

Abstract
OTHER ARTICLES PUBLISHED IN THIS MINI-REVIEW SERIES ON B CELL SUBSETS IN DISEASE
B cells in multiple sclerosis: drivers of disease pathogenesis and Trojan horse for Epstein—Barr virus entry to the central nervous system? Clinical and Experimental Immunology 2012, 167: 1–6. Transitional B cells in systemic lupus erythematosus and Sjögren's syndrome: clinical implications and effects of B cell-targeted therapies. Clinical and Experimental Immunology 2012, 167: 7–14.
Haematopoietic stem cell transplantation (HSCT) is an immunological treatment that has been used for more than 40 years to cure a variety of diseases. The procedure is associated with serious side effects, due to the severe impairment of the immune system induced by the treatment. After a conditioning regimen with high-dose chemotherapy, sometimes in combination with total body irradiation, haematopoietic stem cells are transferred from a donor, allowing a donor-derived blood system to form. Here, we discuss the current knowledge of humoral problems and B cell development after HSCT, and relate these to the current understanding of human peripheral B cell development. We describe how these studies have aided the identification of subsets of transitional B cells and also a robust memory B cell phenotype.
Keywords: B cell, haematopoietic stem cell transplantation, lymphocyte development
Haematopoietic stem cell transplantation
Haematopoietic stem cell transplantation (HSCT) following high-dose treatment with cytotoxic drugs and sometimes total body irradiation (TBI) has become a common clinical practice for treatment of malignant and non-malignant diseases over the past 40 years. Today, HSCT is the only curative treatment for certain inherited disorders, including immune deficiencies, haemoglobinopathies and bone marrow failure syndromes, and it is also used to treat haematological and non-haematological malignancies. Patients with malignant disease receiving HSCT are often heavily pretreated with combinations of cytostatic drugs and already have immune dysfunctions prior to transplantation [1]. Patients transplanted for non-malignant diseases have a wide range of immunodeficiency, ranging from a total lack of adaptive immunity in severe combined immunodeficiency (SCID) patients to essentially normal function in patients with haemoglobinopathies. Before infusion of stem cells, patients are conditioned with a regimen consisting of high doses of cytostatic drugs and sometimes also TBI to obtain a total (myeloablative conditioning) or a subtotal myeloablation (reduced intensity conditioning). In malignant disorders, an intense conditioning regimen contributes to the eradication of remaining neoplastic cells, while for non-malignant disorders a regimen that induces tolerance to the graft is sufficient. The graft consists of CD34+ stem cells obtained from related or unrelated donors mixed with a smaller or larger number of mature immune cells. Stem cells obtained from peripheral blood contain larger number of haematopoietic cells and give faster engraftment, but are also associated with an increased incidence of graft-versus-host disease (GVHD) [2]. The goal for the procedure is a haematopoietic system that produces fully functional erythrocytes, thrombocytes, myeloid cell lineages and lymphocytes. The first three objectives are usually accomplished within the first months. Lymphocyte numbers and functions are, however, impaired for extended periods. Whereas the development of T cell lineages after HSCT has been defined in considerable detail, less is known about B cell development and how it relates to the impaired functional immunity.
The establishment of haematopoietic lineages after HSCT
The production of different cell types from grafted haematopoietic stem cells follows a specific pattern [3,4]. The first cells produced are granulocytes and other cells of myeloid lineages [monocytes, macrophages and dendritic cells (DC)], erythrocytes and thrombocytes. These early cells are sometimes functionally impaired. Monocytes may not produce normal amounts of interleukin (IL)-1 and neutrophil functions (e.g. chemotaxis, phagocytosis and bacterial killing) may be attenuated, in particular in patients developing GVHD [5,6]. In addition, subtype deficiencies have been described within certain cell lineages. Myeloid DC and Langerhans cells are, for example, usually found within the first 6 months, whereas CD123+ plasmacytoid DC are rare even 1 year after transplantation [7,8].
Cells of the adaptive immune system form slower than innate cells after HSCT, and functional deficiencies can be detected years after normal numbers of cells are reached [3,4]. As the numbers of CD8+ T cells increase earlier than CD4+ T cells, the CD4/CD8 ratio is initially reversed [9]. This early expansion is dependent on homeostatic peripheral proliferation of memory T cells, both of rare recipient-derived T cells surviving the conditioning regimen and of donor-derived co-transferred T cells, rather than from thymic production [10,11].
Although this is a polyclonal proliferation, it is not completely antigen-independent – CD8+ T cell clones recognizing viruses present in the body at the time of transplantation, e.g. cytomegalovirus (CMV) or Epstein–Barr virus (EBV) may expand rapidly [12–14]. During the first months after transplantation a narrow and skewed repertoire of T lymphocytes with memory-like phenotype predominates. De novo production of naive cells from the thymus starts later, particularly in adult patients [10]. Therefore, a dominance of oligoclonal T cells with a memory phenotype is observed, sometimes for several years [15]. The generation of naive T cells from donor cells in thymus does not result in full immune reconstitution for at least 1 year after transplantation and is compromised by factors affecting thymic epithelial cells such as irradiation, GVHD and age. Indeed, slow recovery of specific T cells has been shown to have a significant impact on survival of the patients [16].
B cells are rare in peripheral blood during the first months after HSCT and reach close to normal levels within 6–12 months [17]. The CDR3 immunoglobulin (Ig)M spectra, i.e. the distribution of B cells with different lengths of their heavy chain V-D-J regions, are essentially normal with no indication of homeostatic, oligoclonal proliferation within 3–6 months after transplantation [18,19]. Memory B cells expressing CD27 do not expand and subnormal levels are observed during the first 2 years after transplantation [20–22]. Transfer of donor memory B cells that can be reactivated after antigen re-encounter is well documented [23,24]. Low numbers of recipient-derived B cells can also be encountered during the first period after transplantation, especially after reduced intensity conditioning [25]. Because B cell-depleted and non-depleted bone marrow give similar reconstitution in patients who underwent intense myeloablative conditioning, pre-existing mature B cells do not seem to play a major role long-term [17,26,27]. Although only donor-derived B cells circulate in blood, the maintenance of recipient-derived serum antibodies several years after transplantation reflects the resistance and longevity of plasma cells [28–30].
Immunological problems after HSCT
HSCT is associated with major immunological complications. During the first weeks after transplantation, high levels of inflammatory cytokines such as IL-7 and IL-15 induced by the myeloablative conditioning in combination with lymphopenia induce expansion of donor-derived T cells. The presence of specific antigens may favour proliferation of cells directed against mismatched histocompatibility antigens in the host, leading to acute GVHD (aGVHD) [31]. The chronic form of GVHD (cGVHD) can appear later, and donor-derived T cells processed in the thymus are then involved [32]. A certain level of cytotoxic T cell-mediated self-reactivity may still be beneficial in some cases, as the so-called graft-versus-leukaemia reaction (GVL), which reduces the risk of relapse, is mediated by cytotoxic T cells and natural killer (NK) cells from the donor and prevents relapse [33].
