
Background
Planted forests constitute only 7% of the global forested area, but contribute to a significant proportion of overall forest goods and services (e.g. up to 35% of industrial roundwood supply). In the context of climate change, the adaptation of planted forests is essential for a sustainable forestry sector. The adaptation of industrial plantations to present and future environmental conditions (including extreme weather events) depends on several factors, including the genetic diversity of the material used for reforestation and the phenotypic plasticity of individual genotypes. Genetic diversity ensures that forest trees can survive, adapt and evolve under changing environmental conditions [1,2], whereas phenotypic plasticity constitutes a shorter term response to environmental changes at the individual level of particular importance in long-lived organisms, such as trees [2,3].
Eucalyptus is one of the key genera among planted trees. The genus is includes the most important hardwood fibre crops species planted worldwide (19 million hectares according to [4]). Several Eucalyptus species grow rapidly and are highly adaptable. These properties led to their introduction worldwide, at latitudes extending from southern Europe to South Africa. In its natural range (Australia and some nearby islands), Eucalyptus are also found in a diverse spectrum of ecological niches. The genetic diversity of Eucalyptus has been studied extensively and remarkable levels of variation have been detected using neutral markers [5-11] and in genes possibly involved in adaptive traits [12-14]. Phenotypic plasticity is also likely to ensure better adaptation of individual genotypes to changing environmental conditions [15] and is of particular importance in clonal forestry.
Ecophysiological studies have shown that water is the principal factor limiting stem growth in Eucalyptus (e.g. [16,17]). Moreover, some studies have reported that eucalyptus genotypes differ in terms of their capacity for phenotypic modification in response to water deficit [18-20]. Several physiological mechanisms for coping with drought have been described in these species: i) the regulation of transpiration to decrease water loss [21], ii) resource reallocation from the shoot to the root, to increase water uptake [17], and iii) adjustment of osmotic potential [22] or protection against reactive oxygen species, to prevent damage due to stress [20]. Drought tolerance mechanisms have been described in detail at the molecular level for both annual and perennial model plants, such as Arabidopsis [23-25] and Populus [26-28], but little is known about the molecular basis of drought tolerance in Eucalyptus, particularly in field conditions.
Next-generation sequencing (NGS) provides new opportunities for studies of the molecular plasticity in response to water deficit. The high throughput of NGS is particularly useful in non-model organisms for which few genomic resources are available [29]. Moreover, NGS is suitable for transcript profiling, combining the high throughput of serial analysis of gene expression (SAGE) with the functional annotation capacity of EST sequencing [30]. These techniques have been widely used for transcriptome profiling, particularly for studies of biotic [31] and abiotic [24] stress responses, and the characterisation of developmental processes [32]. Considerable sequencing depth can be obtained, making it possible to identify transcriptome expression variation [29].
In plants, the shoot apical meristem (SAM) is a key organ in stem development. The SAM initiates phytomers and regulates shoot growth by integrating several signals, such as hormones (ABA, auxins, cytokinins) and transcription (e.g. homeobox) [33]. When plants are subjected to environmental stimuli, the leaf developmental network is adjusted by changes in shoot apex activation [34]. In Eucalyptus, EST resources have been developed for various tissues, such as roots, leaves and wood-forming tissues [35-38], but a limited number of genomic resources are available for shoot apices, despite the important role of this organ in plant organogenesis.
In this study, we compared transcript profiles in the shoot apices of two eucalyptus genotypes used in industrial plantations, under two watering regimes -- irrigated (IR) versus non-irrigated (NI). The two genotypes differ in their growth rates and ecophysiological characteristics at maturity, with one genotype being more productive and water use-efficient than the other. We used pyrosequencing (Roche 454) to sequence non-normalized cDNA libraries constructed from shoot tip mRNA. After verifying technical reproducibility, we addressed the following questions: i) Are there molecular differences between genotypes, reflected in the contrasting phenotypes, and do these differences affect specific pathways or have a random effect on the transcriptome? ii) Can we detect molecular plasticity in the response to water shortage during the dry season, and which pathways are affected? iii) Does this plasticity differ between genotypes (i.e. is there any genotype-by-environment interaction?), and which genes or pathways reflect these differences?
Methods
Plant material
We compared the response of two eucalyptus genotypes, 1-41 (NCBI Taxonomy ID: 764271) and 18-50 (NCBI Taxonomy ID: 765255), to water shortage during the dry season of 2008. These two genotypes are used in industrial plantations in the Republic of Congo. Hybrid 1-41 (named G1 in the following sections) was obtained by open pollination of E. alba (the male parent is unknown) and the hybrid 18-50 (named G2) was derived from a controlled pollination of E. urophylla (genotype 14-36) by E. grandis (genotype 9-10). These two hybrids differ in their growth rates and water use efficiency (WUE, estimated by isotopic carbon composition) at maturity, G2 being superior than G1.
Field experiment
Trees were vegetatively propagated by rooted cuttings and established in a field experiment in Yanika, Republic of Congo (4°20'S, 11°38'E, 50 m above sea level), in June 2007. Trees were planted in plots of 64 cuttings per genotype and per treatment, including a buffer zone of 40 plants. Two watering regimes were used during the dry season: no irrigation (NI) and irrigation (IR). Trees were watered with sprinklers, to replenish evapotranspiration losses, estimated at 3 mm per day. In order to evaluate the effect of water deficit on above-ground biomass growth and molecular plasticity, plant material was sampled in September 2008, 16 months after the trees were planted. The dry season began approximately on May 15th 2008 -therefore, trees under NI treatment were subjected to four months without rainfall by the time samples were collected.
Soil water content
Volumetric water content (VWC) was measured by time domain reflectometry (TDR; Trase system, Soil moisture, Santa Barbara, CA). Four series of TDR probes per genotype and per treatment were installed horizontally, at six depths (0.15, 0.5, 1, 2, 3 and 4 m). Mean values were calculated from the four replicated measurements at each depth.
Biomass production
We harvested 11 trees per treatment and dissected them into the following compartments: stem, dead branches, living branches and leaves. Each compartment was weighed in the field. Representative subsamples of each compartment were then harvested, and weighed before and after drying at 65 °C to constant weight. Water content was calculated for each of these subsamples and used to estimate total dry biomass for each compartment. Total above-ground biomass (the sum of all the compartment) was analysed by two-way ANOVA, with R (R Development Core Team), according to the following model:
where Xijk is the above-ground biomass in genotype i (G1 or G2), treatment j (NI or IR) and replicate k. a, b and c are the regression coefficients of G, the genotypic effect, T, the treatment effect and G × T the interaction between genotype and treatment, and εijk is the residual.
cDNA synthesis
The experimental design, from tissue sampling to library construction and sequencing, is described in figure 1. Shoot apices were collected from three trees from each genotype and treatment, and immediately frozen in liquid nitrogen. Two RNA extractions of three apices from each tree were performed, as previously described by Reid et al. [39]. RNA samples were treated with Turbo RNase-Free DNase (Ambion, Austin, TX, USA) and purified with the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). RNA concentration and quality were analysed with an Agilent Technologies 2100 Bioanalyser (Agilent Technologies, Mississauga, ON, USA) and a ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE, USA). The three RNA preparations per replicate and per condition (corresponding to three trees), were pooled in equal proportions, providing templates for cDNA libraries S1-S8 (figure 1). Full-length cDNA was obtained from 1 μg of RNA, with the Smart cDNA Library Construction Kit (Clontech, Mountain View, CA, USA), according to the manufacturer's instructions. We amplified the cDNA with PCR Advantage II Polymerase (Clontech, Mountain View, CA, USA), over 16 cycles (7 s at 95 C, 20 s at 66 °C, and 4 min at 72 °C). This cDNA amplification procedure was repeated eight times in separate tubes for each sample, with pooling to give a total of 6 μg of cDNA fragments longer than 1,000 bp quantified with an Agilent Technologies 2100 Bioanalyzer. Eight cDNA libraries (S1-S8) were constructed, giving two biological replicates for the two genotypes (G1, G2) subjected to the two watering regimes (IR, NI).
Figure 1.

Procedure used, from tissue sampling to sequencing. Two genotypes (G1 and G2) were subjected to two watering regimes (IR and NI). Shoot apices from three trees per genotype and per treatment were collected in the field. Total RNA was extracted from three apices per tree. Two replicate RNA extractions were carried out for the construction of two independent replicate cDNA libraries per genotype and per treatment (resulting in 8 templates for cDNA library construction). For sequencing, each cDNA library was tagged, and two independent multiplexes were created by pooling one sample for each genotype and treatment combination. Multiplex #1 was sequenced with two 454-Roche FLX Titanium half-runs, resulting in eight sequencing sets, whereas multiplex #2 was sequenced with one half-run and resulted in four sequencing sets.
