

Abstract
OTHER ARTICLES PUBLISHED IN THIS MINI-REVIEW SERIES ON B CELL SUBSETS IN DISEASE
B cells in multiple sclerosis: drivers of disease pathogenesis and Trojan horse for Epstein—Barr virus entry to the central nervous system? Clinical and Experimental Immunology 2012, 167: 1–6. Reconstitution after haematopoietic stem cell transplantation – revelation of B cell developmental pathways and lineage phenotypes. Clinical and Experimental Immunology 2012, 167: 15–25.
Systemic lupus erythematosus (SLE) and Sjögren's syndrome are autoimmune disorders which are characterized by a disturbed B cell homeostasis which leads ultimately to dysfunction of various organs. One of the B cell subsets that appear in abnormal numbers is the population of transitional B cells, which is increased in the blood of patients with SLE and Sjögren's syndrome. Transitional B cells are newly formed B cells. In mice, transitional B cells undergo selection checks for unwanted specificity in the bone marrow and the spleen in order to eliminate autoreactive B cells from the circulating naive B cell population. In humans, the exact anatomical compartments and mechanisms of the specificity check-points for transitional B cells remain unclear, but appear to be defective in SLE and Sjögren's syndrome. This review aims to highlight the current understanding of transitional B cells and their defects in the two disorders before and after B cell-targeted therapies.
Keywords: belimumab, rituximab, systemic lupus erythematosus, transitional B cells
Transitional B cells
B cell development in the bone marrow occurs continuously throughout life. The diverse antigen-binding repertoire of B cell surface immunoglobulin is generated as B cells develop by a process of gene rearrangement that produces both wanted and unwanted specificities. In mice, there are two known major check-points that filter out autoreactive and to some extent polyspecific B cells from the newly generated cells, thus ensuring that they make a minimal contribution to the mature recirculating B cell population. The first check-point for specificity, in humans as in mice, occurs in the bone marrow, where selection of B cells is achieved mainly by eliminating autoreactive B cells. B cells that react with self-antigens present in the bone marrow will be removed by mechanisms that include anergy [1,2] clonal deletion [3,4] and receptor-editing [5], particularly editing of the immunoglobulin (Ig) light chain. Receptor editing replaces one gene rearrangement with another in immature B cells, often on the same allele. There is evidence of light chain editing in human cells as well as animal models; in human cells, the most V proximal IGKJ segment is more common in the unproductive than the productive rearrangements [6,7]. Positive selection and B cell survival requires the anti-apoptotic cytokine BLyS (B lymphocyte stimulator, also known as BAFF or TNFSF13B) [8]. Moreover, current data suggest that tonic signals via B cell receptors (BCRs) are critical for immature B cells to suppress Rag expression and maintain specific developmental stages [9]. Only a small percentage of transitional B cells manage to pass through the first tolerance check and will then enter the periphery in mice and humans. The second check-point for selection seems to be the spleen and the bone marrow in mice where transitional B cells need to receive survival signals before they differentiate into mature naive B cells [10,11]. The sites and mechanisms involved in human transitional B cell selection are less clearly understood.
The development of transitional B cells in mice has been investigated in more detail, and seems to differ from the differentiation in humans in multiple ways. For example, murine transitional B cells comprise the subclasses T0, T1, T2 and T3, a classification based on the expression of CD23, IgM and the developmental marker AA4. T1 cells are found in the bone marrow, blood and spleen. T2 cells are found in the spleen and are thought to be derived from the T1 cells. T3 cells seem to be an anergic splenic population that does not progress into the mature stage [12–15]. Direct equivalent populations are not apparent in humans, although subsets of human transitional B cells can be distinguished based, for example, on the expression of CD21 [16]. A subset of human B cells with transitional cell phenotype has been reported to be immunosuppressive regulatory B cells (Breg) [17]. Murine transitional B cells eventually enter different splenic compartments and develop into follicular or marginal zone B cells. T1 cells home to the marginal zone (MZ), but are also found in the splenic follicles. It is unknown whether these cells need to pass through the follicles before they enter the MZ [18]. Although the precise lineage scheme is not understood, it is considered that in mice, marginal zone B cells form at a branch in the B cell maturation pathway and they are a subset or separate developmental lineage of naive B cells to those that occupy the follicular mantle. The situation can be confused by the observed presence of memory cells in the marginal zone in mice [19]. These cells may reside in the marginal zone or they may be in transit. In contrast, there is no known direct progression from transitional B cells to marginal zone B cells in humans, where marginal zone B cells have features of memory cells and somatic mutations in their immunoglobulin variable region genes, indicating a history of involvement in a germinal centre response [20].
The uncertainties in current understanding of the pathways of B cell maturation in humans should be considered when the pathogenesis of SLE and primary Sjögren's syndrome (pSS) is studied, because mouse models are unlikely to provide sufficient mechanistic insight into the disturbed specificity checks of human immature B cells in these disorders (Fig. 1).
Fig. 1.