Another major problem after HSCT is the high incidence of severe infections due to the immunocompromised state of the patients [34]. At the very early pre-engraftment stage, leukopenia in combination with mucosal membrane damage induced by the conditioning regimen put the patients at high risk for infections with bacteria and fungi. Post-engraftment, newly produced cells of the innate immune system confer some protection, but cellular immunity is impaired. This stage is associated with viral infections, in particular herpesvirus such as CMV, but fungal infections are also common. At a later phase, cellular and humoral immunity slowly recovers. In some patients this phase only lasts for a year, while in others it may develop into a chronic problem. The development of GVHD requiring treatment with immunosuppressive agents interferes with immunological reconstitution. Recurrent infections with encapsulated bacteria, such as Haemophilus influenzae and Streptococcus pneumonia, are common problems even late after HSCT [35–37]. B cell-mediated immunity is important in protection from these bacteria and a deficiency of the humoral immunity is hence likely to be involved [38].
Humoral immunity after HSCT
Although mature B cells are efficiently depleted during conditioning, the levels of circulating IgG antibodies drop slowly [17,39]. This is due to the long half-life of IgG in serum [40] and the survival of many plasma cells after myelodepletion [30]. Eventually, specific plasma cells disappear, resulting in loss of antibodies against antigens encountered before transplantation [41,42]. Why plasma cells are lost relatively quickly in HSCT patients compared to healthy individuals is not clear, but may be due to cytotoxicity of the conditioning regimen [43], damage to supporting cells in the bone marrow such as eosinophils and stroma [44,45] or depletion of plasma cells of recipient origin by donor-derived T cells [30]. In addition, memory B cells may be needed to replenish the pool of long-lived plasma cells [46,47].
Specific antibodies from long-lived plasma cells are pivotal in sterile immunity after vaccination, through blocking of viruses, bacteria and toxins [48]. The loss of specific antibodies after HSCT necessitates revaccination. Some vaccines, in particular from inactivated viruses and bacteria, give protective responses within a year after grafting [49]. Others, especially carbohydrate-based vaccines, are not efficient until much later [50,51]. Recipient-derived memory B cells sometimes seem to survive the conditioning regimen and give rise to transient monoclonal production of antibodies [30] and functional memory B cells can also be transferred from the donor [23,52]. Thus, regimens where both the donor and the recipient are vaccinated before transplantation have been tested to improve post-transplantation immune responses [53]. As a drawback to this phenomenon, transfer of autoimmune diseases and allergies from the donor has been documented [54].
Antibody subclasses emerge in a distinct order after HSCT, with production of IgM antibodies within a few months, followed by IgG1/IgG3, IgG2/IgG4 and finally IgA [55]. This order recapitulates normal development during the first year of life. Long-term antibody class deficiencies are observed in some patients [56]. B cells from HSCT patients show close to normal in vitro responses to polyclonal EBV activation, while they respond to a lesser degree to pokeweed mitogen (PWM) [57,58]. The in vitro response to PWM requires T cells to be present, while EBV does not [59,60]. T cells from HSCT patients have a decreased ability to support B cell activation by PWM. Intrinsic deficiencies within the B cell compartment that may hinder PWM responses have also been demonstrated [57,58].
Thus, humoral deficiencies after HSCT are common and can be life-long in some patients, due to intrinsic defects in B cells as well as in supporting cells. Low numbers of naive CD4+ T cells in blood have been documented years after transplantation [15]. Follicular dendritic cells (FDC), a non-haematological cell type supporting germinal centre formation, are also damaged [61,62]. These defects severely impair the production of T cell-dependent antibody responses with germinal centre reactions and B memory cell development. From a clinical perspective, the most important problems late after HSCT, even in the absence of cGVHD, are recurrent infections with encapsulated bacteria and poor responses to polysaccharide vaccines [34]. Antibody responses against polysaccharides are initiated typically through T cell-independent pathways, and intrinsic B cell defects may be critically involved in causing these problems.
Human B cell differentiation stages and lineages present in peripheral blood
The current model of human peripheral B cell development involves five major consecutive stages: transitional B cells that have just left the bone marrow but are still unable to respond to antigen, naive B cells that are fully mature but have not encountered antigen, germinal centre B cells in lymphoid organs that are actively engaged in immune responses, memory B cells that have encountered antigen and survive for extended periods and plasma cells that produce soluble antibodies (Fig. 1a). In mice, splenic marginal zone B cells (MZB) and peritoneal B1 B cells represent separate lineages [63]. The presence or not of these in humans will also be discussed below. Germinal centre cells and fully mature plasma cells are only rarely present in blood and have been covered in other recent reviews [64]. They will therefore not be discussed further here.
Fig. 1.
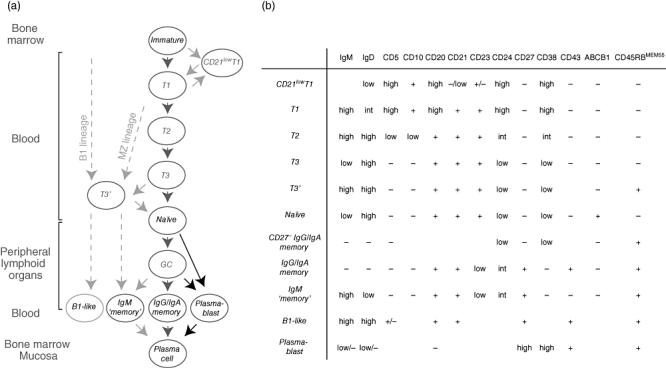
Human B cell development. (a) When immature B cells leave the bone marrow, they go through distinct differentiation stages. Transitional (T1, T2 and T3), naive and memory B cells as well as plasmablasts are found in blood. Black arrows indicate differentiation pathways well supported by experimental data, grey arrows pathways less well-defined or only proposed pathways. Recent data suggest that immunoglobulin (Ig)M+CD27+ cells may not be memory cells, but rather the human counterpart to marginal zone B or B1 lineages. T3′ cells lack CD27- but express CD45RBMEM55 and may be upstream cells in these lineages or cells downstream from T3 cells. (b) The different B cell stages can be distinguished from each other based on expression of cell surface markers. The subtypes express or lack expression, as indicated. Blank spaces indicate that the expression of this marker has not, to our knowledge, been described in the literature.
Transitional cells
Developmentally, mouse transitional B cells are positioned between bone marrow immature B cells and mature peripheral B cells. Induction of apoptosis following BCR receptor signalling makes this stage an important checkpoint for removal of autoreactive specificities [65]. Analogous cells were described more recently in humans based on functional characteristics and expression of several cell surface markers [65–67]. Although rare in adult blood, making up fewer than 5% of the B cells, increased frequencies of transitional B cells are found in cord blood, after bone marrow transplantation, and in patients with systemic lupus erythematosus (SLE), X-linked lymphoproliferative syndrome ((XLP) or common variable immunodeficiency (CVID) [66,67]. When first described in humans, transitional cells were divided into two populations, T1 and T2, based on expression of transitional markers [67,68] (Figs 1b and 2a). It was recently shown that CD21 expression divides the T1 population into two. CD21low T1 cells were suggested to be more immature than CD21+ T1 cells [69]. Because most anergic human B cells that have encountered antigen down-regulate expression of CD21 [70], it is possible that CD21low T1 cells are anergic. An alternative definition of human transitional B cells is that they are CD27-IgM+ and lack the ABCB1 transporter protein [71] (Fig. 2b). Based on these two ways of defining transitional cells, Palanichamy et al. found that cells lacking ABCB1 but otherwise conforming to the phenotype of naive cells were enriched after B cell depletion with rituximab [72]. These cells appeared to be at a stage between T2 and naive cells, and were designated T3 cells. We found recently that T3 cells can be divided further into two populations based on CD45RBMEM55 expression (Fig. 2b; Bemark et al., submitted; see below). Thus, in humans, three consecutive stages of transitional cells are now recognized – T1, T2 and T3 – and T1 and T3 cells can be divided further based on expression of CD21 and CD45RBMEM55, respectively (Fig. 1a,b).