Library construction, 454-sequencing, sequence quality control and assembly
We nebulised 5 μg of each cDNA sample to a mean fragment size of 650 bp and ligated it to an adaptor, according to standard procedures [40]. Each cDNA library was tagged with Multiplex Identifiers (MID) barcode adaptors, and two independent multiplexes were created by pooling one sample from each genotype and treatment. Multiplex #1 comprised samples S1, S3, S5 and S7, whereas muliplex #2 comprised samples S2, S4, S6 and S8. One half-run (run A) of sequencing was initially carried out for multiplex #1 on a GS-FLX Titanium platform (454 Life Science, Brandford, CT, USA) at Cogenics (Meylan, France). Two half-runs of sequencing for multiplex #1 (run B) and multiplex #2 (run C) were then performed by Agencourt (Beverly, MA, USA) on a GS-FLX Titanium sequencer. Base calling with GS-FLX System software generated 353,344 high-quality reads for the first half-run and in 785,322 reads for the second complete run. Sequences were deposited at the NCBI short-read archive (SRA) under accession number SRA012867.2 (Figure 2). The 454-sequencing reads (1,138,666 from this study and 1,041,876 from Novaes et al. [13]) were screened by cross_match (http://bozeman.mbt.washington.edu/phrap.docs/phrap.html) for primers and adaptors and then masked. For each 454-sequencing read, the longest non-masked region was extracted and further trimmed with SeqClean (http://compbio.dfci.harvard.edu/tgi/). The shorter regions were discarded to eliminate potential chimeras. Sequences were assembled as previously described [41], with TGICL [42], using the 12 sets of sequencing data from this study and the four sets of sequencing data obtained for E. grandis [accession number SRA001122] by Novaes et al. [13]. In parallel, all reads were stored in the NG6 system (http://vm-bioinfo.toulouse.inra.fr/ng6/, project: BIOGECO eucalyptus) and three kinds of analysis were performed for each of the 16 sequencing sets, as previously described [41]: i) BLAST search for E. coli, phage and yeast contaminants, ii) read quality analysis and iii) removal of sequences that were too long or too short, sequences with an excess of errors (more than 4% of N), low-complexity sequences and duplicated reads, using Pyrocleaner program. Only unigene elements (UE) resulting from sequences generated in this experiment (the E.spp sequencing set) were used for digital gene expression analysis.
Figure 2.

Pipeline used for sequence analysis. White boxes correspond to sequence generation, red boxes correspond to library quality control, assembly and annotation and green boxes correspond to abundance and functional analysis. The software suite or program used is indicated in the upper part of the box and the results of each step of the analysis are shown under the box. UE: unigene element; "G": genetic effect, "T": treatment effect and "GxT": genotype by treatment interaction effect. *E. grandis. sff files were obtained from Novaes et al. (2008). α: available at http://genotoul-contigbrowser.toulouse.inra.fr:9092/Eucalyptus/index.html β: available at http://vm-bioinfo.toulouse.inra.fr/ng6/, user = euca; password = euca33bio!
Digital gene expression analysis
Contigs with less than 10 reads for the 12 sequencing sets generated in this study were eliminated from further statistical analyses. For the 14,460 remaining contigs, the numbers of reads per sequencing set and per contig were used to assess gene abundance. Two types of statistical analysis were performed. First, pairwise comparisons were carried out between genotypes (G2 vs. G1 sequencing sets, irrespective of treatment) and between treatments (IR vs. NI sequencing sets, irrespective of genotype). Four additional comparisons were carried out for each genotype and each treatment, as follows: IR vs. NI for genotypes G1 and G2, G1 vs. G2 for treatments IR and NI. Statistical tests, based on the use of the MARS method in the DEGseq package [43] were performed to assess differential expression [43]. Second, two-way analysis of variance (ANOVA) was performed on contigs, making use of the three replicates (run A, B and C) per treatment to estimate random variation and test the genotype (G), treatment (T) and genotype-treatment interaction (GxT) effects. Transcript abundance was normalized by dividing the number of reads by the sequence length of the contigs and the total number of sequences in each sequencing set. Contigs with a q-value <0.05 in the DEGseq test (i.e. after false discovery rate corrections) [44] and with p-values <0.05 after ANOVA were considered to be differentially expressed and were extracted for further analysis. The 14,460 genes analysed were classified into four classes: not significantly differentially expressed (NS), and showing genotype ("G" contigs), treatment ("T" contigs) or genotype × treatment ("GxT" contigs) effects. For the comparison of expression levels, we used log2-transformed fold-changes between contig abundances in the various contrasts obtained in the DEGseq analysis.
Functional annotation of differentially expressed genes
Contigs were assigned a putative function by BLASTX [45], using various public databases: UniProtKB/Swiss-Prot (release 57.1), RefSeq Protein (release 34), Pfam (release 23.0), with an e-value cut-off value of 10e-5. Sequences were also compared to TIGR's assemblies of Arabidopsis_thaliana (release 14), Helianthus_annuus (release 6), Populus (release 4), Picea (release 3) and Vitis vinifera (release 6), with an e-value cut-off of 10e-2. Gene Ontology terms were assigned via the UniprotKB accession and clustered with Blast2GO [46]. The differential distributions of each class of effect (T, G, GxT and NS) between Biological Processes, Molecular Functions and Cellular Components were assessed using Fisher's exact tests, with a significance threshold of 0.05. Pathway analysis was carried out with Mapman [47]. Differentially expressed contigs were assigned to functional categories (or bins) by Mercator (http://mapman.gabipd.org/web/guest/mercator). A dedicated pathway map was created to represent most of these contigs. The Wilcoxon rank sum test was used to identify differentially regulated bins.
Results
Monitoring of soil water content
A factorial design including two genotypes (G1 and G2) and two water regimes (irrigated IR vs. non-irrigated NI) was established in a field trial in one of the main areas of eucalyptus plantation in the Republic of Congo. The experiment was evaluated over a period of two years. Soil water content (SWC) was monitored throughout the experiment at six depths (0.15-4 m), to assess water availability in different experimental conditions. In this study, we focus on the effect of water availability in the second dry season (after four months without rainfall) on biomass production and the transcriptome.
In the NI treatment, SWC varied from 4.5% to 8%, and no significant difference was found between the two genotypes (Figure 3). SWC values were close to wilting point (pF 4.2), i.e., when plants ceased to be able to absorb soil water. In the IR treatment, SWC ranged from 11% to 17%, and exceeded field capacity (pF 2.0), indicating that water was not a limiting factor for tree growth. SWC was also higher in the area surrounding genotype G2 than in genotype G1, except at two depths (1 m and 4 m). This result suggests that the two genotypes absorb water preferentially from different depths, possibly because their root system develops differently.
Figure 3.

Profile of soil water content (SWC). Means and standard errors of SWC at six depths, for both genotypes (G1 and G2), subjected to two water regimes (irrigated: IR and not irrigated: NI). SWC at wilting point (pF4.2) and at field capacity (pF2) were calculated from data obtained at a site close to the experimental field (Laclau, personal communication).
Effect of water deficit on biomass production
Genotype (p < 0.0001) and treatment (p < 0.001) had significant effects on above-ground biomass production (stem, dead branches, living branches and leaves) (Figure 4). Mean biomass was much higher for genotype G2 (12.8 kg) than for genotype G1 (8.6 kg) confirming earlier findings [48]. Not surprisingly, mean biomass was higher for the IR treatment (11.8 kg) than for the NI treatment (9.7 kg). A two-way ANOVA showed that the GxT interaction effect was not significant (Additional file 1), but relative biomass loss was nonetheless lower for genotype G2 (12%) than for genotype G1 (24%). These results suggest that the growth of genotype G2 is less affected by water shortage compared to G1.
Figure 4.

Above-ground biomass. Box plot of above-ground biomass calculated for 11 trees per treatment, for both genotypes (G1 and G2), subjected to two water regimes (irrigated: IR and not irrigated: NI). Centre line and outside edge of each box indicate the median and range of inner quartile around the median, respectively; vertical lines on the two sides of the box represent the first and the ninth decile, respectively. Letters indicate the groups obtained in Bonferroni tests for multiple pairwise comparisons.
Sequencing of the Eucalyptus shoot apex transcriptome
We chose to sequence the shoot apex transcriptome to study the molecular response of eucalyptus to water deficit, due to its role in shoot organogenesis [49]. Shoot apices were pooled from three trees in four sets of conditions (2 genotypes × 2 treatments). Three half-runs (A, B, C) of 454-Roche FLX Titanium sequencing provided 353,344 (run A), 405,223 (run B) and 380,099 (run C) reads, for a total of 1,138,666 sequences (398 Mb). The mean read length was 334 bp for run A, 369 bp for run B and 344 bp for run C (Table 1). Reads were slightly shorter for run A, with a higher abundance of reads comprised between 400-420 bp in length, whereas runs B and C were characterised by reads of 460-480 bp in length (Additional file 2). To increase contig length of the assembly, we combined all the reads (1,138,666) generated in this study (E. spp sequencing set) with other GS-20 and GS-FLX 454 reads (1,041,876 reads) from various organs of E. grandis (E. grandis sequencing set; [13]). Figure 2 summarises the various stages in sequence analysis, corresponding to data quality control, read assembly, annotation and abundance analysis. After removal of vector and adaptor sequences, 1,994,741 reads were available for assembly. Assembly with TGICL generated 231,715 unigene elements (UE) comprising 80,854 contigs and 150,861 singletons. Removal of low-quality sequences and duplicated reads with the NG6 platform resulted in a total of 202,279 UE (69,584 contigs and 132,690 singletons), from which 129,993 UE (49,748 contigs and 80,245 singletons) were identified in the E. spp sequencing set (this study) and used for digital gene expression analysis (Table 2).