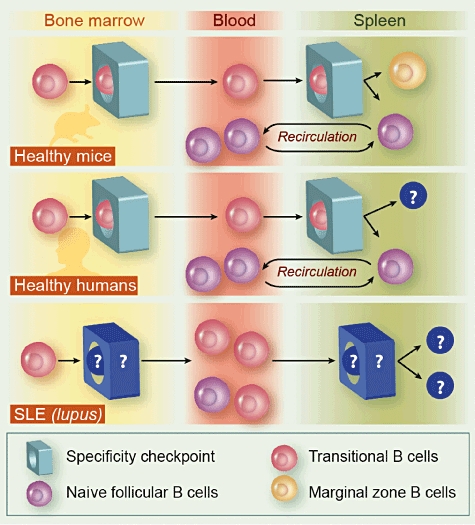
Specificity checks for transitional B cells in mice and humans. In mice, transitional B cells emerge from the bone marrow, where they need to pass their first specificity check. They enter the spleen and after further checks they enter the marginal zone or reside in the splenic follicles as naive follicular B cells. The latter can circulate between blood and lymphoid tissues. In humans, the second check-point is less well understood. Although it is believed that it lies within the spleen, there is little evidence for this in humans. The maturation of transitional cells into marginal zone B cells that have mutated immunoglobulin variable region genes normally indicative of germinal centre transit is uncertain. Patients with systemic lupus erythematosus (SLE) have an increased proportion of autoreactive B cells which presumably have not been checked for specificity. Although the defective check-point is unknown, it has been observed that transitional B cells emerge from the bone marrow and accumulate in the blood in SLE constituting a relatively large proportion of blood B cells. Whether a further maturation defect lies in the spleen or elsewhere remains unknown.
Increased frequencies of transitional B cells in autoimmune disorders
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of various autoantibodies and immune complexes and results in multiple organ damage. Although the pathogenesis of the condition is not fully understood, it has become clear that B cells play a key role. B cells do not only mediate the pathogenesis by the production of autoantibodies, they also contribute to the condition at the levels of abnormal cytokine secretion [21], deregulated signalling upon B cell receptor (BCR) engagement [22], reduction in the suppressor Breg B cell subset [17] and increased expression of the co-stimulatory molecule CD86 on CD19+ peripheral B cells [23]. The main characteristic of the disturbed B cell homeostasis in SLE is the expansion of peripheral CD27high plasmablasts [24,25], which also correlates with disease activity and the titre of autoantibodies [26]. Furthermore, SLE is associated with naive (CD19+CD27-) B cell lymphopenia [25], and an increased proportion of CD24high CD38high transitional B cells [27,28]. The frequency of CD19+CD27+ memory B cells seems to be largely unaffected in SLE patients with active and inactive disease, although the total number of memory B cells is decreased in SLE patients compared to healthy controls [25]. In the blood of healthy patients, transitional B cells account for 2–3% of all B cells [28,29]. In contrast, SLE patients have these cells in an increased frequency of approximately 6·7% [28]. The proportion of transitional B cells does not correlate with disease activity and other pathological features of SLE (e.g. titre of autoantibodies) [28]. Due to the general lymphopenia in SLE, the absolute number of transitional B cells is not different to that of healthy controls. SLE patients have a severe defect in the B cell tolerance check which results in high numbers of autoreactive mature naive B cells which subsequently give rise to autoantibody-producing plasma cells [30], as summarized in [31]. The defect is most likely to occur at the transition stage between new bone marrow emigrants and mature naive B cells in the periphery. The same level (∼40–50%) of autoreactive antibodies was detected in new bone marrow emigrants of SLE patients and healthy controls. However, while the naive mature B cells in healthy controls show a drastic reduction in autoreactive antibody expression (4·3%), this cell subset in SLE patients remains high with regard to the production of autoreactive Abs (between 25 and 31%) [30]. The nature of the second tolerance check-point prior to maturation from transitional B cell stage in humans and the breakdown of this check-point in SLE that allows the persistence of autoreactive transitional B cells are not understood. Therefore, the population of transitional B cells in SLE patients needs further investigation, which might also reveal whether and how these cells contribute to disease pathogenesis.