Fig. 2.
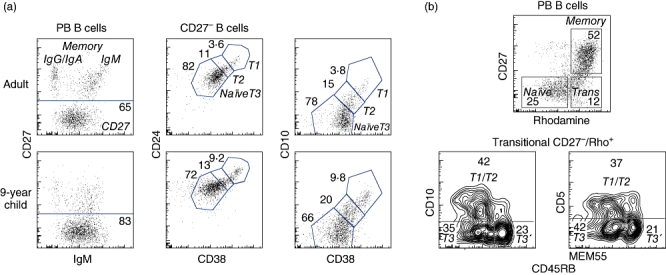
Identification of peripheral blood B differentiation stages based on expression of surface markers. In (a) peripheral blood cells were stained with antibodies against CD19, CD27, immunoglobulin (Ig)M, CD24, CD38 and CD10. To the left, CD19+ B cells were gated into IgM+CD27+, class-switched memory B cells and CD27- cells. The CD27- cells were divided further into T1, T2 and T3/naive cells based on expression of CD24 and CD38 (middle panels) or CD10 and CD38 (right panels). Typical results obtained using peripheral blood from five adults and four children are shown. In (b), peripheral blood cells were preincubated with the dye Rhodamine 123 followed by antibodies against CD19, CD27, CD5, CD10 and CD45RBMEM55. CD19+ B cells were divided into naive, memory and transitional cells based on expression of CD27 and extrusion of the dye (upper panel). The transitional cells could be divided further into T1/T2 cells expressing CD5 and CD10, and two distinct populations (T3 and T3′) that lacked these based on expression of CD45RBMEM55 (lower panels). The data represent typical data from three different healthy donors tested.
Naive cells
Naive cells constitute more than half of the blood B cells in healthy adults [73]. They express low levels of IgM, high levels of IgD and lack expression of CD27. In addition, they are the only blood B cells that express the ABCB1 transporter [71]. Activation requires binding of antigen to cell-surface antibodies, CD40 signals from CD4+ T cells as well as cytokine stimulation. After activation, some naive B cells will differentiate to early IgM-producing plasma cells while others enter B cell follicles, where they form germinal centres [74]. By regulating the class-switch recombination process, cytokines derived from T cells present during B cell activation determine which antibody class the B cells will produce. After random mutagenesis and selection of high-affinity B cell clones, mutated high-affinity class-switched B cells leave the germinal centre to become long-lived memory cells or long-lived plasma cells.
Memory B cells
Memory B cells are easier to activate than naive cells, and induce specific IgG production rapidly after antigen re-encounter [75]. Markers to distinguish naive B cells from memory B cells with mutated antibody were lacking for a long time. The discovery that CD27 was expressed on mutated human peripheral blood B cells was therefore important [73]. When the expression of both CD27 and IgM is determined on B cells, two populations of similar sizes can be identified: IgM- class-switched memory and IgMhigh memory cells. The latter are sometimes separated further based on high or low IgD expression. Whereas it is generally agreed that the IgM- cells are class-switched post-germinal centre memory cells, the origin of IgMhighCD27+ cells is controversial. It is either argued that IgM+ memory cells are bona fide memory B cells generated early during germinal centre formation [76,77] or B cells diversified in the absence of an immune response that are similar to cells found in the marginal zone of the spleen [78,79] (see below). In addition to these populations, other minor memory B cell populations have been described. These include CD27+IgM-IgD+ cells and CD27- class-switched cells [71,73,80]. Recently, our group found that all memory B cell populations, but not naive cells, express CD45RBMEM55, possibly making it a better marker for memory B cells than CD27 [81].
Plasmablasts and plasma cells
Cells immediately preceding plasma cells in development, so-called plasmablasts, home to bone marrow or mucosal surfaces through the blood to develop into plasma cells. They can be identified by their expression of high levels of CD27 and CD38 and lack of CD20. Normally, these cells represent a minor population in peripheral blood, but after infection or vaccination the number of antigen-specific plasmablasts in blood increases rapidly and then disappears quickly [82]. Few fully mature plasma cells are found in blood.
Marginal zone and B1 B cells
The human subsets defined above, except for IgMhighCD27+ B cells, are similar to developmental stages described for mouse follicular B2 cells. In mice, two separate B cell lineages associated with innate immunity exist; peritoneal B1 B cells and marginal zone B cells (MZB), that have distinct developmental pathways [63]. These are sometimes referred to as innate-like B cells, as they have a separate repertoire of unmutated antibody specificities and respond to antigens in the absence of T cell help. Mouse MZB share their early differentiation with follicular B2 cells, but are selected during the transitional stage to enter the marginal zone of the spleen. B1 B cells are generated early during ontogeny and home to the peritoneum where they form a self-renewing compartment [83]. If these subsets are also present in humans has been debated, specially whether human MZB and B1 cells are present among the IgMhighCD27+ cells in blood.
Weill and colleagues suggested in 2004 that non-class-switched CD27+ human blood B cells are the circulating counterpart of cells present in the splenic marginal zone rather than memory cells generated in germinal centres [84]. Subsequently, the same group showed that these subsets may diversify their antibody genes through somatic hypermutation in the absence of typical immune responses [85]. In favour of a non-memory origin, mass sequencing of antibody genes did not reveal related sequences between IgM+ and IgM- CD27+ B cells in human blood, and found little evidence for clonal expansion among IgM-CD27+ B cells [79]. Reports have also suggested that the function of circulating IgMhighCD27+ B cells and marginal zone human B cells may be similar to the one described for MZB in mice, i.e. to respond to polysaccharides in T independent response [78]. Mouse MZB are non-mutated, mainly sessile B cells, clearly differentiating them from human circulating IgMhighCD27+ B cells, and many groups still prefer to interpret them as memory cells [76,77].
Unlike the situation in mice, only a small percentage of cells isolated from the human peritoneum express B cell markers, making up a population of less than 105 cells in healthy adults [86]. Of these, some express CD5 [87], a marker for B1 cells in mice, but it is questionable if these are B1 cells, as the largest CD5-expressing human B cell population is transitional cells. B cells in the human peritoneum do not seem to contribute to mucosal IgA-producing plasma cells, a feature associated with mouse B1 cells [88]. This does not exclude the possibility of B1-like cells in other organs. Griffins et al. described recently a small population of unmutated IgM+ B cells present in both cord blood and healthy adults that expressed CD27 together with CD43 and shared functional characteristics with mouse B1 cells [89]. Although this is to be confirmed by other groups, it may explain the importance of IgMhighCD27+ B cells in the responses to encapsulated bacteria [38].