Table 1.
Summary statistics for the three 454-sequencing half-runs
| Sample | Genotype | Treatment | Sequencing set | Runs | # of Reads | # of Reads | # of bp | Average length of reads (bp) | Average length of reads (bp) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | G1 | IR | 58,921 | 19,313,318 | 328 | ||||
| 3 | G2 | IR | SS2: G2IR_A | 92,165 | 30,365,092 | 329 | |||
| 5 | G1 | NI | SS3: G1-NI_A | run A | 353,344 | 95,500 | 31,350,808 | 328 | 334 |
| 7 | G2 | NI | SS4: G2NI_A | 106,758 | 37,126,644 | 348 | |||
| 1 | G1 | IR | SS5: G1IR_B | 139,137 | 51,085,203 | 367 | |||
| 3 | G2 | IR | SS6: G2IR_B | run B | 405,223 | 112,051 | 41,110,040 | 367 | |
| 5 | G1 | NI | SS7: G1-NI_B | 59,907 | 21,833,839 | 364 | 369 | ||
| 7 | G2 | NI | SS8: G2NI_B | 94,128 | 35,374,789 | 376 | |||
| 2 | G1 | IR | SS9: G1IR_C | 93,651 | 32,338,261 | 345 | |||
| 4 | G2 | IR | SS10: G2IR_C | 90,511 | 31,595,445 | 349 | |||
| 6 | G1 | NI | SS11: G1NI_C | run C | 380,099 | 83,530 | 27,929,481 | 334 | 344 |
| 8 | G2 | NI | SS12: G2NI_C | 112,407 | 38,802,862 | 345 | |||
| TOTAL | 1.5 run GS-FLX Titanium | 1,138,666 | 1,138,666 | 398,225,782 | 350 | 350 | |||
Table 2.
Assembly statistics from TGICL (I), and figures obtained after Pyrocleaner analysis (II), for the set of sequences reported here (III)
| TGICL (I) | Pyrocleaner (II) | E. spp (III) | |
|---|---|---|---|
| # Contigs | 80,854 | 69,584 | 49,748 |
| # reads in contigs | 1,843,806 | 1,386,859 | 851,751 |
| Average length of all contigs (bp) | 552 | 608 | 734 |
| # Large Contigs >500 bp | 34,076 | 33,962 | 32,694 |
| Average length of large contigs | 900 | 901 | 912 |
| # Singletons | 150,861 | 132,695 | 80,245 |
| # UE | 231,715 | 202,279 | 129,993 |
Contribution of the three half-run replicates
In total, 90,579 UE (70% of E. spp UE) did not have sequence similarity to the E. grandis sequence reads from Novaes et al. [13]. Most of these sequences were singletons (71,761), although some were contigs (18,818), corresponding to 27% of the E. spp contigs. A BLAST homology search (cut-off: 10-10) of published eucalyptus databases (ESTs from GenBank available on April 2010; 454-ESTs generated by Novaes et al., [13]; 454-ESTs from JGI from E. globulus xylem and leaf tissues; Illumina contigs generated by Mizrachi et al., [38]) showed that 21,401 UE (comprising 3,066 contigs and 18,335 singletons) did not match any known sequence. Thus, the resource described here greatly extends the list of genes known to be expressed in Eucalyptus, which will be critical for the annotation of the genome sequence. Due to the smaller number of reads in run A, the total number of UE including reads generated from run A was also smaller (58,763) than that generated from the other two runs (67,467 in run B and 67,756 in run C). Each supplementary half-run produced between 16 and 21% new contigs for a second half-run, and between 5 and 7% for a third half-run (Additional file 3). Vega-Arreguin et al. [50] reported similar trends in maize, with a plateau of gene representation reached after the third successive GS-20 454-sequencing run. The number of reads generated was therefore considered sufficient to sample most expressed genes.
The Pearson's correlation coefficients (figure 5) obtained for read frequencies in the 12 sequencing sets from E. spp (87% on average) and the four sequencing sets from E. grandis (86% on average) were similar. However, the mean correlation was much weaker (52%) between the E. spp and E. grandis sequencing sets, suggesting that different fractions of the transcriptome had been sampled from these two studies and/or that gene expression differed between both sequencing sets. As expected, correlations were stronger between replicates (92%; illustrated by squares in figure 5) than between different samples (86%) in E. spp sequencing sets, suggesting a high level of technical repeatability.
Figure 5.

Pearson correlation coefficient for read frequencies (within contigs) between sequencing sets. SS1-SS12 are sequencing sets from E.spp; SRR001658-SRR001661 are sequencing sets from E. grandis (Novaes et al., 2008). The colour scale indicates the strength of the correlation, from red (strongest correlations) to green (weakest correlations). All the correlation coefficients were significant, with a p-value <0.001.
Within conditions, correlations between the G1 and G2 sequencing sets were robust and similar between treatments: 90% for the IR treatment and 88% for the NI treatment, suggesting that these two genotypes displayed similar patterns of gene expression when placed in the same environmental conditions. Correlations between the IR and NI sequencing sets were slightly weaker and a stronger contrast was observed within genotypes: on average, 86% for genotype G1 and 82% for genotype G2. The weaker correlations obtained for G2 suggest that this genotype had a more pronounced response to water deficit than genotype G1.
Homology search
BLAST search (e-value cut-off of 10e-5) results are summarised in Table 3. A functional annotation was obtained more frequently for contigs - 70% of the contigs harboured similarity to sequences in protein databases, and 75% to sequences in nucleic acid databases. In contrast, for singletons only 39% and 9% of the contigs had similarity to protein and nucleic acid sequences, respectively. These differences were expected, given the longer mean length of the contigs (734 bp) compared to singletons (319 bp), their lower abundance and the fact that singletons are more likely to be sequencing artefacts. A larger number of nucleic acid sequences had similarity to Arabidopsis (55% of annotated sequences), followed by Vitis (25%), and Populus (6%). The greater similarity to Arabidopsis genes may be due to the closer phylogenetic relationship of Eucalyptus to Arabidopsis (both belong to the eurosid II phylogenetic clade) than to Populus (eurosid I, [51]). The Arabidopsis genome has also been annotated in greater detailed than the Populus genome. Interestingly, the similar characteristics of eucalyptus and poplar in terms of growth habits do not translated into higher similarity of the sequences transcribed.
Table 3.
Annotation results for protein hits, nucleic acid hits, and Gene Ontologies (GO): Biological Process (BP), Cellular Component (CC) and Molecular Function (MF)
| E.spp Protein hits | SSequencing set | Nucleic acid hits | GO-BP | GO-CC | GO-MF | |
|---|---|---|---|---|---|---|
| # UE | 129,993 | 66,135 (51%) | 44,652 (34%) | 32,835 (25%) | 30,836 (24%) | 33,222 (26%) |
| # Contigs | 49,748 | 34,951 (70%) | 37,383 (75%) | 18,091 (36%) | 17,242 (35%) | 18,355 (37%) |
| # Singletons | 31,184 (39%) | 80,245 | 7,269 (9%) | 14,744 (18%) | 13,594 (17%) | 1,4867 (19%) |
% of sequences related to E. spp. libraries is shown in brackets.
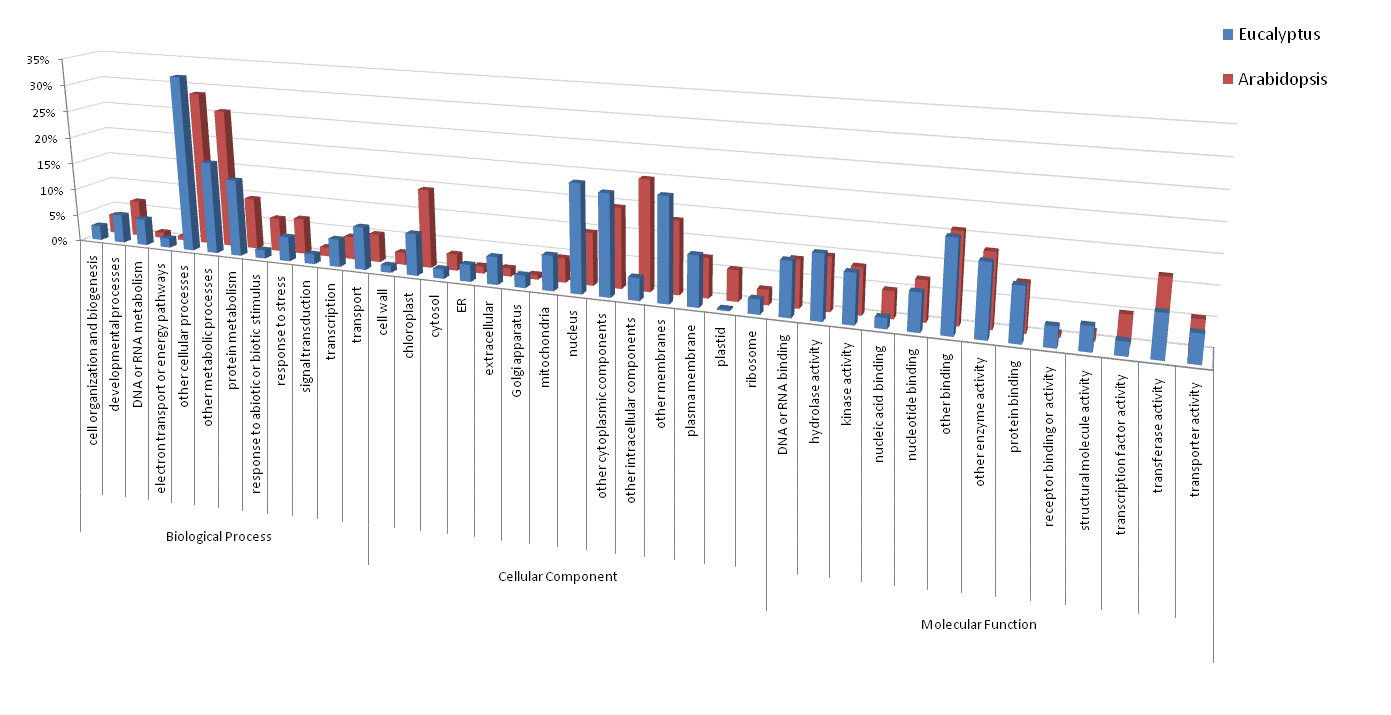
According to Gene Ontology (GO) classification, 38,190 UE (25% of the E.spp sequencing set UE) were associated with at least one biological process (BP), molecular function (MF) or cellular component (CC). The proportions of UE annotated in each category were generally similar to those obtained in Arabidopsis (Additional file 4), suggesting that the E.spp sequencing sets are appropriate for the analysis of gene expression on a broad range of functional categories.