Primary Sjögren's syndrome
Primary Sjögren's syndrome (pSS), also called ‘autoimmune exocrinopathy’, is an autoimmune disorder of unknown origin characterized by chronic inflammation of exocrine glands, with the lachrymal and salivary glands affected in particular. However, pSS is a systemic disorder with extraglandular manifestations and can be associated with other autoimmune diseases, including rheumatoid arthritis, SLE or progressive systemic sclerosis (summarized in [32]). Characteristic features of pSS include focal inflammatory infiltrates in glands, which are comprised of B and T lymphocytes as well as macrophages and dendritic cells [33]. Furthermore, pSS patients present with B cell abnormalities, hypergammaglobulinaemia and various autoantibodies [34]. One-quarter of pSS patients have ectopic development of B cell proliferations which resemble germinal centres (GC), and have been linked to B cell lymphoma in pSS [35]. Some of the ectopic B cell proliferations are genuine GCs which include autoreactive B cells. These results were published by Le Pottier et al., who phenotyped B cells within GC-like structures in salivary glands of pSS patients [36]. Investigation of the B cell populations in the peripheral blood of pSS patients demonstrated a disturbed pattern of B cell subsets. While CD27+IgD+ and CD27+IgM+ memory cells were reduced in frequency and total cell number [37], pSS patients had increased numbers of CD27- naive B cells [29]. The disturbed B cell subsets in the blood of pSS patients might be due partially to retention or migration of memory B cells in sites of glandular inflammation [37,38]. The thorough phenotyping of the B cells infiltrating the salivary glands of pSS patients revealed the presence of a possibly more developed subset of transitional B cells, so-called ‘TII B cells’[39], which were identified as CD19+IgD+CD38-IgM+CD21+CD23+cells. These cells lack expression of CD38, which is present on transitional B cells that emigrated from the bone marrow into the circulation. The same study demonstrated elevated levels of BLyS within the B cell infiltrates of pSS patients [38]. Previously, BLyS has also been detected in increased levels in the serum of pSS patients [40]. These data suggest that autoreactive transitional B cells which have not been removed during the tolerance check are present in the infiltrates, where they receive survival signals via BLyS and contribute to the pathology within the affected glands.
B cell-targeted therapies and their effect on transitional B cells
Deregulation of B cell development and function resulting in defective tolerance are a central feature of SLE and pSS and led to the introduction of anti-B cell antibodies as new biological therapies for moderate to severe cases of these disorders [41]. Therapy with belimumab, a humanized monoclonal antibody against soluble BLyS is based on the finding that BLys is found in high concentrations in serum of SLE patients [42]. BLyS is part of the tumour necrosis factor (TNF) superfamily secreted by myeloid cells, and provides a key signal for B cell survival [43,44]. Belimumab was approved for the therapy of SLE in March 2011 by the Food and Drugs Administration (FDA) in the United States. This was a landmark approval which made belimumab the first drug to be licensed for use in SLE in almost 50 years. The two large Phase III trials (BLISS-52 and BLISS-76) of moderately active non-nephritis SLE patients treated with belimumab met their primary efficacy end-point of clinical superiority compared to placebo plus standard of care [45,46]. The analysis of this heterogeneous population of 1684 SLE patients with non-renal moderate to severe active SLE was performed using the FDA-approved ‘SLE Responder Index’. The SLE Responder Index rates in all seropositive SLE patients treated with belimumab, irrespective of the dose, were 46% at week 52 and 49% at week 56. This made a difference of 29–35% when compared with placebo (P < 0·05). In the intention-to-treat analysis at 160 weeks, the rates of SLE Responder Index were 55%.
The consequences of blocking BLyS function on B cell subsets have been studied in animal models and in humans. Early studies in murine SLE demonstrated that blocking BLyS mainly reduced the number of transitional T2 cells as well as the number of follicular and marginal zone B cells. The transitional T1 cells were, however, unaffected in this mouse model and autoantibodies were still produced [47]. This might be explained by the fact that APRIL (a proliferation-inducing ligand) delivers the homeostatic signal to long-lived plasma cells when BLyS is absent [48]. Memory B cells may differ in mice and humans in their expression of receptors for BLyS. BLyS has three receptors: transmembrane activator and calcium modulator ligand interactor (TACI), B cell maturation antigen (BCMA) and BAFF-receptor (BAFF-R). Whereas human memory B cells have been reported to express all three receptors, murine memory B cells express TACI, but only low levels of BCMA and BAFF-R [47]. This implies that species differences in the persistence of memory following the blocking of BLyS function might be expected, potentially affecting memory in humans more profoundly than in mice.
The long-term changes in circulating B cell numbers after treatment with belimumab or placebo were studied in 17 patients with moderately active SLE in a large Phase II trial [49]. In this subgroup, 13 patients had received belimumab. On day 532 after treatment began, only IgM levels had dropped in some patients but no reduction in IgG and antibodies against dsDNA (double-stranded DNA) was seen. Total B cell numbers had decreased due to a reduction of naive and transitional B cells, whose decrease occurred by day 84 after treatment began and were at a stable level of 88% and 75%, respectively, below the baseline at day 532. Of interest in the context of the expression of receptors for BLyS [47] class-switched CD27+IgD- memory B cell numbers were not affected by BLyS blockage [49]. Therefore, inhibition of BLyS mainly affects newly formed B cells and reduces the number of transitional B cells but does not reduce anti-dsDNA in the majority of patients [49,50]. Therefore, the properties of the newly formed B cells that are efficiently depleted by inhibition of BLyS are identified as potentially important cells in the disease process in SLE. In contrast, the established autoreactive B cells and their plasma cell progeny which continue to release autoantibodies during belimumab therapy appear less involved.
To summarize, BLyS inhibition induces slow, selective B cell depletion with attrition occurring specifically in transitional, naive B cell lines and the CD27- isotype-switched memory population with effector surface phenotype but not in conventional CD27+ memory cells or antibody-secreting cells [49] (Fig. 2). It remains unclear whether belimumab therapy might be able to restore tolerance at the critical transitional B cell check-point for newly formed B cells, which is thought to be BLyS-dependent based on animal models, or whether a change of intrinsic properties of both transitional and naive B cells underlies the success of this therapeutic approach [43,51–53].