Thus, although it is not proven indisputably that humans have B cell lineages distinct from follicular B cells, several recent studies suggest that this may be the case. In particular, these studies have suggested that such cells can be identified among circulating IgMhighCD27+ human B cells. The IgMhighCD27+ population may hence not be homogeneous but rather made up of several subtypes of cells with distinct characteristics, possibly explaining some of the conflicting data with regard to the origin and function of this subtype.
CD45RBMEM55 is expressed differentially during B cell differentiation
We demonstrated recently that CD45RBMEM55 is expressed differentially during B cell differentiation [81]. Although CD45 is expressed on all haematopoietic cells, it has three exons, RA, RB and RC, which are expressed differentially in many of them during differentiation as a consequence of regulated splicing [90]. The splicing pattern of CD45 does not change during peripheral human B cell development, with the longest CD45RABC form being dominant at all stages. However, the expression of the CD45RBMEM55 epitope changes through developmentally regulated glycosylation in human B cells, with high expression on essentially all CD27+ B cells but not on naive or transitional B cells [81]. The CD45RBMEM55 epitope is also present on a minor population of CD27- B cells that express high levels of IgM (Fig. 2b). These cells are present in increased numbers in young children and in cord blood, and constitute 25–50% of all B cells even 1 year after HSCT, indicating that these are immature B cells (Bemark et al., submitted). Interestingly, these IgMhighCD27-CD45RBMEM55+ cells lack transitional cell markers such as CD5 and CD10 but, in similar with transitional cells, do not express ABCB1.
Development of B cell subtypes after HSCT
Some recent studies have investigated the development of B cell subsets after HSCT following the classifications outlined above. These have found low numbers of class-switched and IgM+ CD27-expressing memory B cells in peripheral blood for extended periods after transplantation [20,91–94]. In contrast, the number of transitional cells was high [66,94,95]. Studies performed before the current schedule of B cell stages, however, also determined the expression of many cell surface markers on B cells (Table 1). These studies can now be reconciled with the current understanding of B cell development. Many of these early studies found increased expression of CD5, CD38 and IgM and lowered levels of CD62L expressing cells, all features of transitional B cells. In addition, few class-switched cells were found, in line with a delay in memory cell formation.
Table 1.
Cell surface markers showing abnormal expression on B cells after haematopoietic stem cell transplantation (HSCT)
| Antigen | Number of cells* | Comments | References |
|---|---|---|---|
| mIgM | A | High levels of membrane bound IgM on B cells post-HSCT | [98–100] |
| mIgD | A | Slightly increased levels of membrane-bound IgD post-HSCT | [98] |
| Few mIgD- class-switched cells 1 year after transplantation | [21] | ||
| CD1c | + | Increased numbers first year post-HSCT | [97] |
| CD5 | + | Increased numbers first year post-HSCT | [96–98,101] |
| Increased expression of IgM, CD20 and HLA-DR on CD5+ B cells | [101] | ||
| Normal expression of CD11a, CD44, CD54 and CD62L on CD5+ B cells | [102] | ||
| N | Normal numbers first year post-HSCT | [103] | |
| CD10 | + | Increased numbers post-HSCT | [95,104,105] |
| N | Normal numbers post-HSCT | [97,103,106] | |
| CD11a | − | Decreased numbers of CD5- B cells that express CD11a 4 months after transplantation | [102] |
| CD21 | − | Decreased numbers first half-year post-GSCT | [27,93,98,99,105] |
| N | Normal numbers of cells first 10 months post-HSCT | [97,103] | |
| CD23 | − | Low numbers first half-year post-HSCT, then normal or slightly increased numbers of cells | [26] |
| + | Increased numbers of cells first 10 months, then normal numbers | [97,103,104] | |
| N | Normal numbers early and late post-HSCT | [98] | |
| CD27 | − | Low numbers post-HSCT. Both IgM+ and class-switched cells affected | [20,91–94] |
| CD38 | + | Increased numbers first half-year post-HSCT | [97–99,106] |
| CD44 | − | Low numbers of CD5- B cells that express CD44 4 months after transplantation | [102] |
| CD54 | − | Low numbers of CD5- B cells that express CD54 4 months after transplantation | [102] |
| CD62L | – | Low numbers first 10 months | [98,103] |
| Low numbers of CD5- B cells express CD62L 4 months after transplantation | [102] | ||
| N | Normal numbers post-HSCT | [97] | |
| HLA-DR | N | Normal numbers post-HSCT | [97,106] |
The number of B cells in blood expressing the respective marker: A: all B cells; N: normal expression compared to healthy controls; −: decreased percentage of B cells in blood compared to healthy control; +: increased percentage of B cells in blood compared to healthy control; HLA-DR: human leucocyte antigen D-relayed; Ig: immunoglobulin.
It should, however, be pointed out that phenotypic features of transitional human B cells change after HSCT (Fig. 3c; Bemark et al.; submitted). Although IgMhigh cells in blood constitute the majority of B cells 1 year after transplantation, fewer than half of these express other transitional markers (Fig. 3a–c). Many IgMhighCD27- B cells instead express CD45RBMEM55, identifying them as the T3′ population described above. The slow development of CD27+ B cells after HSCT is of interest (Fig. 3b). The establishment of class-switched memory cells mimics the production of class-switched antibodies in serum, and most probably the ability to form germinal centres in peripheral lymphoid organs. With the proposed role of IgM+CD27+ B cells in response to encapsulated bacteria, their slow occurrence may be linked to infections after HSCT. Furthermore, the suggestion that IgMhighCD27+ B cells are from a separate lineage to follicular B cells indicate that these lineages may establish slowly or not at all after HSCT.
Fig. 3.
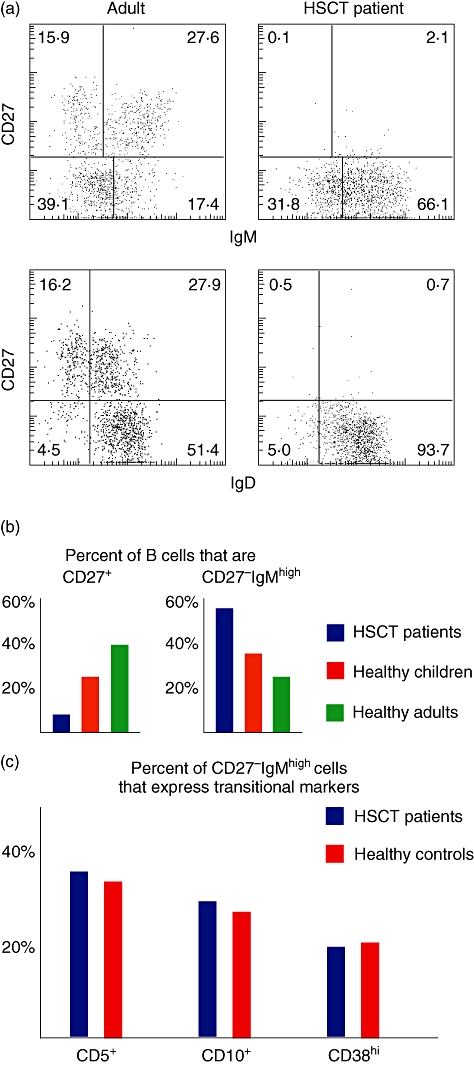
Subtyping of B cells in children who have undergone haematopoietic stem cell transplantation (HSCT). (a) In healthy adults, the CD19+ B cells can be divided into immunoglobulin (Ig)MhighIgDlowCD27+ and IgM-IgD-CD27+ memory cells and IgMlowIgDhighCD27- naive cells, with few cells being IgMhighCD27-. In paediatric patients who have undergone HSCT, IgMhighIgDhighCD27- B cells is a major population even 1 year after transplantation when normal numbers of B cells has been reached in blood. In (b) are shown the mean frequency of CD27+ B cells and IgMhighCD27+ B cells in healthy children (n = 9), healthy adults (n = 3) or children who have undergone HSCT 1 year prior to the analysis (n = 10). (c) Few of the IgMhighCD27+ B cells express markers typical for transitional cells in healthy controls (n = 4) or paediatric HSCT patients (n = 4).