Transcript abundance analysis
After removing contigs represented by fewer than 10 reads in all the E.spp sequencing sets, 14,460 contigs remained for abundance analysis. Two statistical tests were performed in series to detect differences in the expression levels of these 14,460 contigs, among the four experimental conditions (G1IR, G1NI, G2IR and G2NI). First, a DEG-seq test [43] identified 1,651 differentially expressed contigs (FDR ≤ 5%). A two-way ANOVA was then performed to assess the effects of the two main factors (G and T) and their interaction (GxT) on the number of reads per contig. This analysis identified 1,445 contigs with at least one significant effect (p-value ≤ 5%; figure 6, additional file 5). With an error rate of 5%, only 83 false positives are expected among the 1,445 contigs.
Figure 6.

Differentially expressed contigs. Venn diagram indicating the number of differentially expressed contigs showing a G, T and/or GxT effect, for the 14,460 contigs analysed.
Most of the differentially expressed contigs (1,280) showed a genotype effect ("G contigs"), with 624 "G contigs" overexpressed in genotype G1 (positive log2-transformed fold-change between contig abundance in G1 vs. G2) vs. 656 "G contigs" underexpressed in G1 ( negative log2-transformed fold-change; figure 7A). Of the 656 contigs overexpressed in genotype G2, 289 (44%) were expressed only in that genotype (with no corresponding reads in the G1 sequencing sets) whereas only 55 contigs (9%) showed the reverse trend, suggesting that G2 may express of a larger set of genes or different splicing variants.
Figure 7.

Fold-change (FC) distribution. Log2-transformed FC were calculated with DEGseq and plotted for each class of effect. (A) Genotype effect: contigs overexpressed in genotype G1 are presented with positive values (+), whereas contigs underexpressed in genotype G1 have negative values (-). Similarly, (B) treatment effects: overexpressed in IR (+) and under expressed in IR (-); (C) genotype × treatment effect for genotype G1: IR (+) and NI (-); (D) genotype × treatment effect for genotype G2: overexpressed in IR (+) and underexpressed in IR (-); (E) genotype × treatment effect for treatment IR: overexpressed for G1 (+) and underexpressed for G1 (-); (F) genotype × treatment effect for treatment NI: overexpressed for G1 (+) and underexpressed for G1 (-). The number of contigs overexpressed is shown in brackets.
A total of 155 contigs showing a treatment effect ("T contigs"; figure 7B) were identified with similar numbers overexpressed in the two treatments (81 in IR and 74 in NI). Thirteen "T contigs" were expressed only in NI conditions, whereas all the "T contigs" overexpressed in IR conditions were also found in NI sequencing sets, suggesting that few "specific genes" are upregulated in response to water deficit but that the set of genes expressed in favourable conditions is also expressed at a lower level in stressed plants.
Finally, 274 contigs corresponded to "GxT" contigs. The larger number of "GxT" contigs than of "T contigs" suggests that some of the observed molecular plasticity is under genetic control. Only 11 "GxT contigs" displayed significant differential expression between the IR and NI conditions in genotype G1 (figure 7C), whereas 112 "GxT contigs" displayed such behaviour in genotype G2 (figure 7D), suggesting a more pronounced response in G2. Similarly, 48 "GxT contigs" were differentially expressed between the two genotypes in IR conditions (figure 7E), whereas 228 "GxT contigs" were differentially expressed between the two genotypes for the NI treatment (figure 7F). These results suggest that, despite the rather similar expression patterns for the two genotypes in IR conditions, water deficit induced a molecular response specific to each genotype, reflecting different strategies to respond to water shortage during the dry season.
Blast2GO functional analysis
Gene Ontology (GO) analysis was performed on 9,058 contigs (of 14,460 contigs containing more than 10 reads). These contigs were assigned to biological processes (BP), with 11 main subcategories (7,593 contigs), cellular compartments (CC), with five main subcategories (7,347 contigs), and molecular functions (MF), with eight main subcategories (7,638 contigs). Consistent with the number of annotations per category (BP, CC, MF), two subcategories were found to be strongly represented: > 65% of the contigs were assigned to cellular and metabolic processes for BP, more than 40% were assigned to cells and 85% to organelles for CC, and > 40% were assigned to binding and catalytic activity for MF. Figure 8 shows the distribution of contigs between these subcategories as a function of the four classes of effects ("NS", "G", "T", and "GxT" contigs). The homogeneity of the relative abundance of contigs between the "significant" classes ("G", "T", or "GxT") and the "not significant" class ("NS") in each GO category was assessed with Fisher's exact tests. In BP, "G" contigs were overrepresented in four subcategories (response to stimulus, developmental process, death and multiorganism process). "T" contigs were overrepresented in only one subcategory (response to stimulus). Finally, "GxT" contigs were overrepresented in three subcategories (response to stimulus, death and developmental process). These differences in relative abundance suggest that genes related to defence reactions are the main contributors to differences between significant and "NS" contigs. For CC, only one subcategory (extracellular region) presented a higher relative abundance for all three significant effects. For MF, "G" contigs were overrepresented in two subcategories (structural molecule activity and molecular transducer activity), whereas "T" contigs were overrepresented in only one subcategory (molecular transducer activity); for "GxT" contigs, two other subcategories (catalytic activity and antioxidant activity) presented higher relative abundances than "NS".
Figure 8.

Distribution of Gene Ontology categories between effect classes. Percentage of contigs annotated for Biological Process, Cellular Component and Molecular Function GO category (level 2). Non-significant contigs (NS) and contigs displaying genotype (G), treatment (T), or genotype × treatment (G*T) effects are shown. The numbers in brackets are the numbers of annotated contigs per class of contigs and GO category. *Indicates distributions significantly different between NS and other classes of effect, in Fisher's exact tests with a threshold of 0.05.
Analysis of metabolic pathways with MapMan
Of the 1,445 contigs displaying significant differential expression, the 1,280 "G contigs" did not enable characterisation of specific molecular processes (i.e. did not show any clear co-regulation with genes of the same biosynthesis pathway). For 95% of these contigs, many different genes from different molecular processes were activated, depending on the experimental condition (Additional file 6). In some instances (5% of "G" contigs), some bins presented specific overexpression in one genotype (Additional file 7). For example, contigs related to ethylene biosynthesis and cell organisation were overexpressed mainly in genotype G2, whereas contigs related to photosynthesis, nitrilases, calcium signalling and pathogenesis-related protein bins were overexpressed in genotype G1. Serine proteases (9 contigs) were expressed more strongly in genotype G1 (p-value = 0.038), whereas ubiquitin E3-encoding proteins (23 contigs) were expressed more strongly in genotype G2 (p-value = 0.096), suggesting that proteolysis occurred via different pathways in the two genotypes.
Analysis of the metabolic pathways for "T" contigs was limited because of the small number of contigs (155), which were distributed in several bins (Additional file 8). However, the expression of genes related to carbohydrate degradation and ethylene biosynthesis were found to be stronger for the NI treatment, whereas the expression of genes related to ribosomal protein synthesis and cell development appeared to be stronger for the IR treatment (Additional file 7).