Fig. 2.

B cell-directed antibodies are emerging therapies in the management of systemic lupus erythematosus (SLE). Belimumab binds BLyS, a factor which induces B cell proliferation and immunoglobulin secretion, resulting in reduced B cell survival. Belimumab therapy reduces the number of blood transitional B cells, but seems to be unable to reduce autoantibodies. Rituximab targets CD20, a surface molecule present on all B cell stages with the exception of plasma cells. The treatment with rituximab leads to a general B cell depletion and to an increase in transitional B cells during reconstitution. It is not clear yet whether rituximab influences the levels of autoantibodies. The antibody epratuzumab blocks CD22, a transmembrane molecule associated with the B cell receptor, and mediates a moderate B cell depletion which includes transitional B cells. Epratuzumab treatment does not reduce levels of autoantibodies in SLE patients.
Rituximab is a chimeric mouse/human monoclonal antibody against CD20 and depletes the body of all B cells stages that express this marker; that is, mature B cells and B cell precursors from the pre-B cell stage onwards [54,55]. Repopulation after rituximab therapy bears many similarities with the B cell repopulation observed after bone marrow transplantation [56]. During reconstitution following rituximab therapy in SLE patients, the number of CD24++CD38++ transitional B cells in blood increased to about 19% (compared to 5% in the SLE cohort and 4% in healthy control subjects in this study) [57]. Long-term responders had higher numbers of transitional B cells and lower levels of anti-dsDNA than short-term responders. Rituximab treatment led to a long-standing reduction of peripheral memory B cells with a delayed recovery of blood memory B cells compared to memory B cells in tissue. The same group further demonstrated normalization of initially disturbed B cell populations following rituximab therapy in SLE patients [58]. These included naive IgD+CD27- B cell lymphopenia and expansion of circulating CD272+ plasmablasts. Improvement in anti-dsDNA levels occurred in only 50% of investigated patients, although disease activity improved in all patients [54]. These data suggest that B cell depletion with anti-CD20 and the subsequent reconstitution might result in a fresh B cell compartment with improved selection for autoreactivity (Fig. 2). However, the exact mechanisms in this process remain elusive. Looney and co-workers analysed B cell depletion with high sensitivity flow cytometry and concluded that good responders to rituximab had very significant depletion of B cells compared to average or non-responders [54]. This early finding, suggesting the potential benefit of B cell-targeted therapy in SLE, has been confirmed in more recent studies and extrapolated further to advocate the use of other B cell-targeting drugs.
Several open-label prospective and retrospective studies have demonstrated the effectiveness of rituximab in the management of moderately severe to severe SLE with statistically significant improvement in the British Isles Lupus Assessment Group (BILAG) score [59–62] and Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score [63–65]. In 2009, Ramos-Calas et al. published a review of studies involving 188 SLE patients managed with rituximab since 2005 [66]. In this paper 171 patients had a clinical response to rituximab, and although 46 relapsed, 80% of those relapsed patients who were retreated with rituximab had a clinical response [66]. In addition, studies of SLE patients with severe refractory disease have further highlighted the effectiveness of rituximab in this population of patients [65,67–76]. Although the rituximab Phase III randomized controlled trials, the EXPLORER trial (non-renal SLE patients) [77] and the LUNAR trial (lupus nephritis patients) [78] did not demonstrate superiority to standard care in all patients treated with rituximab, it is worth noting the positive results that emerged. B cell-depleting therapy was associated with statistically significant improvement in the serological markers of disease activity in the SLE patients in the EXPLORER trial such as complement C3 levels and dsDNA antibodies [79,80]. Factors that may have contributed to the negative outcome of the EXPLORER and LUNAR trials have been debated and include the fact that both trials set very high standards for primary and secondary end-points; however, the follow-up period was short, thus limiting the time period available to ascertain differences objectively in therapeutic arms. In addition, there was significant concomitant use of high-dose corticosteroids in both the standard therapy and rituximab arms, thus influencing the outcome of the trials.
The first double-blind study of rituximab in pSS achieved the primary end-point (20% improvement in fatigue visual analogue score, VAS) in the 17 patients studied. However, there was no significant benefit documented in other clinical manifestations of pSS such as Schirmer's test (a test which determines if a patient suffers from dry eyes) or unstimulated salivary flow rate, thus demonstrating no benefit to sicca symptoms despite adequate B cell depletion [81,82]. An extended follow-up study of patients with pSS in the Netherlands reported favourable results, with improvement in the VAS for dry mouth and an increase in the stimulated submandibular/sublingual salivary flow post-rituximab treatment, although the benefits were temporary, i.e. less than 48 weeks [83]. The role of B cell depletion for the treatment of extraglandular manifestations of pSS such as peripheral neuropathy and inflammatory arthritis is yet to be established and further research is required [83].