Interestingly, many phenotypic changes encountered on B cells after HSCT are more pronounced in patients developing GVHD, and the B cell reconstitution is, in this case, also delayed [91–94,96–99]. Thus, monitoring of phenotypic changes on B cells following HSCT may not only give clues about the maturity of the immune system, but may also give early indications if GVHD is developing.
Concluding remarks
HSCT patients provide a rare opportunity to study early peripheral cell development through consecutive blood samples from a single donor during reconstitution, allowing refinement of our knowledge of B cell subset development and complexity. Deficiencies associated with poor B cell responses can result in long-term problems. Therefore, monitoring the development of B cell subsets may prove clinically relevant, giving insights into how deficiencies develop and to tailor the care of the patients. Such measurements could, for example, monitor infection sensitivity, development of GVHD, determine when vaccinations are best performed and track responses to vaccines or infections. Absolute and relative frequencies of immature, naive, class-switched memory B cells or plasma blasts in blood are candidates for such predictions. In addition, as the true identity of IgMhighCD27+ B cells is now being unravelled, measurements of these may give insights into why some HSCT patients experience repeated infections with encapsulated bacteria. Few recent clinical studies have been performed where the presence or absence of B cell subtypes in HSCT patients are linked to clinical problem, and future studies that address these issues are therefore called for.
Acknowledgments
This work has been supported by grants from the Swedish Children's Cancer Foundation, the Sahlgrenska University Hospital Foundation, The Swedish Research Council and the Mucosal Immunobiology and Vaccine Centre (MIVAC).
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Ek T, Mellander L, Andersson B, Abrahamsson J. Immune reconstitution after childhood acute lymphoblastic leukemia is most severely affected in the high risk group. Pediatr Blood Cancer. 2005;44:461–8. doi: 10.1002/pbc.20255. [DOI] [PubMed] [Google Scholar]
- 2.Torelli GF, Lucarelli B, Iori AP, et al. The immune reconstitution after an allogeneic stem cell transplant correlates with the risk of graft-versus-host disease and cytomegalovirus infection. Leuk Res. 2011;35:1124–6. doi: 10.1016/j.leukres.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Storek J, Geddes M, Khan F, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol. 2008;30:425–37. doi: 10.1007/s00281-008-0132-5. [DOI] [PubMed] [Google Scholar]
- 4.Williams KM, Gress RE. Immune reconstitution and implications for immunotherapy following haematopoietic stem cell transplantation. Best Pract Res Clin Haematol. 2008;21:579–96. doi: 10.1016/j.beha.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahdev I, O'Reilly R, Black P, Heller G, Hoffmann M. Interleukin-1 production following T-cell-depleted and unmodified marrow grafts. Pediatr Hematol Oncol. 1996;13:55–67. doi: 10.3109/08880019609033372. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerli W, Zarth A, Gratwohl A, Speck B. Neutrophil function and pyogenic infections in bone marrow transplant recipients. Blood. 1991;77:393–9. [PubMed] [Google Scholar]
- 7.Chklovskaia E, Nowbakht P, Nissen C, Gratwohl A, Bargetzi M, Wodnar-Filipowicz A. Reconstitution of dendritic and natural killer-cell subsets after allogeneic stem cell transplantation: effects of endogenous flt3 ligand. Blood. 2004;103:3860–8. doi: 10.1182/blood-2003-04-1200. [DOI] [PubMed] [Google Scholar]
- 8.Collin MP, Hart DNJ, Jackson GH, et al. The fate of human Langerhans cells in hematopoietic stem cell transplantation. J Exp Med. 2006;203:27–33. doi: 10.1084/jem.20051787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gratama JW, Naipal A, Oljans P, et al. T lymphocyte repopulation and differentiation after bone marrow transplantation. Early shifts in the ratio between T4+ and T8+ T lymphocytes correlate with the occurrence of acute graft-versus-host disease. Blood. 1984;63:1416–23. [PubMed] [Google Scholar]
- 10.Dumont-Girard F, Roux E, van Lier RA, et al. Reconstitution of the T-cell compartment after bone marrow transplantation: restoration of the repertoire by thymic emigrants. Blood. 1998;92:4464–71. [PubMed] [Google Scholar]
- 11.van Leeuwen JE, van Tol MJ, Joosten AM, Wijnen JT, Khan PM, Vossen JM. Mixed T-lymphoid chimerism after allogeneic bone marrow transplantation for hematologic malignancies of children is not correlated with relapse. Blood. 1993;82:1921–8. [PubMed] [Google Scholar]
- 12.Chalandon Y, Degermann S, Villard J, et al. Pretransplantation CMV-specific T cells protect recipients of T-cell-depleted grafts against CMV-related complications. Blood. 2006;107:389–96. doi: 10.1182/blood-2005-07-2746. [DOI] [PubMed] [Google Scholar]
- 13.Lucas KG, Small TN, Heller G, Dupont B, O'Reilly RJ. The development of cellular immunity to Epstein–Barr virus after allogeneic bone marrow transplantation. Blood. 1996;87:2594–603. [PubMed] [Google Scholar]
- 14.Reusser P, Riddell SR, Meyers JD, Greenberg PD. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: pattern of recovery and correlation with cytomegalovirus infection and disease. Blood. 1991;78:1373–80. [PubMed] [Google Scholar]
- 15.Rufer N, Helg C, Chapuis B, Roosnek E. Human memory T cells: lessons from stem cell transplantation. Trends Immunol. 2001;22:136–41. doi: 10.1016/s1471-4906(00)01849-4. [DOI] [PubMed] [Google Scholar]
- 16.Cavazzana-Calvo M, André-Schmutz I, Dal Cortivo L, Neven B, Hacein-Bey-Abina S, Fischer A. Immune reconstitution after haematopoietic stem cell transplantation: obstacles and anticipated progress. Curr Opin Immunol. 2009;21:544–8. doi: 10.1016/j.coi.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Baumgartner C, Morell A, Hirt A, et al. Humoral immune function in pediatric patients treated with autologous bone marrow transplantation for B cell non-Hodgkin's lymphoma. The influence of ex vivo marrow decontamination with anti-Y 29/55 monoclonal antibody and complement. Blood. 1988;71:1211–17. [PubMed] [Google Scholar]
- 18.Gokmen E, Raaphorst FM, Boldt DH, Teale JM. Ig heavy chain third complementarity determining regions (H CDR3s) after stem cell transplantation do not resemble the developing human fetal H CDR3s in size distribution and Ig gene utilization. Blood. 1998;92:2802–14. [PubMed] [Google Scholar]