Different patterns were observed for "GxT" contigs (figure 9): i) Some pathways (25% of "GxT" contigs) displayed similar patterns in the two genotypes, but with responses of different magnitudes (i.e. scale plasticity, as defined by Lynch and Walsh [52]). Photosystem components tended to be overrepresented in genotype G2 in the IR condition, whereas few differences were observed between conditions for genotype G1. Conversely, genes related to cell organisation and PR-proteins were more likely to be overexpressed in the NI condition in genotype G2 compared to genotype G1, ii) Interestingly, most of the pathways (75% of "GxT"contigs) displayed opposite trends in the two genotypes (i.e. a re-ranking interaction effect [52]). For example, in the NI treatment, genes related to phenylpropanoid biosynthesis, auxin biosynthesis, heat stress, and light signalling were overexpressed in genotype G1, while they where underexpressed in genotype G2. Conversely, genes related to global primary metabolism (particularly starch degradation and ribosomal protein synthesis) and receptor kinases were overexpressed in genotype G2, but underexpressed in genotype G1 in the NI condition (additional file 7). This result strongly suggests that different metabolic pathways and genes were activated in these two genotypes in response to water shortage. Thus, these two genotypes exhibit different molecular strategies to cope with water deficit during the dry season.
Figure 9.

Distribution of "GxT" contigs between metabolic pathways. Each square represents the log2-transformed fold-change of abundance between irrigated (IR) and non-irrigated (NI) treatments for one contig: for genotype G1 (A) and G2 (B). Contigs in blue were overexpressed for the IR treatment and contigs in red were overexpressed for the NI treatment.
Discussion
Gene discovery and expression analysis by NGS
Next-generation sequencing (NGS) technologies are becoming the method of choice for large-scale transcriptome analysis, even for non-model species (e.g. [29,41], reviewed in [53]). Several technologies have been developed, differing essentially in the number of reads generated and read length (reviewed in [54]), making it possible to catalogue the genes expressed and to monitor gene expression.
In Eucalyptus, Mizrachi et al. [38] generated 3.93 Gbp of short reads (36-60 bp) with sequencing-by-synthesis technology from Illumina, and assembled this information de novo into 18,894 contigs (Illumina-contigs) longer than 200 bp (22.1 Mbp in total). In this study, we obtained 0.398 Gbp of sequences with longer reads (mean of 350 bp), which were assembled into 48,950 contigs (454-sequencing contigs) with more than 200 bp each (36.5 Mbp in total). We were thus able to assemble more reads, i.e. 9.2% of the sequencing set, that is a much higher rate than the 0.56% reported by Mizrachi et al. [38]. BLAST searches for sequence similarities between the two datasets showed that 86% of the contigs were common to both studies (42,550 454-sequencing contigs matched 16,278 Illumina contigs). However, each Illumina contig matched a mean of five 454-sequencing contigs, indicating that UE detected with our approach were probably confounded in the short-read assembly. In addition, the short Illumina contigs may represent domains shared by multiple proteins, confirming the difficulty involved in assembling short reads into transcriptional units [55]. Alternatively we can not rule out the fact that genes were split in multiple contigs in the 454 assembly because of the lack of coverage compared to Illumina's short reads. Finally, we found that 36,985 454-UE did not match any previously described eucalyptus ESTs, and that 43% of these UE (18% of the 454-sequencing contigs) displayed no match with any nucleic acid or protein sequence published for any other species. Therefore, our 454-sequencing data considerably enriched the Eucalyptus EST collection.
RNA-Seq is also an interesting approach to obtain a comprehensive digital gene expression profile for specific tissues, cell types or developmental processes. In this study, the high degree of repeatability observed for the three replicates made it possible to test G, T and GxT effects with a statistical support. We were able to monitor the expression of 14,460 UE and to identify 1,445 UE displaying at least one significant effect. Some technical biases, such as non-linear amplification and a lack of sequencing depth may have resulted in a lack of precision in the prediction of gene expression by 454-sequencing (Additional file 9). Short read-based sequencing approaches provide ample read coverage and generally give better predictions of gene expression [56,57]. Thus, a combination of long and short reads may be seen as a reasonable strategy for the analysis of gene expression [55,58,59]. With this combined strategy, long-read sequencing can be used to establish a comprehensive catalogue of transcriptional units, while short reads mapped onto this assembly provide greater sequencing depth, improving predictions of abundance.
Behaviour of variance components at the phenotypic and molecular levels
We observed that phenotypic and molecular variation are accounted for principally by genotypic differences. Indeed, above-ground biomass and contig abundance were influenced principally by genotype. Above-ground biomass was, on average, 49% higher for G2 than for G1, and most of the differentially expressed contigs (1,280 of 1,455) in this study presented a genotypic effect. The number of contigs overexpressed in one or the other of the genotypes was similar - 624 and 656 contigs were overexpressed in G1 and G2, respectively. No particular genotypic signature in terms of functional categories or pathways was detected. The two genotypes differed strongly in phenotype (not only it terms of growth potential, but also in terms of leaf morphology, stomatal distribution and water use efficiency), but it remains unclear whether these differences in transcript abundance were responsible for trait variation, neutral, or simply involved in reproductive isolation between parental species. Indeed, differences in gene expression between species have been reported in the field of ecological genomics [60,61] and interpreted as a mechanism of speciation. In our study, both genotypes were hybrid combinations between different species. This may have increased the number of differences between their transcriptomes. Further investigations about the role of gene expression in ecological speciation is an important question, particularly for eucalyptus, in which species complexes are common [62].
The variance accounted for by genotype-by-environment interaction (GEI) at the phenotypic and molecular level was also significant in this study. The NI treatment resulted in a significantly lower above-ground biomass, and this difference was greater for the least productive of the two genotypes, G1 (24%), than for G2 (12%). The G and T effects accounted for 56% and 13% of the above-ground biomass variation, respectively, whereas GxT effects accounted for only 0.2% of the variance. At the molecular level, we also found a higher proportion of genes displaying G effects (1,280 contigs, 8.8% of the contigs screened) but, surprisingly, we found fewer genes displaying T effects (155 contigs, 1.1% of the contigs screened) than GxT effects (274 contigs, 1.9% of the contigs screened).
While only 11 contigs were differentially expressed between the two treatments in genotype G1, 112 contigs showed differential expression in genotype G2 (4 contigs displayed differential expression for both genotypes). Moreover, when the whole E. spp sequencing set was screened, genotype G2 presented a larger number of specific contigs (10.5% of the E.spp sequencing set) than G1 (5.5% of the E.spp sequencing set). These results suggest that a larger set of genes is activated in genotype G2, leading to the triggering of specific responses to water deficit. This higher molecular sensitivity of genotype G2 may confer advantages ultimately resulting in a greater capacity to cope with water deficit during the dry season and, therefore, in stronger growth capacity (table 4).
Table 4.
Summary of phenotypic and molecular plasticity evidenced for the two studied genotypes (G1 and G2), between irrigated (IR) and non irrigated (NI) treatments
| G1 | G2 | ||
|---|---|---|---|
| IR → NI | IR → NI | ||
| Above-ground biomass | decrease | stable | |
| Transcript abundance | Hormones | + | -- |
| Secondary messengers | + (light signalling) | -- (light signalling) ++ (receptor kinases) | |
| Other stress related genes | + | -- (heat-shock proteins) ++ (PR proteins) | |
| Transcription factors | stable | ++ | |
| Cellular protection | + | -- | |
| Damages repair | + (carbohydrates secondary metabolism), | ++ (cell organisation, lipids, proteins) -- (secondary metabolism) | |
| Cellular activity maintenance | stable | -- (photosynthesis, transport) | |
Abbreviations: +/- means that in a given category more than 50% of the genes are over/underexpressed in NI.++/-- means that in a given category more than 50% of the genes are over/underexpressed in NI with a log2-tranformed fold change <-0.8 or >0.8.
Genes displaying GEI effects reflect differences in signal perception and response strategy
Contigs displaying GEI effects could be classified into two groups according to the function of the proteins encoded [63]: i) regulatory proteins responsible for drought signal transduction and response triggering, and ii) functional proteins involved in cell protection, damage repair and the maintenance of cell activity.
Regarding regulatory functions, the genes involved in the biosynthesis of hormones, such as ethylene and auxins in particular (aldo/keto reductase, proteins of the ethylene-responsive family, 2-oxoglutarate-dependent dioxygenase) were mostly overexpressed in genotype G1 and underexpressed in G2, in the NI condition. Genes acting as second messengers in the transduction of hormonal signal to stomatal guard cells [64,65] also displayed GEI effects: in the NI condition, genes involved in calcium signalling were predominantly overexpressed in G1, whereas the response of G2 to drought preferentially involved receptor kinases. We also identified other signal transducers, such as light-induced proteins and heat-shock proteins, which may be related to other types of stress induced by water deficit, including osmotic stress due to pH variations and oxidative stress due to the accumulation of reactive oxygen species (ROS) [66,67]. These pathways were mostly overexpressed in G1 and underexpressed in G2 during the dry season. Pathogenesis-related (PR) protein genes were overexpressed in G2, but displayed a less clear-cut pattern of expression in G1. PR proteins were initially reported to be induced by hormones or ROS in response to biotic stress [68,69], but they have also been shown to be involved in other abiotic stresses [70]. Lee et al. [69] also suggested that PR proteins may be used as storage proteins when growth is limited by environmental factors. Some transcription factors responded strongly to water shortage in genotype G2. Two, in particular, encoded factors homologous to AtMYB12 and AtMYB85, which have been shown to regulate secondary metabolism (flavonoid and lignin biosynthesis, respectively) in Arabidopsis [71]. These results suggest that water shortage induces different cellular stress cascades, perceived differently by the two genotypes.