The humanized anti-CD22 monoclonal antibody (mAb) epratuzumab leads to a moderate B cell depletion [84]. CD22 is an adhesion molecule and co-receptor for BCR, where it attenuates BCR signalling. Binding of the antibody leads to internalization and phosphorylation of CD22 [85,86]. Epratuzumab infusion resulted in a reduction of CD27- transitional and naive B cells which lasted about 4 months and reduced CD22 expression on the remaining CD27- B cells. The number of CD27+ memory B cells was less affected, explained partly by only low expression of CD22 [84]. Besides reducing CD27- B cells, epratuzumab seemed to decrease the expression of the adhesion molecules CD62L, β7 integrin and increased expression of β1 integrin on blood B cells in SLE patients. Another effect observed was the increased migratory capacity of CD27- B cells towards the chemokine CXCL12 in vitro[87] (Fig. 2). Epratuzumab had no effect on autoantibody concentrations in SLE patients; however, the majority of patients' disease activity improved [88]. Clinical improvement was also reported for pSS patients after epratuzumab infusion. However, nor were changes in autoantibody levels seen in these patients [89]. In conclusion, the exact mechanism by which epratuzumab modulates B cells in SLE remains unclear, but it seems likely that it affects activation status and migratory properties of CD27- B cells. EMBLEM is a 12-week Phase IIB placebo-controlled trial of epratuzumab at different doses in six treatment arms. Preliminary results of treatment response using the BILAG index and SLEDAI have shown positive results in both clinical and statistical analysis [90]. Further research is in the pipeline for this novel humanized IgG1 anti-CD22 monoclonal antibody.
Conclusion
Connective tissue disorders such as SLE and pSS are heterogeneous conditions which are manifested by a broad spectrum of clinical presentations, severity and prognosis. The pathogenesis of SLE and pSS is rooted in a disturbed B cell homeostasis, and therefore in-depth understanding and knowledge of the immunological mechanisms that result in autoimmune connective tissue disease, in particular the subanalysis of B cell biology, would facilitate the development of appropriately targeted, safe therapies. The emergence of novel B cell-targeting therapies including belimumab, rituximab and epratuzumab will not only revolutionize the management of complex disorders such as SLE and pSS, but will also aid the identification of disease-relevant B cell subsets.
Acknowledgments
A.V. is funded by the Medical Research Council UK.
Disclosure
The authors declare no conflict of interest.
References
- 1.Brink R, Goodnow CC, Crosbie J, et al. Immunoglobulin M and D antigen receptors are both capable of mediating B lymphocyte activation, deletion, or anergy after interaction with specific antigen. J Exp Med. 1992;176:991–1005. doi: 10.1084/jem.176.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–9. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 3.Nemazee D, Buerki K. Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc Natl Acad Sci USA. 1989;86:8039–43. doi: 10.1073/pnas.86.20.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–6. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 5.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brauninger A, Goossens T, Rajewsky K, Kuppers R. Regulation of immunoglobulin light chain gene rearrangements during early B cell development in the human. Eur J Immunol. 2001;31:3631–7. doi: 10.1002/1521-4141(200112)31:12<3631::aid-immu3631>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 7.Su W, Gordon JN, Barone F, et al. Lambda light chain revision in the human intestinal IgA response. J Immunol. 2008;181:1264–71. doi: 10.4049/jimmunol.181.2.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harless SM, Lentz VM, Sah AP, et al. Competition for BLyS-mediated signaling through Bcmd/BR3 regulates peripheral B lymphocyte numbers. Curr Biol. 2001;11:1986–9. doi: 10.1016/s0960-9822(01)00598-x. [DOI] [PubMed] [Google Scholar]
- 9.Tze LE, Schram BR, Lam KP, et al. Basal immunoglobulin signaling actively maintains developmental stage in immature B cells. PLoS Biol. 2005;3:e82. doi: 10.1371/journal.pbio.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cariappa A, Chase C, Liu H, Russell P, Pillai S. Naive recirculating B cells mature simultaneously in the spleen and bone marrow. Blood. 2007;109:2339–45. doi: 10.1182/blood-2006-05-021089. [DOI] [PubMed] [Google Scholar]
- 11.Loder F, Mutschler B, Ray RJ, et al. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167:6834–40. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 13.Merrell KT, Benschop RJ, Gauld SB, et al. Identification of anergic B cells within a wild-type repertoire. Immunity. 2006;25:953–62. doi: 10.1016/j.immuni.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava B, Quinn WJ, 3rd, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. J Exp Med. 2005;202:1225–34. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teague BN, Pan Y, Mudd PA, et al. Cutting edge: transitional T3 B cells do not give rise to mature B cells, have undergone selection, and are reduced in murine lupus. J Immunol. 2007;178:7511–5. doi: 10.4049/jimmunol.178.12.7511. [DOI] [PubMed] [Google Scholar]