- 19.Omazic B, Lundkvist I, Mattsson J, Permert J, Nasman-Bjork I. Memory B lymphocytes determine repertoire oligoclonality early after haematopoietic stem cell transplantation. Clin Exp Immunol. 2003;134:159–66. doi: 10.1046/j.1365-2249.2003.02260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avanzini MA, Locatelli F, Dos Santos C, et al. B lymphocyte reconstitution after hematopoietic stem cell transplantation: functional immaturity and slow recovery of memory CD27+ B cells. Exp Hematol. 2005;33:480–6. doi: 10.1016/j.exphem.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Storek J, Witherspoon RP, Storb R. Reconstitution of membrane IgD− (mIgD–) B cells after marrow transplantation lags behind the reconstitution of mIgD+ B cells. Blood. 1997;89:350–1. [PubMed] [Google Scholar]
- 22.Suzuki I, Milner EC, Glas AM, et al. Immunoglobulin heavy chain variable region gene usage in bone marrow transplant recipients: lack of somatic mutation indicates a maturational arrest. Blood. 1996;87:1873–80. [PubMed] [Google Scholar]
- 23.Lausen BF, Hougs L, Schejbel L, Heilmann C, Barington T. Human memory B cells transferred by allogenic bone marrow transplantation contribute significantly to the antibody repertoire of the recipient. J Immunol. 2004;172:3305–18. doi: 10.4049/jimmunol.172.5.3305. [DOI] [PubMed] [Google Scholar]
- 24.Wimperis JZ, Gottlieb D, Duncombe AS, Heslop HE, Prentice HG, Brenner MK. Requirements for the adoptive transfer of antibody responses to a priming antigen in man. J Immunol. 1990;144:541–7. [PubMed] [Google Scholar]
- 25.Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, et al. Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell transplant recipients. Blood. 2002;99:1442–8. doi: 10.1182/blood.v99.4.1442. [DOI] [PubMed] [Google Scholar]
- 26.Bengtsson M, Smedmyr B, Festin R, Oberg G, Simonsson B, Tötterman TH. B lymphocyte regeneration in marrow and blood after autologous bone marrow transplantation: increased numbers of B cells carrying activation and progression markers. Leuk Res. 1989;13:791–7. doi: 10.1016/0145-2126(89)90092-1. [DOI] [PubMed] [Google Scholar]
- 27.Pedrazzini A, Freedman AS, Andersen J, et al. Anti-B-cell monoclonal antibody-purged autologous bone marrow transplantation for B-cell non-Hodgkin's lymphoma: phenotypic reconstitution and B-cell function. Blood. 1989;74:2203–11. [PubMed] [Google Scholar]
- 28.Okudaira H, Ishizaka K. Reaginic antibody formation in the mouse. XI. Participation of long-lived antibody-forming cells in persistent antibody formation. Cell Immunol. 1981;58:188–201. doi: 10.1016/0008-8749(81)90160-x. [DOI] [PubMed] [Google Scholar]
- 29.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–72. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 30.van Tol MJ, Gerritsen EJ, de Lange GG, et al. The origin of IgG production and homogeneous IgG components after allogeneic bone marrow transplantation. Blood. 1996;87:818–26. [PubMed] [Google Scholar]
- 31.Auletta JJ, Cooke KR. Bone marrow transplantation: new approaches to immunosuppression and management of acute graft-versus-host disease. Curr Opin Pediatr. 2009;21:30–8. doi: 10.1097/MOP.0b013e3283207b2f. [DOI] [PubMed] [Google Scholar]
- 32.Wingard JR, Majhail NS, Brazauskas R, et al. Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J Clin Oncol. 2011;29:2230–9. doi: 10.1200/JCO.2010.33.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Popat U, Carrum G, Heslop HE. Haemopoietic stem cell transplantation for acute lymphoblastic leukaemia. Cancer Treat Rev. 2003;29:3–10. doi: 10.1016/s0305-7372(02)00092-0. [DOI] [PubMed] [Google Scholar]
- 34.Tomblyn M, Chiller T, Einsele H, et al. Guidelines for preventing infectious complications among hematopoietic cell transplant recipients: a global perspective. Bone Marrow Transplant. 2009;44:453–558. doi: 10.1038/bmt.2009.254. [DOI] [PubMed] [Google Scholar]
- 35.Atkinson K, Storb R, Prentice RL, et al. Analysis of late infections in 89 long-term survivors of bone marrow transplantation. Blood. 1979;53:720–31. [PubMed] [Google Scholar]
- 36.Engelhard D, Cordonnier C, Shaw PJ, et al. Early and late invasive pneumococcal infection following stem cell transplantation: a European Bone Marrow Transplantation survey. Br J Haematol. 2002;117:444–50. doi: 10.1046/j.1365-2141.2002.03457.x. [DOI] [PubMed] [Google Scholar]
- 37.Winston DJ, Schiffman G, Wang DC, et al. Pneumococcal infections after human bone-marrow transplantation. Ann Intern Med. 1979;91:835–41. doi: 10.7326/0003-4819-91-6-835. [DOI] [PubMed] [Google Scholar]
- 38.Kruetzmann S, Rosado MM, Weber H, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003;197:939–45. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Storek J, Joseph A, Espino G, et al. Immunity of patients surviving 20 to 30 years after allogeneic or syngeneic bone marrow transplantation. Blood. 2001;98:3505–12. doi: 10.1182/blood.v98.13.3505. [DOI] [PubMed] [Google Scholar]
- 40.Cohen S, Freeman T. Metabolic heterogeneity of human gamma-globulin. Biochem J. 1960;76:475–87. doi: 10.1042/bj0760475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ljungman P, Wiklund-Hammarsten M, Duraj V, et al. Response to tetanus toxoid immunization after allogeneic bone marrow transplantation. J Infect Dis. 1990;162:496–500. doi: 10.1093/infdis/162.2.496. [DOI] [PubMed] [Google Scholar]
- 42.Wahren B, Gahrton G, Linde A, et al. Transfer and persistence of viral antibody-producing cells in bone marrow transplantation. J Infect Dis. 1984;150:358–65. doi: 10.1093/infdis/150.3.358. [DOI] [PubMed] [Google Scholar]
- 43.Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci USA. 2008;105:4802–7. doi: 10.1073/pnas.0800555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auletta JJ, Cooke KR, Solchaga LA, Deans RJ, van't Hof W. Regenerative stromal cell therapy in allogeneic hematopoietic stem cell transplantation: current impact and future directions. Biol Blood Marrow Transplant. 2010;16:891–906. doi: 10.1016/j.bbmt.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chu VT, Fröhlich A, Steinhauser G, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151–9. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 46.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 47.Gray D. A role for antigen in the maintenance of immunological memory. Nat Rev Immunol. 2002;2:60–5. doi: 10.1038/nri706. [DOI] [PubMed] [Google Scholar]
- 48.Lycke N, Bemark M. Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol. 2010;3:556–66. doi: 10.1038/mi.2010.54. [DOI] [PubMed] [Google Scholar]
- 49.Witherspoon RP, Kopecky K, Storb RF, et al. Immunological recovery in 48 patients following syngeneic marrow transplantation or hematological malignancy. Transplantation. 1982;33:143–9. doi: 10.1097/00007890-198202000-00008. [DOI] [PubMed] [Google Scholar]