Stress signal transducers interact to trigger the regulation of gene expression for the maintenance of three main functions: cell protection, damage repair and the maintenance of cell activity. Our results suggest that more genes related to cell protection were involved in the response to water shortage in genotype G1 than in genotype G2. Protection against drought stress seems to involve mostly carbohydrates, with 11 contigs displaying GEI effects, and, to a lesser extent, polyamines, which may modulate some ion channels [72].
By contrast to the trends observed for genes related to cell protection, more genes related to damage repair seem to be expressed during the dry season in genotype G2 than in genotype G1, particularly those related to cell organisation. The overexpression, during NI treatment, of genes related to primary metabolism, including carbohydrate, lipid and protein synthesis and degradation, suggests that resources are reallocated for the repair of cell structures or the formation of new structures in drought stressed plants. The patterns of expression of secondary metabolism genes differed between the two genotypes as well. As an example, G1 displayed a higher number of genes related to terpenoids and flavonoids synthesis (that may protect against oxidative stress) overexpressed in the NI treatment compared to genotype G2. However, the contrasts between NIR and IR treatments were much higher for G2. Conversely, genes related to lignin biosynthesis (e.g. CCoAOMT) were overexpressed in the IR condition only for G2.
Gene related to photosynthesis were found to be under-expressed in G2 subjected to NI treatment, whereas no variation was found in G1. Other metabolic processes, such cell development and transport, controlled by genes encoding water or sugar channels, decreased in NI treatment, particularly in G2. These results confirm the trends observed by Alexandersson et al. [73] in Arabidopsis. These authors studied the expression of 18 genes encoding aquaporins and showed that most of these genes were downregulated in leaves subjected to a gradual water deficit. Interestingly, cell activity seemed to be more reduced at the transcriptional level for G2, although this genotype grew more strongly. It is possible that, during water deficit, genotype G2 reduces its rate of photosynthesis and reallocates resources (as suggested by changes in primary metabolism) to preserve its cell structures and ability to resume growth when conditions become more favourable.
Evolutionary implication behind GEI
We found that 31 of the 274 contigs displaying a GxT effect were absent from the G1 sequencing set (11.3%), whereas only two such contigs were absent in G2 (0.7%). Unfortunately, of 31 contigs absent in G1, 16 could not been assigned to a homolog gene in Arabidopsis. The others corresponded to genes related to cell organisation (ankyrins), ethylene synthesis, protein metabolism, PR proteins and receptor kinases. These genes may be considered non-essential for tree development, and are therefore unlikely to be subject to selection constraints. Landry et al. [74] found an overrepresentation of non-essential genes (the deletion of which is not lethal) among genes displaying GEI in Saccharomyces cerevisiae. They proposed two hypotheses to account for the activity of these genes being compensated in cells: i) metabolic buffering: non-coded metabolites may be rerouted through the metabolic network, and ii) genetic buffering: paralogous genes may supply the missing function. We showed in the results section (Additional file 5) that differences between genotypes may be accounted for by the preferential expression of different members or splicing forms of genes from the same family. This observation may confirm the hypothesis of genetic buffering.
Scale plasticity was observed for 146 of the 274 differentially expressed contigs: genotype ranks were conserved between treatments but one genotype reacted more or less strongly to the environmental variation. Conversely, 90 contigs showed a change in ranking between genotypes (rank plasticity). Landry et al. [74] hypothesised that these two types of GEI would have different effects on the evolution of plastic traits. In the case of scale plasticity, whatever the environment, selection would result in the same favoured genotype, whereas in the case of rank plasticity, different genotypes would be selected in different environments. In the present study, we found differentially expressed genes presenting both scale plasticity (62%) and rank plasticity (38%), indicating different types of reaction norms on which natural selection would act on.
Conclusions
We showed that next-generation sequencing is a powerful tool for transcriptome screening: with 398 Mb of sequence, we were able to assemble ESTs into 69,584 contigs, with remaining 80,245 singletons, and to determine the relative abundance of 14,460 contigs each comprising more than 10 reads. Large differences between genotypes, in terms of phenotypic behaviour and transcriptome regulation, were observable. Differences in gene expression between the two genotypes appear to affect the whole transcriptome, rather than specific pathways. The genotype-specific response to water shortage (i.e. GxT effect) was more pronounced than the response common to both genotypes (i.e. T effect). The genes displaying genetically controlled plasticity were found to belong to a number of different pathways essentially related to signal transduction and primary metabolism. The more productive genotype, G2, express a larger set of genes, leading to the triggering of specific molecular responses. Moreover, GxT interaction results principally from a lack of molecular response in genotype G1, together with a strong response of genotype G2 (table 4). The ability to regulate more actively its transcriptome might be a key component in the maintenance of biomass in water deficit conditions.
Finally, although this study provides clues to the way in which different genotypes activate their transcriptomes when subjected to water deficit, more research is required to understand the molecular mechanisms involved during the dry season. First, there is a need to characterize reaction norm in a broader genetic background [75]. Second, epigenetics or post-transcriptional regulation mechanisms that are well known to interfere with abiotic stress responses [76,77] deserve specific investigations.
Authors' contributions
EV: participated in the field work, performed the molecular work, carried out the statistical analysis and drafted the manuscript. CK and CN performed the bioinformatic work (assembly and annotation). EN and MK provided E. grandis sequencing sets and participated in the statistical analysis. CP participated in co-ordination of the molecular, bioinformatic and statistical work and in drafting the manuscript. JMG participated in the design of the study, co-ordination of the field, molecular, bioinformatic and statistical work and drafting the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Material
Results of the ANOVA for above-ground biomass.
Distribution of read length for the three half-runs.
Increasing coverage with successive runs. Number of contigs represented in each half-run or combination of several half-runs. Performing a second half-run increased contig coverage by an average of 18%, and a third half run increased coverage by an average of 6%.
Comparison of the distribution of Gene Ontology (GO) categories between Eucalyptus spp unigene elements (UE) and Arabidopsis annotated unigenes. Proportion of each GO category (Biological Process, Cellular Component and Molecular Function) found in the E. spp sequencing set and in the annotated Arabidopsis genome.
{kind=link}
Differentially expressed contigs displaying significant T and GxT effects. Each contig showing T or GxT is associated with a functional category (as defined by Mercator : http://mapman.gabipd.org/web/guest). Log2-transformed fold change between abundance in irrigated and non irrigated libraries are indicated for each genotype, as well as p-value of T and GxT effects analyzed by the ANOVA.
Distribution of "G" contigs between functional pathways. Each square represents the log2-transformed fold-change of abundance between genotypes 1-41 and 18-50 for one contig. Contigs in green were overexpressed in genotype 1-41 and contigs in blue were overexpressed in genotype 18-50.
Significant categories for the Wilcoxon rank sum test, according to Mapman analysis for the pairwise comparison of differentially expressed contigs displaying genotype (G), treatment (T) and genotype × treatment (GxT) effects. ** Categories differentially expressed at an error rate threshold of 0.05 * Categories differentially expressed at an error rate threshold of 0.1
Distribution of "T" contigs between several functional pathways. Each square represents the log2-transformed fold-change of abundance between irrigated (IR) and non-irrigated (NI) treatments for one contig. Contigs in blue were overexpressed for the IR treatment and contigs in red were overexpressed for the NI treatment.
Supporting information: validation of digital profiles by analyzing expression by RT-qPCR on 36 genes.
Contributor Information
Emilie Villar, Email: villar@cirad.fr.
Christophe Klopp, Email: Christophe.Klopp@toulouse.inra.fr.
Céline Noirot, Email: Celine.Noirot@toulouse.inra.fr.
Evandro Novaes, Email: novaes@agro.ufg.br.
Matias Kirst, Email: mkirst@ufl.edu.
Christophe Plomion, Email: plomion@pierroton.inra.fr.
Jean-Marc Gion, Email: gion@cirad.fr.
Acknowledgements and Funding
We thank Andréas Ndeko, André Mabiala, Joël Polidori and Dr. Aubain R. Saya (CRDPI, Rep. of Congo) for setting up the experimental trial. This article is part of the PhD thesis of Emilie Villar, supervised by Jean-Marc Gion and Christophe Plomion. EV was supported by CIRAD. This work was supported by grants from CIRAD, ATP project "Plasticité phénotypique des pérennes sous contrainte hydrique au champ", and the Conseil Régional d'Aquitaine ("ABIOGEN" project no.° Presage 32973). Some of the experiments (RT-qPCR) presented here were performed at the Genome Transcriptome facility of Bordeaux (grants from the Conseil Régional d'Aquitain nos. 20030304002FA and 20040305003FA and from the European Union, FEDER no. 2003227).
References
- Krutovskii KV, Neale DB. Forest genomics for conserving adaptive genetic diversity. Forest Genetic Resources. 2001. pp. 6–8.