- 16.Suryani S, Fulcher DA, Santner-Nanan B, et al. Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood. 2010;115:519–29. doi: 10.1182/blood-2009-07-234799. [DOI] [PubMed] [Google Scholar]
- 17.Blair PA, Norena LY, Flores-Borja F, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;32:129–40. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Chopin M, Quemeneur L, Ripich T, Jessberger R. SWAP-70 controls formation of the splenic marginal zone through regulating T1B-cell differentiation. Eur J Immunol. 2010;40:3544–56. doi: 10.1002/eji.201040556. [DOI] [PubMed] [Google Scholar]
- 19.Liu YJ, Oldfield S, MacLennan IC. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur J Immunol. 1988;18:355–62. doi: 10.1002/eji.1830180306. [DOI] [PubMed] [Google Scholar]
- 20.Weill JC, Weller S, Reynaud CA. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–85. doi: 10.1146/annurev.immunol.021908.132607. [DOI] [PubMed] [Google Scholar]
- 21.Tsokos GC, Wong HK, Enyedy EJ, Nambiar MP. Immune cell signaling in lupus. Curr Opin Rheumatol. 2000;12:355–63. doi: 10.1097/00002281-200009000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest. 1996;98:2549–57. doi: 10.1172/JCI119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bijl M, Horst G, Limburg PC, Kallenberg CG. Expression of costimulatory molecules on peripheral blood lymphocytes of patients with systemic lupus erythematosus. Ann Rheum Dis. 2001;60:523–6. doi: 10.1136/ard.60.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada Y, Kawano MM, Huang N, et al. Identification of early plasma cells in peripheral blood and their clinical significance. Br J Haematol. 1996;92:184–91. doi: 10.1046/j.1365-2141.1996.300835.x. [DOI] [PubMed] [Google Scholar]
- 25.Odendahl M, Jacobi A, Hansen A, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 26.Jacobi AM, Odendahl M, Reiter K, et al. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:1332–42. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- 27.Cuss AK, Avery DT, Cannons JL, et al. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. 2006;176:1506–16. doi: 10.4049/jimmunol.176.3.1506. [DOI] [PubMed] [Google Scholar]
- 28.Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. 2005;105:4390–8. doi: 10.1182/blood-2004-11-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnhorst JO, Bjorgan MB, Thoen JE, Natvig JB, Thompson KM. Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjogren's syndrome. J Immunol. 2001;167:3610–18. doi: 10.4049/jimmunol.167.7.3610. [DOI] [PubMed] [Google Scholar]
- 30.Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yurasov S, Hammersen J, Tiller T, Tsuiji M, Wardemann H. B-cell tolerance checkpoints in healthy humans and patients with systemic lupus erythematosus. Ann NY Acad Sci. 2005;1062:165–74. doi: 10.1196/annals.1358.019. [DOI] [PubMed] [Google Scholar]
- 32.Fox RI. Sjogren's syndrome. Lancet. 2005;366:321–31. doi: 10.1016/S0140-6736(05)66990-5. [DOI] [PubMed] [Google Scholar]
- 33.Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in Sjogren's syndrome. J Autoimmun. 2010;34:400–7. doi: 10.1016/j.jaut.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Hansen A, Lipsky PE, Dorner T. B cells in Sjogren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther. 2007;9:218. doi: 10.1186/ar2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salomonsson S, Jonsson MV, Skarstein K, et al. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren's syndrome. Arthritis Rheum. 2003;48:3187–201. doi: 10.1002/art.11311. [DOI] [PubMed] [Google Scholar]
- 36.Le Pottier L, Devauchelle V, Fautrel A, et al. Ectopic germinal centers are rare in Sjogren's syndrome salivary glands and do not exclude autoreactive B cells. J Immunol. 2009;182:3540–7. doi: 10.4049/jimmunol.0803588. [DOI] [PubMed] [Google Scholar]
- 37.Hansen A, Odendahl M, Reiter K, et al. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2002;46:2160–71. doi: 10.1002/art.10445. [DOI] [PubMed] [Google Scholar]
- 38.Daridon C, Pers JO, Devauchelle V, et al. Identification of transitional type II B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2006;54:2280–8. doi: 10.1002/art.21936. [DOI] [PubMed] [Google Scholar]
- 39.Chung JB, Silverman M, Monroe JG. Transitional B cells: step by step towards immune competence. Trends Immunol. 2003;24:343–9. doi: 10.1016/s1471-4906(03)00119-4. [DOI] [PubMed] [Google Scholar]
- 40.d'Arbonneau F, Pers JO, Devauchelle V, Pennec Y, Saraux A, Youinou P. BAFF-induced changes in B cell antigen receptor-containing lipid rafts in Sjogren's syndrome. Arthritis Rheum. 2006;54:115–26. doi: 10.1002/art.21478. [DOI] [PubMed] [Google Scholar]