- 50.Giebink GS, Warkentin PI, Ramsay NK, Kersey JH. Titers of antibody to pneumococci in allogeneic bone marrow transplant recipients before and after vaccination with pneumococcal vaccine. J Infect Dis. 1986;154:590–6. doi: 10.1093/infdis/154.4.590. [DOI] [PubMed] [Google Scholar]
- 51.Lortan JE, Vellodi A, Jurges ES, Hugh-Jones K. Class- and subclass-specific pneumococcal antibody levels and response to immunization after bone marrow transplantation. Clin Exp Immunol. 1992;88:512–9. doi: 10.1111/j.1365-2249.1992.tb06480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lutz E, Ward KN, Szydlo R, Goldman JM. Cytomegalovirus antibody avidity in allogeneic bone marrow recipients: evidence for primary or secondary humoral responses depending on donor immune status. J Med Virol. 1996;49:61–5. doi: 10.1002/(SICI)1096-9071(199605)49:1<61::AID-JMV10>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 53.Tomblyn M, Chiller T, Einsele H, et al. Guidelines for preventing infectious complications amonghematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15:1143–238. doi: 10.1016/j.bbmt.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hallstrand TS, Sprenger JD, Agosti JM, Longton GM, Witherspoon RP, Henderson WR., Jr Long-term acquisition of allergen-specific IgE and asthma following allogeneic bone marrow transplantation from allergic donors. Blood. 2004;104:3086–90. doi: 10.1182/blood-2004-05-1775. [DOI] [PubMed] [Google Scholar]
- 55.Gerritsen EJ, van Tol MJ, Lankester AC, et al. Immunoglobulin levels and monoclonal gammopathies in children after bone marrow transplantation. Blood. 1993;82:3493–502. [PubMed] [Google Scholar]
- 56.Aucouturier P, Barra A, Intrator L, et al. Long lasting IgG subclass and antibacterial polysaccharide antibody deficiency after allogeneic bone marrow transplantation. Blood. 1987;70:779–85. [PubMed] [Google Scholar]
- 57.Lum LG, Seigneuret MC, Orcutt-Thordarson N, Noges JE, Storb R. The regulation of immunoglobulin synthesis after HLA-identical bone marrow transplantation: VI. differential rates of maturation of distinct functional groups within lymphoid subpopulations in patients after human marrow grafting. Blood. 1985;65:1422–33. [PubMed] [Google Scholar]
- 58.Korsmeyer SJ, Elfenbein GJ, Goldman CK, Marshall SL, Santos GW, Waldmann TA. B cell, helper T cell, and suppressor T cell abnormalities contribute to disordered immunoglobulin synthesis in patients following bone marrow transplantation. Transplantation. 1982;33:184–90. doi: 10.1097/00007890-198202000-00015. [DOI] [PubMed] [Google Scholar]
- 59.Keightley RG, Cooper MD, Lawton AR. The T cell dependence of B cell differentiation induced by pokeweed mitogen. J Immunol. 1976;117:1538–44. [PubMed] [Google Scholar]
- 60.Kirchner H, Tosato G, Blaese RM, Broder S, Magrath IT. Polyclonal immunoglobulin secretion by human B lymphocytes exposed to Epstein–Barr virus in vitro. J Immunol. 1979;122:1310–3. [PubMed] [Google Scholar]
- 61.Dilly SA, Sloane JP. Cellular composition of the spleen after human allogeneic bone marrow transplantation. J Pathol. 1988;155:151–60. doi: 10.1002/path.1711550212. [DOI] [PubMed] [Google Scholar]
- 62.Sale GE, Alavaikko M, Schaefers KM, Mahan CT. Abnormal CD4 : CD8 ratios and delayed germinal center reconstitution in lymph nodes of human graft recipients with graft-versus-host disease (GVHD): an immunohistological study. Exp Hematol. 1992;20:1017–21. [PubMed] [Google Scholar]
- 63.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–57. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson SM, Wilson PC, James JA, Capra JD. Human B cell subsets. Adv Immunol. 2008;98:151–224. doi: 10.1016/S0065-2776(08)00405-7. [DOI] [PubMed] [Google Scholar]
- 65.Carsetti R, Köhler G, Lamers MC. Transitional B cells are the target of negative selection in the B cell compartment. J Exp Med. 1995;181:2129–40. doi: 10.1084/jem.181.6.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cuss AK, Avery DT, Cannons JL, et al. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. 2006;176:1506–16. doi: 10.4049/jimmunol.176.3.1506. [DOI] [PubMed] [Google Scholar]
- 67.Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. 2005;105:4390–8. doi: 10.1182/blood-2004-11-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee J, Kuchen S, Fischer R, Chang S, Lipsky PE. Identification and characterization of a human CD5+ pre-naive B cell population. J Immunol. 2009;182:4116–26. doi: 10.4049/jimmunol.0803391. [DOI] [PubMed] [Google Scholar]
- 69.Suryani S, Fulcher DA, Santner-Nanan B, et al. Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood. 2010;115:519–29. doi: 10.1182/blood-2009-07-234799. [DOI] [PubMed] [Google Scholar]
- 70.Andrews S, Wilson P. The anergic B cell. Blood. 2010;115:4976. doi: 10.1182/blood-2010-03-276352. [DOI] [PubMed] [Google Scholar]
- 71.Wirths S, Lanzavecchia A. ABCB1 transporter discriminates human resting naive B cells from cycling transitional and memory B cells. Eur J Immunol. 2005;35:3433–41. doi: 10.1002/eji.200535364. [DOI] [PubMed] [Google Scholar]
- 72.Palanichamy A, Barnard J, Zheng B, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. J Immunol. 2009;182:5982–93. doi: 10.4049/jimmunol.0801859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679–89. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–8. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 75.Bemark M, Bergqvist P, Stensson A, Holmberg A, Mattsson J, Lycke NY. A unique role of the cholera toxin A1-DD adjuvant for long-term plasma and memory B cell development. J Immunol. 2011;186:1399–410. doi: 10.4049/jimmunol.1002881. [DOI] [PubMed] [Google Scholar]
- 76.Seifert M, Küppers R. Molecular footprints of a germinal center derivation of human IgM+(IgD+)CD27+ B cells and the dynamics of memory B cell generation. J Exp Med. 2009;206:2659–69. doi: 10.1084/jem.20091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tangye SG, Good KL. Human IgM+CD27+ B cells: memory B cells or ‘memory’ B cells? J Immunol. 2007;179:13–19. doi: 10.4049/jimmunol.179.1.13. [DOI] [PubMed] [Google Scholar]
- 78.Weill J-C, Weller S, Reynaud C-A. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–85. doi: 10.1146/annurev.immunol.021908.132607. [DOI] [PubMed] [Google Scholar]
- 79.Wu Y-C, Kipling D, Leong HS, Martin V, Ademokun AA, Dunn-Walters DK. High-throughput immunoglobulin repertoire analysis distinguishes between human IgM memory and switched memory B-cell populations. Blood. 2010;116:1070–8. doi: 10.1182/blood-2010-03-275859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fecteau JF, Côté G, Néron S. A new memory CD27–IgG+ B cell population in peripheral blood expressing VH genes with low frequency of somatic mutation. J Immunol. 2006;177:3728–36. doi: 10.4049/jimmunol.177.6.3728. [DOI] [PubMed] [Google Scholar]