- Hamrick JL. Response of forest trees to global environmental changes. Forest Ecology and Management. 2004;197:323–335. doi: 10.1016/j.foreco.2004.05.023. [DOI] [Google Scholar]
- Jump AS, Penuelas J. Running to stand still: adaptation and the response of plants to rapid climate change. Ecology Letters. 2005;8:1010–1020. doi: 10.1111/j.1461-0248.2005.00796.x. [DOI] [PubMed] [Google Scholar]
- Eucalyptus universalis. Global cultivated forests map. 2008. http://www.git-forestry.com/
- McGowen MH, Wiltshire RJE, Potts BM, Vaillancourt RE. The origin of Eucalyptus vernicosa, a unique shrub eucalypt. Biological Journal of the Linnean Society. 2001;74:397–405. [Google Scholar]
- Jones RC, Steane DA, Potts BM, Vaillancourt RE. Microsatellite and morphological analysis of Eucalyptus globulus populations. Canadian Journal of Forest Research-Revue Canadienne De Recherche Forestiere. 2002;32:59–66. doi: 10.1139/x01-172. [DOI] [Google Scholar]
- Holman JE, Hughes JM, Fensham RJ. A morphological cline in Eucalyptus: a genetic perspective. Molecular Ecology. 2003;12:3013–3025. doi: 10.1046/j.1365-294X.2003.01970.x. [DOI] [PubMed] [Google Scholar]
- Tripiana V, Bourgeois M, Verhaegen D, Vigneron P, Bouvet JM. Combining microsatellites, growth, and adaptive traits for managing in situ genetic resources of Eucalyptus urophylla. Canadian Journal of Forest Research-Revue Canadienne De Recherche Forestiere. 2007;37:773–785. doi: 10.1139/X06-260. [DOI] [Google Scholar]
- Payn KG Dvorak WS Janse BJH Myburg AA Microsatellite diversity and genetic structure of the commercially important tropical tree species Eucalyptus urophylla, endemic to seven islands in eastern Indonesia Tree Genetics & Genomes 20084519–530. 10.1007/s11295-007-0128-722065253 [DOI] [Google Scholar]
- Butcher PA, Williams ER. Variation in outcrossing rates and growth in Eucalyptus camaldulensis from the Petford Region, Queensland; Evidence of outbreeding depression. Silvae Genetica. 2002;51:6–12. [Google Scholar]
- Shepherd M, Sexton TR, Thomas D, Henson M, Henry RJ. Geographical and historical determinants of microsatellite variation in Eucalyptus pilularis. Canadian Journal of Forest Research-Revue Canadienne De Recherche Forestiere. 2010;40:1051–1063. doi: 10.1139/X10-049. [DOI] [Google Scholar]
- McKinnon GE, Potts BM, Steane DA, Vaillancourt RE. Population and phylogenetic analysis of the cinnamoyl coA reductase gene in Eucalyptus globulus (Myrtaceae) Australian Journal of Botany. 2005;53:827–838. doi: 10.1071/BT04195. [DOI] [Google Scholar]
- Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff RR, Kirst M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. Bmc Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulheim C, Yeoh SH, Maintz J, Foley WJ, Moran GF. Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. Bmc Genomics. 2009;10:452. doi: 10.1186/1471-2164-10-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan SE. Phenotypic plasticity for plant development, function and life history. Trends in Plant Science. 2000;5:537–542. doi: 10.1016/S1360-1385(00)01797-0. [DOI] [PubMed] [Google Scholar]
- Stape JL, Binkley D, Ryan MG. Production and carbon allocation in a clonal Eucalyptus plantation with water and nutrient manipulations. Forest Ecology and Management. 2008;255:920–930. doi: 10.1016/j.foreco.2007.09.085. [DOI] [Google Scholar]
- Campion JM, Nkosana M, Scholes MC. Biomass and N and P pools in above- and below-ground components of an irrigated and fertilised Eucalyptus grandis stand in South Africa. Australian Forestry. 2006;69:48–57. [Google Scholar]
- Eyles A, Pinkard EA, Mohammed C. Shifts in biomass and resource allocation patterns following defoliation in Eucalyptus globulus growing with varying water and nutrient supplies. Tree Physiology. 2009;29:753–764. doi: 10.1093/treephys/tpp014. [DOI] [PubMed] [Google Scholar]
- Tatagiba SD, Pezzopane JEM, Reis EFd, Penchel RM. Performance of six clones of eucalyptus in response to substrate water availability. Engenharia na Agricultura. 2009;17:179–189. [Google Scholar]
- Shvaleva AL, Silva FCE, Breia E, Jouve L, Hausman JF, Almeida MH, Maroco JP, Rodrigues ML, Pereira JS, Chaves MM. Metabolic responses to water deficit in two Eucalyptus globulus clones with contrasting drought sensitivity. Tree Physiology. 2006;26:239–248. doi: 10.1093/treephys/26.2.239. [DOI] [PubMed] [Google Scholar]
- Pita P, Pardos JA. Growth, leaf morphology, water use and tissue water relations of Eucalyptus globulus clones in response to water deficit. Tree Physiology. 2001;21:599–607. doi: 10.1093/treephys/21.9.599. [DOI] [PubMed] [Google Scholar]
- Merchant A, Peuke AD, Keitel C, Macfarlane C, Warren CR, Adams MA. Phloem sap and leaf delta C-13, carbohydrates, and amino acid concentrations in Eucalyptus globulus change systematically according to flooding and water deficit treatment. Journal of Experimental Botany. 2010;61:1785–1793. doi: 10.1093/jxb/erq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb A, Pereira A. Screening Arabidopsis Genotypes for Drought Stress Resistance. Plant Reverse Genetics: Methods and Protocols. 2011. pp. 191–198. [DOI] [PubMed]
- Deyholos MK. Making the most of drought and salinity transcriptomics. Plant Cell and Environment. 2010;33:648–654. doi: 10.1111/j.1365-3040.2009.02092.x. [DOI] [PubMed] [Google Scholar]
- Lefebvre V, Kiani SP, Durand-Tardif M. A Focus on Natural Variation for Abiotic Constraints Response in the Model Species Arabidopsis thaliana. International Journal of Molecular Sciences. 2009;10:3547–3582. doi: 10.3390/ijms10083547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Bogeat-Triboulot MB, Tisserant E, Balzergue S, Martin-Magniette ML, Lelandais G, Ningre N, Renou JP, Tamby JP, Thiec Dl, Hummel I. Comparative transcriptomics of drought responses in Populus: a meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genomics. 2010;11:630. doi: 10.1186/1471-2164-11-630. (12 November 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta M, Giovannelli A, Sebastiani F, Camussi A, Racchi ML. Transcriptome changes in the cambial region of poplar (Populus alba L.) in response to water deficit. Plant Biology. 2010;12:341–354. doi: 10.1111/j.1438-8677.2009.00320.x. [DOI] [PubMed] [Google Scholar]
- Hamanishi ET, Raj S, Wilkins O, Thomas BR, Mansfield SD, Plant AL, Campbell MM. Intraspecific variation in the Populus balsamifera drought transcriptome. Plant Cell and Environment. 2010;33:1742–1755. doi: 10.1111/j.1365-3040.2010.02179.x. [DOI] [PubMed] [Google Scholar]
- Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Molecular Ecology. 2008;17:1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x. [DOI] [PubMed] [Google Scholar]
- Torres TT, Metta M, Ottenwalder B, Schlotterer C. Gene expression profiling by massively parallel sequencing. Genome Research. 2008;18:172–177. doi: 10.1101/gr.6984908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat A, DiLoreto DS, Zhang Y, Smith C, Baier K, Powell WA, Wheeler N, Sederoff R, Carlson JE. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biology. 2009;9:51. doi: 10.1186/1471-2229-9-51. (9 May 2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagna F, D'Agostino N, Torchia L, Servili M, Rao R, Pietrella M, Giuliano G, Chiusano ML, Baldoni L, Perrotta G. Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Genomics. 2009;10:399. doi: 10.1186/1471-2164-10-399. (26 August 2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traas J, Vernoux T. The shoot apical meristem: the dynamics of a stable structure. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences. 2002;357:737–747. doi: 10.1098/rstb.2002.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Juez E, Dillon E, Magyar Z, Khan S, Hazeldine S, de Jager SM, Murray JAH, Beemster GTS, Bogre L, Shanahan H. Distinct light-initiated gene expression and cell cycle programs in the shoot apex and cotyledons of Arabidopsis. Plant Cell. 2008;20:947–968. doi: 10.1105/tpc.107.057075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller G Marchal T SanClemente H Navarro M Ladouce N Wincker P Couloux A Teulieres C Marque C Development and functional annotation of an 11,303-EST collection from Eucalyptus for studies of cold tolerance Tree Genetics & Genomes 20095317–327. 10.1007/s11295-008-0184-722065253 [DOI] [Google Scholar]
- Rengel D, San Clemente H, Servant F, Ladouce N, Paux E, Wincker P, Couloux A, Sivadon P, Grima-Pettenati J. A new genomic resource dedicated to wood formation in Eucalyptus. BMC Plant Biology. 2009;9:36. doi: 10.1186/1471-2229-9-36. (27 March 2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria DA, Mamani EMC, Pappas MR, Pappas GJ, Grattapaglia D. A Selected Set of EST-Derived Microsatellites, Polymorphic and Transferable across 6 Species of Eucalyptus. Journal of Heredity. 2010;101:512–520. doi: 10.1093/jhered/esq024. [DOI] [PubMed] [Google Scholar]