- 41.Traczewski P, Rudnicka L. Treatment of systemic lupus erythematosus with epratuzumab. Br J Clin Pharmacol. 2010;71:175–82. doi: 10.1111/j.1365-2125.2010.03767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pers JO, Daridon C, Devauchelle V, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann NY Acad Sci. 2005;1050:34–9. doi: 10.1196/annals.1313.004. [DOI] [PubMed] [Google Scholar]
- 43.Cancro MP. The BLyS/BAFF family of ligands and receptors: key targets in the therapy and understanding of autoimmunity. Ann Rheum Dis. 2006;65(Suppl. 3):iii34–6. doi: 10.1136/ard.2006.058412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancro MP, D'Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest. 2009;119:1066–73. doi: 10.1172/JCI38010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarra S, Illianova E, Bae SC, et al. Belimumab, a BLyS-specific inhibitor, reduced disease activity, flares and steroid use in patients with seropositive SLE: BLISS-52 study. Ann Rheum Dis. 2010;69(Suppl. 3):555. [Google Scholar]
- 46.van Vollenhoven RF, Zamani O, Wallace DJ, et al. Belimumab, a BLyS-specific inhibitor, reduces disease activity and severe flares in seropositive SLE patients: BLISS-76 study. Ann Rheum Dis. 2010;69(Suppl. 3):74. [Google Scholar]
- 47.Ramanujam M, Wang X, Huang W, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116:724–34. doi: 10.1172/JCI26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benson MJ, Dillon SR, Castigli E, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180:3655–9. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- 49.Jacobi AM, Huang W, Wang T, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;62:201–10. doi: 10.1002/art.27189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furie R, Stohl W, Ginzler EM, et al. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: a phase I trial in patients with systemic lupus erythematosus. Arthritis Res Ther. 2008;10:R109. doi: 10.1186/ar2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lesley R, Xu Y, Kalled SL, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–53. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 52.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Ann Rev Immunol. 2003;21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 53.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 54.Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–9. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 55.Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–45. [PubMed] [Google Scholar]
- 56.Bemark M, Holmqvist J, Abrahamsson J, Mellgren K. Translational Mini-Review Series: B cell subsets in disease. Reconstitution after hematopoietic stem cell transplantation – revelation of B cell developmental pathways. Clin Exp Immunol. 2012;167:15–25. doi: 10.1111/j.1365-2249.2011.04469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anolik JH, Barnard J, Owen T, et al. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007;56:3044–56. doi: 10.1002/art.22810. [DOI] [PubMed] [Google Scholar]
- 58.Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum. 2004;50:3580–90. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- 59.Leandro MJ, Cambridge G, Edwards JC, Ehrenstein MR, Isenberg DA. B-cell depletion in the treatment of patients with systemic lupus erythematosus: a longitudinal analysis of 24 patients. Rheumatology (Oxf) 2005;44:1542–5. doi: 10.1093/rheumatology/kei080. [DOI] [PubMed] [Google Scholar]
- 60.Ng KP, Cambridge G, Leandro MJ, Edwards JC, Ehrenstein M, Isenberg DA. B cell depletion therapy in systemic lupus erythematosus: long-term follow-up and predictors of response. Ann Rheum Dis. 2007;66:1259–62. doi: 10.1136/ard.2006.067124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reynolds JA, Toescu V, Yee CS, Prabu A, Situnayake D, Gordon C. Effects of rituximab on resistant SLE disease including lung involvement. Lupus. 2009;18:67–73. doi: 10.1177/0961203308094653. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka Y, Yamamoto K, Takeuchi T, et al. A multicenter phase I/II trial of rituximab for refractory systemic lupus erythematosus. Mod Rheumatol. 2007;17:191–7. doi: 10.1007/s10165-007-0565-z. [DOI] [PubMed] [Google Scholar]
- 63.Albert D, Dunham J, Khan S, et al. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis. 2008;67:1724–31. doi: 10.1136/ard.2007.083162. [DOI] [PubMed] [Google Scholar]
- 64.Jonsdottir T, Gunnarsson I, Risselada A, Henriksson EW, Klareskog L, van Vollenhoven RF. Treatment of refractory SLE with rituximab plus cyclophosphamide: clinical effects, serological changes, and predictors of response. Ann Rheum Dis. 2008;67:330–4. doi: 10.1136/ard.2007.079095. [DOI] [PubMed] [Google Scholar]
- 65.Tokunaga M, Saito K, Kawabata D, et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007;66:470–5. doi: 10.1136/ard.2006.057885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: a systematic review of off-label use in 188 cases. Lupus. 2009;18:767–76. doi: 10.1177/0961203309106174. [DOI] [PubMed] [Google Scholar]
- 67.Diaz-Lagares C, Garcia-Hernandez FJ, De Ramon E, et al. Treatment of severe and/or refractory SLE with rituximab: analysis of 128 patients. Ann Rheum Dis. 2010;69(Suppl. 3):550. [Google Scholar]