- 81.Koethe S, Zander L, Koster S, et al. Pivotal advance: CD45RB glycosylation is specifically regulated during human peripheral B cell differentiation. J Leukoc Biol. 2011;90:5–19. doi: 10.1189/jlb.0710404. [DOI] [PubMed] [Google Scholar]
- 82.Wrammert J, Smith K, Miller J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–71. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghosn EEB, Sadate-Ngatchou P, Yang Y, Herzenberg LA, Herzenberg LA. Distinct progenitors for B-1 and B-2 cells are present in adult mouse spleen. Proc Natl Acad Sci USA. 2011;108:2879–84. doi: 10.1073/pnas.1019764108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weller S, Braun MC, Tan BK, et al. Human blood IgM ‘memory’ B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104:3647–54. doi: 10.1182/blood-2004-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weller S, Mamani-Matsuda M, Picard C, et al. Somatic diversification in the absence of antigen-driven responses is the hallmark of the IgM+ IgD+ CD27+ B cell repertoire in infants. J Exp Med. 2008;205:1331–42. doi: 10.1084/jem.20071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kubicka U, Olszewski WL, Tarnowski W, Bielecki K, Ziolkowska A, Wierzbicki Z. Normal human immune peritoneal cells: subpopulations and functional characteristics. Scand J Immunol. 1996;44:157–63. doi: 10.1046/j.1365-3083.1996.d01-297.x. [DOI] [PubMed] [Google Scholar]
- 87.Donze HH, Lue C, Julian BA, Kutteh WH, Kantele A, Mestecky J. Human peritoneal B-1 cells and the influence of continuous ambulatory peritoneal dialysis on peritoneal and peripheral blood mononuclear cell (PBMC) composition and immunoglobulin levels. Clin Exp Immunol. 1997;109:356–61. doi: 10.1046/j.1365-2249.1997.4541352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boursier L, Farstad IN, Mellembakken JR, Brandtzaeg P, Spencer J. IgVH gene analysis suggests that peritoneal B cells do not contribute to the gut immune system in man. Eur J Immunol. 2002;32:2427–36. doi: 10.1002/1521-4141(200209)32:9<2427::AID-IMMU2427>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 89.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70–. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Horgan KJ, Tanaka Y, Luce GE, van Seventer GA, Nutman TB, Shaw S. CD45RB expression defines two interconvertible subsets of human CD4+ T cells with memory function. Eur J Immunol. 1994;24:1240–3. doi: 10.1002/eji.1830240536. [DOI] [PubMed] [Google Scholar]
- 91.Corre E, Carmagnat M, Busson M, et al. Long-term immune deficiency after allogeneic stem cell transplantation: B-cell deficiency is associated with late infections. Haematologica. 2010;95:1025–9. doi: 10.3324/haematol.2009.018853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.D'Orsogna LJ, Wright MP, Krueger RG, et al. Allogeneic hematopoietic stem cell transplantation recipients have defects of both switched and igm memory B cells. Biol Blood Marrow Transplant. 2009;15:795–803. doi: 10.1016/j.bbmt.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 93.Greinix HT, Pohlreich D, Kouba M, et al. Elevated numbers of immature/transitional CD21− B lymphocytes and deficiency of memory CD27+ B cells identify patients with active chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2008;14:208–19. doi: 10.1016/j.bbmt.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 94.Sarantopoulos S, Stevenson KE, Kim HT, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009;113:3865–74. doi: 10.1182/blood-2008-09-177840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marie-Cardine A, Divay F, Dutot I, et al. Transitional B cells in humans: characterization and insight from B lymphocyte reconstitution after hematopoietic stem cell transplantation. Clin Immunol. 2008;127:14–25. doi: 10.1016/j.clim.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 96.Antin JH, Ault KA, Rappeport JM, Smith BR. B lymphocyte reconstitution after human bone marrow transplantation. Leu-1 antigen defines a distinct population of B lymphocytes. J Clin Invest. 1987;80:325–32. doi: 10.1172/JCI113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Small TN, Keever CA, Weiner-Fedus S, Heller G, O'Reilly RJ, Flomenberg N. B-cell differentiation following autologous, conventional, or T-cell depleted bone marrow transplantation: a recapitulation of normal B-cell ontogeny. Blood. 1990;76:1647–56. [PubMed] [Google Scholar]
- 98.Storek J, Ferrara S, Ku N, Giorgi JV, Champlin RE, Saxon A. B cell reconstitution after human bone marrow transplantation: recapitulation of ontogeny? Bone Marrow Transplant. 1993;12:387–98. [PubMed] [Google Scholar]
- 99.Velardi A, Cucciaioni S, Terenzi A, et al. Acquisition of Ig isotype diversity after bone marrow transplantation in adults. A recapitulation of normal B cell ontogeny. J Immunol. 1988;141:815–20. [PubMed] [Google Scholar]
- 100.Elfenbein GJ, Bellis MM, Ravlin HM, Santos GW. Phenotypically immature B mu cells in the peripheral blood after bone marrow grafting in man. Exp Hematol. 1982;10:551–9. [PubMed] [Google Scholar]
- 101.Ault KA, Antin JH, Ginsburg D, et al. Phenotype of recovering lymphoid cell populations after marrow transplantation. J Exp Med. 1985;161:1483–502. doi: 10.1084/jem.161.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parra C, Roldán E, Brieva JA. Deficient expression of adhesion molecules by human CD5− B lymphocytes both after bone marrow transplantation and during normal ontogeny. Blood. 1996;88:1733–40. [PubMed] [Google Scholar]
- 103.Kagan JM, Champlin RE, Saxon A. B-cell dysfunction following human bone marrow transplantation: functional–phenotypic dissociation in the early posttransplant period. Blood. 1989;74:777–85. [PubMed] [Google Scholar]
- 104.Eyrich M, Leiler C, Lang P, et al. A prospective comparison of immune reconstitution in pediatric recipients of positively selected CD34+ peripheral blood stem cells from unrelated donors vs recipients of unmanipulated bone marrow from related donors. Bone Marrow Transplant. 2003;32:379–90. doi: 10.1038/sj.bmt.1704158. [DOI] [PubMed] [Google Scholar]
- 105.Uckun FM, Haissig S, Ledbetter JA, et al. Developmental hierarchy during early human B-cell ontogeny after autologous bone marrow transplantation using autografts depleted of CD19+ B-cell precursors by an anti-CD19 pan-B-cell immunotoxin containing pokeweed antiviral protein. Blood. 1992;79:3369–79. [PubMed] [Google Scholar]
- 106.Koehne G, Zeller W, Stockschlaeder M, Zander AR. Phenotype of lymphocyte subsets after autologous peripheral blood stem cell transplantation. Bone Marrow Transplant. 1997;19:149–56. doi: 10.1038/sj.bmt.1700624. [DOI] [PubMed] [Google Scholar]