

Abstract
OTHER THEMES PUBLISHED IN THIS IMMUNOLOGY IN THE CLINIC REVIEW SERIES
Allergy, Host Responses, Cancer, Autoinflammatory Diseases, Type 1 diabetes and viruses.
Historically, the development of type 2 diabetes has been considered not to have an autoimmune component, in contrast to the autoimmune pathogenesis of type 1 diabetes. In this review we will discuss the accumulating data supporting the concept that islet autoreactivity and inflammation is present in type 2 diabetes pathogenesis, and the islet autoimmunity appears to be one of the factors associated with the progressive nature of the type 2 diabetes disease process.
Keywords: autoimmunity, diabetes, inflammation/inflammatory mediators including eicosanoids, T cells
The immune system is a collection of highly regulated processes designed to promote protective immunity against insults from pathogenic organisms and neoplasias. These highly regulated processes (adaptive and innate immune systems) encompass both stimulatory and regulatory pathways aimed at turning on and off appropriate responses designed to rid the host of the assailant without producing long-term damage to the host. To accomplish the eradication of pathogenic organisms, the host mounts an inflammatory insult. The developing inflammation serves to protect a defined region of infected or damaged tissue by recruiting cells necessary to resolve the insult while isolating the area to prevent the spread of inflection. Regulatory mechanisms have evolved in the host to down-regulate and control the immune response and tissue inflammation [1]. However, inflammation sometimes fails to subside and this unresolved inflammation may become chronic. Chronic inflammation has been attributed to the development of inflammatory diseases such as atherosclerosis [2]. Moreover, intriguing evidence is accumulating which indicates that unresolved chronic inflammation may play a role in the initiation, promotion, malignant conversion and metastasis of several human cancers [3,4]. Allowing inflammatory responses to assist in eradicating pathogenic mechanisms while forbidding establishment of chronic inflammatory conditions and subsequent development of inflammatory disease is one of the multiple facets of the normally functioning immune system.
Another facet of the normal immune system is the recognition of self versus altered self. Cancer arises from the host's own tissues, thus immune recognition and eradication of cancer from the host requires that the immune system must recognize an altered version of self. In other words, the immune system must allow generation of autoreactivity to occur to eliminate the cancer cells. Results of studies in cancer immunology are challenging the old concept that the immune system is tightly regulated, not allowing for reactivity to self. Instead, new concepts illustrate that the immune system is not so tightly regulated to prevent reactivity to self; rather, the normal immune repertoire consists of both T cells and B cells capable of recognizing self [5–9]. However, under most normal circumstances the immune system's regulatory mechanisms are effective in maintaining control over the autoreactive cells preventing the development of autoimmune disease while maintaining the immunosurveillance necessary to avoid establishment of malignancies.
A delicate balance exists in the multi-faceted normal immune system encompassing effector mechanisms designed to initiate inflammatory and autoreactivity balanced against regulatory mechanisms designed to control both inflammatory and autoimmune responses and protect the host from subsequent damage. Some of the challenges for medicine are to induce potent tumour immunity (autoreactivity) balanced against the risk of development of autoimmune disease and to establish effective inflammatory responses to rid the host of assaulting pathogens without allowing for chronic inflammatory conditions which may lead to subsequent inflammatory disease. Another emerging area of intriguing data points to the ageing immune system as a potential cause of chronic inflammatory and/or autoimmune disease development. As the host ages the immune system, like many organ systems, experiences either diminished or loss of functional capacity. This concept of autoimmunity proposes that the failure of control mechanisms as the host ages may be a primary risk factor for autoimmune disease development in older individuals [9]. Inflammatory and autoimmune responses are therefore part of the normal and protective capabilities of the host's immune system. However, when does the inflammation become chronic, escalating from an inflammatory condition to an inflammatory disease, or when does the autoreactivity become autoimmune disease? In the remainder of this review, we will focus on the concepts of inflammatory and autoimmune responses in association with the development of type 2 diabetes.
Diabetes mellitus is a spectrum of diseases encompassing type 1 (T1D) and type 2 (T2D) diabetes [10–12]. The diagnosis of T1D versus T2D is commonly made using criteria such as age at onset, abruptness of hyperglycaemic symptoms, presence of ketosis, degree of obesity and the perceived need for insulin replacement. The pathogenesis of T1D is believed to be a cell-mediated autoimmune disease because T cells, but not autoantibodies, are necessary to transfer disease in animal models and human T1D [13–15]. T2D accounts for approximately 90–95% of patients with diabetes, with individuals having disease pathogenesis ranging from predominantly insulin resistance with relative insulin deficiency to primarily an insulin secretory defect with accompanying insulin resistance. Historically, T2D has been considered to be a metabolic disease of the ageing individual and has not been considered to be autoimmune.
Recently, many notable discoveries have provided evidence to support the concept of immune system involvement in obesity and type 2 diabetes development [16–19]. Chronic inflammation of the visceral adipose tissue is believed to be involved in the pathogenesis of insulin resistance and subsequent development of T2D, with multiple groups demonstrating an increase in visceral adipose T cell subsets [20–23]. In fact, proinflammatory T cells present in visceral fat are believed to be involved in the initial establishment of adipose inflammation preceding the infiltration of monocytes into the adipose tissue [20]. Regulatory T cells have been shown to be highly enriched in the abdominal fat of normal mice but reduced significantly in the abdominal fat of insulin-resistant mouse models of obesity [24]. Deiuliis et al. [25] reported that obesity in mice and humans actually results in adipose T regulatory cell depletion. In fact, induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice [26]. Moreover, Meijer K et al. [27] reported that human adipocytes express a number of cytokines and chemokines that are able to induce inflammation and activate CD4+ cells independent of macrophages. These results suggest that the primary event in the sequence leading to chronic inflammation in adipose tissue is a metabolic change in adipocytes inducing production of immunological mediators, and presentation of potential antigens by adipocytes leading to activation of adipose tissue macrophages and other immune cells. Furthermore, many studies, both cross-sectional and prospective, have demonstrated elevated levels of circulating acute phase proteins as well as cytokines and chemokines in patients with T2D, supporting the concept that T2D is an inflammatory disease [28–31].