- 68.Faria RM, Isenberg DA. Three different B-cell depletion (anti-CD20 monoclonal antibodies) treatments for severe resistant systemic lupus erythematosus. Lupus. 2010;19:1256–7. doi: 10.1177/0961203310373107. [DOI] [PubMed] [Google Scholar]
- 69.Jonsdottir T, Sundelin B, Zickert A, Welin Henriksson E, van Vollenhoven RF, Gunnarsson I. Lupus nephritis patients treated with rituximab: results from long term follow-up. Ann Rheum Dis. 2010;69(Suppl. 3):553. doi: 10.1093/rheumatology/kes348. [DOI] [PubMed] [Google Scholar]
- 70.Kur-Zalewska J, Rud U, Sulek M, Tlustochowicz M, Tlustochowicz M. Rituximab in patients with SLE and inadequate response to traditional immunosuppressive therapy. Ann Rheum Dis. 2010;69(Suppl. 3):685. [Google Scholar]
- 71.Marenco J, Fernandez-Nebro A, Lopez-Longo FJ, et al. Effectiveness and safety of rituximab in patients with SLE refractory to standard therapy. Ann Rheum Dis. 2010;69(Suppl. 3):554. [Google Scholar]
- 72.Ng KP, Leandro MJ, Edwards JC, Ehrenstein MR, Cambridge G, Isenberg DA. Repeated B cell depletion in treatment of refractory systemic lupus erythematosus. Ann Rheum Dis. 2006;65:942–5. doi: 10.1136/ard.2005.044487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramos-Casals M, Garcia-Hernandez FJ, de Ramon E, et al. Off-label use of rituximab in 196 patients with severe, refractory systemic autoimmune diseases. Clin Exp Rheumatol. 2010;28:468–76. [PubMed] [Google Scholar]
- 74.Terrier B, Amoura Z, Ravaud P, et al. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum. 2010;62:2458–66. doi: 10.1002/art.27541. [DOI] [PubMed] [Google Scholar]
- 75.Torgashina A, Soloviev SK, Alexandrova EN, Radenska SG, Nasonov EL. Efficacy of rituximab in lupus nephritis: data from Russian Register. Ann Rheum Dis. 2010;69(Suppl. 3):686. [Google Scholar]
- 76.Vital EM, Dass S, Buch MH, et al. Treatment of SLE with B cell depletion: predicting response and managing loss of response. Ann Rheum Dis. 2010;69(Suppl. 3):558. [Google Scholar]
- 77.Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–33. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furie RA, Looney JR, Rovin B, et al. Efficacy and safety of rituximab in patients with proliferative lupus nephritis: results from the randomized, double-blind phase III LUNAR study. Ann Rheum Dis. 2010;69(Suppl. 3):549. [Google Scholar]
- 79.Furie R, Rovin B, Appel G, et al. Effect of rituximab on anti-double stranded DNA antibody and C3 levels and relationship to response: results from the LUNAR trial. Ann Rheum Dis. 2010;69(Suppl. 3):550. [Google Scholar]
- 80.Tew GW, Rabbee N, Wolslegel K, et al. Baseline autoantibody profiles predict normalization of complement and anti-dsDNA autoantibody levels following rituximab treatment in systemic lupus erythematosus. Lupus. 2010;19:146–57. doi: 10.1177/0961203309350752. [DOI] [PubMed] [Google Scholar]
- 81.Dass S, Bowman SJ, Vital EM, et al. Reduction of fatigue in Sjogren syndrome with rituximab: results of a randomised, double-blind, placebo-controlled pilot study. Ann Rheum Dis. 2008;67:1541–4. doi: 10.1136/ard.2007.083865. [DOI] [PubMed] [Google Scholar]
- 82.Looney RJ. Will targeting B cells be the answer for Sjogren's syndrome? Arthritis Rheum. 2007;56:1371–7. doi: 10.1002/art.22604. [DOI] [PubMed] [Google Scholar]
- 83.Meijer JM, Pijpe J, Vissink A, Kallenberg CG, Bootsma H. Treatment of primary Sjogren syndrome with rituximab: extended follow-up, safety and efficacy of retreatment. Ann Rheum Dis. 2009;68:284–5. doi: 10.1136/ard.2008.092601. [DOI] [PubMed] [Google Scholar]
- 84.Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008;67:450–7. doi: 10.1136/ard.2007.075762. [DOI] [PubMed] [Google Scholar]
- 85.Kawasaki N, Rademacher C, Paulson JC. CD22 regulates adaptive and innate immune responses of B cells. J Innate Immun. 2011;3:411–19. doi: 10.1159/000322375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sato S, Jansen PJ, Tedder TF. CD19 and CD22 expression reciprocally regulates tyrosine phosphorylation of Vav protein during B lymphocyte signaling. Proc Natl Acad Sci USA. 1997;94:13158–62. doi: 10.1073/pnas.94.24.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Daridon C, Blassfeld D, Reiter K, et al. Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R204. doi: 10.1186/ar3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dorner T, Kaufmann J, Wegener WA, Teoh N, Goldenberg DM, Burmester GR. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther. 2006;8:R74. doi: 10.1186/ar1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Steinfeld SD, Tant L, Burmester GR, et al. Epratuzumab (humanised anti-CD22 antibody) in primary Sjogren's syndrome: an open-label phase I/II study. Arthritis Res Ther. 2006;8:R129. doi: 10.1186/ar2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kalunian K, Wallace DJ, Petri M, et al. BILAG-measured improvement in moderately and severely affected body systems in patients with SLE by epratuzumab: results from EMBLEM, a phase IIB study. Annals Rheum Dis. 2010;69(Suppl. 3):553. [Google Scholar]