- Mizrachi E, Hefer CA, Ranik M, Joubert F, Myburg AA. De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics. 2010;11:681. doi: 10.1186/1471-2164-11-681. (1 December 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid KE, Olsson N, Schlosser J, Peng F, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biology. 2006;6:11. doi: 10.1186/1471-2229-6-11. 6 (Nov.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Provost Gl, Leger V, Klopp C, Noirot C, Frigerio JM, Salin F, Salse J, Abrouk M, Murat F. et al. Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: oak. BMC Genomics. 2010;11:650. doi: 10.1186/1471-2164-11-650. (23 November 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Huang XQ, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M. A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant Cell and Environment. 2009;32:1211–1229. doi: 10.1111/j.1365-3040.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- Safou-Matondo R, Deleporte P, Laclau JP, Bouillet JP. Hybrid and clonal variability of nutrient content and nutrient use efficiency in Eucalyptus stands in Congo. Forest Ecology and Management. 2005;210:193–204. doi: 10.1016/j.foreco.2005.02.049. [DOI] [Google Scholar]
- Halperin W. Organogenesis at Shoot Apex. Annual Review of Plant Physiology and Plant Molecular Biology. 1978;29:239–262. [Google Scholar]
- Vega-Arreguin JC, Ibarra-Laclette E, Jimenez-Moraila B, Martinez O, Vielle-Calzada JP, Herrera-Estrella L, Herrera-Estrella A. Deep sampling of the Palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics. 2009;10:299. doi: 10.1186/1471-2164-10-299. (6 July 2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Chase MW, Mort ME, Albach DC, Zanis M, Savolainen V, Hahn WH, Hoot SB, Fay MF. et al. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Botanical Journal of the Linnean Society. 2000;133:381–461. [Google Scholar]
- Lynch M, Walsh JB. Genetics and Analysis of Quantitative Traits. Sunderland, MA; 1998. [Google Scholar]
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2010. aop. [DOI] [PMC free article] [PubMed]
- Metzker ML. APPLICATIONS OF NEXT-GENERATION SEQUENCING Sequencing technologies - the next generation. Nature Reviews Genetics. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Zhou X, Su Z, Sammons RD, Peng YH, Tranel PJ, Stewart CN, Yuan JS. Novel software package for cross-platform transcriptome analysis (CPTRA) Bmc Bioinformatics. 2009;10(Suppl 11):S16. doi: 10.1186/1471-2105-10-S11-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall PK, Leebens-Mack J, Chanderbali AS, Barakat A, Wolcott E, Liang HY, Landherr L, Tomsho LP, Hu Y, Carlson JE. et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics. 2009;10:347. doi: 10.1186/1471-2164-10-347. (1 August 2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrousian M. Next-Generation Sequencing Techniques for Eukaryotic Microorganisms: Sequencing-Based Solutions to Biological Problems. Eukaryotic Cell. 2010;9:1300–1310. doi: 10.1128/EC.00123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty MJ, Barker GL, Brennan AC, Edwards KJ, Abbott RJ, Hiscock SJ. Changes to gene expression associated with hybrid speciation in plants: further insights from transcriptomic studies in Senecio. Philosophical Transactions of the Royal Society B-Biological Sciences. 2008;363:3055–3069. doi: 10.1098/rstb.2008.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey SA, Collin H, Nosil P, Rogers SM. The role of gene expression in ecological speciation. Year in Evolutionary Biology. 2010;1206:110–129. doi: 10.1111/j.1749-6632.2010.05765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts BM, Dungey HS. Interspecific hybridization of <i>Eucalyptus</i>: key issues for breeders and geneticists. New Forests. 2004;27:115–138. doi: 10.1023/A:1025021324564. [DOI] [Google Scholar]
- Shinozaki K, Yamaguchi-Shinozaki K. Gene networks involved in drought stress response and tolerance. Journal of Experimental Botany. 2007;58:221–227. doi: 10.1093/jxb/erl164. [DOI] [PubMed] [Google Scholar]
- Hong-Bo S, Li-Ye C, Ming-An S. Calcium as a versatile plant signal transducer under soil water stress. Bioessays. 2008;30:634–641. doi: 10.1002/bies.20770. [DOI] [PubMed] [Google Scholar]
- Schroeder JI, Kwak JM, Allen GJ. Guard cell abscisic acid signalling and engineering drought hardiness in plants. Nature. 2001;410:327–330. doi: 10.1038/35066500. [DOI] [PubMed] [Google Scholar]
- Hutin C, Nussaume L, Moise N, Moya I, Kloppstech K, Havaux M. Early light-induced proteins protect arabidopsis from photooxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4921–4926. doi: 10.1073/pnas.0736939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: Signaling for suicide and survival. Journal of Cellular Physiology. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Kitajima S, Sato F. Plant pathogenesis-related proteins: Molecular mechanisms of gene expression and protein function. Journal of Biochemistry. 1999;125:1–8. doi: 10.1093/oxfordjournals.jbchem.a022244. [DOI] [PubMed] [Google Scholar]
- Lee BR, Jung WJ, Lee BH, Avice JC, Ourry A, Kim TH. Kinetics of drought-induced pathogenesis-related proteins and its physiological significance in white clover leaves. Physiologia Plantarum. 2008;132:329–337. doi: 10.1111/j.1399-3054.2007.01014.x. [DOI] [PubMed] [Google Scholar]
- Przymusinski R, Rucinska R, Gwozdz EA. Increased accumulation of pathogenesis-related proteins in response of lupine roots to various abiotic stresses. Environmental and Experimental Botany. 2004;52:53–61. doi: 10.1016/j.envexpbot.2004.01.006. [DOI] [Google Scholar]
- Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends in Plant Science. 2010;15:573–581. doi: 10.1016/j.tplants.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Takahashi Y, Berberich T, Imai A, Takahashi T, Michael AJ, Kusano T. A protective role for the polyamine spermine against drought stress in Arabidopsis. Biochemical and Biophysical Research Communications. 2007;352:486–490. doi: 10.1016/j.bbrc.2006.11.041. [DOI] [PubMed] [Google Scholar]
- Alexandersson E, Fraysse L, Sjovall-Larsen S, Gustavsson S, Fellert M, Karlsson M, Johanson U, Kjellbom P. Whole gene family expression and drought stress regulation of aquaporins. Plant Molecular Biology. 2005;59:469–484. doi: 10.1007/s11103-005-0352-1. [DOI] [PubMed] [Google Scholar]
- Landry CR, Oh J, Hartl DL, Cavalieri D. Genome-wide scan reveals that genetic variation for transcriptional plasticity in yeast is biased towards multi-copy and dispensable genes. Gene. 2006;366:343–351. doi: 10.1016/j.gene.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Hodgins-Davis A, Townsend JP. Evolving gene expression: from G to E to G × E. Trends in Ecology & Evolution. 2009;24:649–658. doi: 10.1016/j.tree.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnusamy V, Zhu JK. Epigenetic regulation of stress responses in plants. Current Opinion in Plant Biology. 2009;12:133–139. doi: 10.1016/j.pbi.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucotelli E, Mastrangelo AM, Crosatti C, Guerra D, Stanca AM, Cattivelli L. Abiotic stress response in plants: When post-transcriptional and post-translational regulations control transcription. Plant Science. 2008;174:420–431. doi: 10.1016/j.plantsci.2008.02.005. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results of the ANOVA for above-ground biomass.
Distribution of read length for the three half-runs.
Increasing coverage with successive runs. Number of contigs represented in each half-run or combination of several half-runs. Performing a second half-run increased contig coverage by an average of 18%, and a third half run increased coverage by an average of 6%.
Comparison of the distribution of Gene Ontology (GO) categories between Eucalyptus spp unigene elements (UE) and Arabidopsis annotated unigenes. Proportion of each GO category (Biological Process, Cellular Component and Molecular Function) found in the E. spp sequencing set and in the annotated Arabidopsis genome.
Differentially expressed contigs displaying significant T and GxT effects. Each contig showing T or GxT is associated with a functional category (as defined by Mercator : http://mapman.gabipd.org/web/guest). Log2-transformed fold change between abundance in irrigated and non irrigated libraries are indicated for each genotype, as well as p-value of T and GxT effects analyzed by the ANOVA.
Distribution of "G" contigs between functional pathways. Each square represents the log2-transformed fold-change of abundance between genotypes 1-41 and 18-50 for one contig. Contigs in green were overexpressed in genotype 1-41 and contigs in blue were overexpressed in genotype 18-50.
Significant categories for the Wilcoxon rank sum test, according to Mapman analysis for the pairwise comparison of differentially expressed contigs displaying genotype (G), treatment (T) and genotype × treatment (GxT) effects. ** Categories differentially expressed at an error rate threshold of 0.05 * Categories differentially expressed at an error rate threshold of 0.1
Distribution of "T" contigs between several functional pathways. Each square represents the log2-transformed fold-change of abundance between irrigated (IR) and non-irrigated (NI) treatments for one contig. Contigs in blue were overexpressed for the IR treatment and contigs in red were overexpressed for the NI treatment.
Supporting information: validation of digital profiles by analyzing expression by RT-qPCR on 36 genes.