The diagnosis of T2D involves insulin resistance as one of the components in the diabetes disease process. In recent years, the contribution of several proinflammatory cytokines such as interleukin (IL)-1β[32–34], IL-6 [35] and tumour necrosis factor (TNF)-α[36,37] have been implicated in disrupting insulin signalling, causing insulin resistance. In fact, neutralizing TNF-α in rats provided an early suggestion that inflammatory mediators were associated with the development of insulin resistance [36]. Irrespective of the initiation trigger for the chronic inflammation, the involvement of chronic inflammation in the development of insulin resistance and subsequent development of T2D is now widely accepted.
One risk factor for development of autoimmune disease is chronic inflammation [38,39]. Moreover, obesity, which is a phenotypic risk factor for T2D development, has been demonstrated to predispose patients to several autoimmune disorders, including inflammatory bowel disease (IBD) and psoriasis [40,41]. Proinflammatory CD4+ T cells in adipose tissue have been demonstrated to stimulate the development of CD8+ T cells [17,22]. These observations are important, as the CD8+ T cells are generally considered to be the cells capable of lysing cells, both foreign and self, in the development of inflammation and autoreactive responses [42–46]. Until recently, the development of autoinflammatory and autoimmune diseases was believed to rely on the stimulation of a subset of CD4+ proinflammatory cells designated as T helper type 1 (Th1). However, with the discovery of IL-23 it has now become apparent that other immune system players are implicated in autoimmune disease development. One of the immune system culprits is the cytokine IL-17. IL-17 has been demonstrated to be produced by a new T cell subset designated Th17. The Th17 T cells have been implicated directly in the pathogenesis of both inflammatory and autoimmune diseases [47–49]. Moreover, obesity and chronic inflammation have been demonstrated to promote selectively an expansion of the Th17 T cell subset [50]. The increased Th17 bias, the increases in CD8+ T cell subsets and establishment of an inflammatory milieu may represent the link between inflammation, T2D and subsequent development of islet autoimmune disease in T2D patients.
Another component of diabetes disease development is the resulting pancreatic lesion. The pancreatic lesion in patients with diabetes encompasses a spectrum of diminished or destroyed capability of the pancreatic islets to produce insulin. In the pancreas of T1D patients the β cells are destroyed selectively by the immune system in an autoimmune attack, whereas the pancreatic lesion of phenotypic T2D patients has been believed historically to be a metabolic defect, resulting in diminished secretory capability. However, recently the pancreas of T2D patients have been demonstrated to be infiltrated by immune cells [17–19]. These studies suggest that immune-mediated islet damage may be a component of more than just classic T1D. β cell destruction and damage caused by soluble immune mediators occurs most probably in the pathogenesis of both T1D and T2D. In T1D, the invading immune cells produce cytokines such as IL-1β, TNF-α and interferon (IFN)-γ. These cytokines have been demonstrated to directly induce β cell apoptosis [51]. In T2D, the circulating IL-6 and IL-1β have also been associated with β cell apoptosis [52]. Moreover, elevated levels of IL-1β, IL-6 and C-reactive protein (CRP) are predictive of T2D development [28–31]. Treatment of T2D patients with IL-1ra to block the effects of IL-1β improves β cell function and diabetes control [32]. With the backdrop of obesity-associated chronic inflammation, enrichment of T cell populations indicated in autoimmune pathologies, an associated inflammatory pancreatic islet lesion, circulating, and within the pancreatic lesion, soluble β cell apoptotic mediators, and the infiltration of the immune cells not only into the adipose tissue but also into the pancreas, the stage is set for cell-mediated islet autoimmune development in T2D. When does islet autoreactivity become autoimmune disease?
The levels of circulating soluble inflammatory mediators have been shown to be similar among diabetic and non-diabetic obese subjects [31], and cannot be used to predict the efficacy of anti-inflammatory treatments directed at stimulating insulin secretion, decreasing insulin resistance or preventing development of T2D [30–33]. The decline in β cell function observed over time in most T2D patients demonstrates the progressive nature of the T2D disease process [50]. This decline in β cell function during diabetes pathogenesis has been demonstrated to be diminished or halted with diabetes drugs with secondary anti-inflammatory properties [Reichow et al., unpublished data]. What is the target of the anti-inflammatory actions of these drugs which demonstrate efficacy in the treatment of T2D? Could one of the mechanisms responsible for the subsequent drop in pancreatic insulin output over time observed in T2D patients be cell-mediated islet autoimmune destruction? Could the autoreactive T cells present in normal individuals become autoreactive effector cells capable of initiating islet autoimmune disease in T2D patients within the chronic inflammatory mileu associated with obesity and T2D?
In 1996 our laboratory developed a T cell assay, cellular immunoblotting, with excellent sensitivity and specificity for measuring islet-specific T cell responses in autoimmune diabetes [54,55]. We have utilized cellular immunoblotting to measure islet-reactive T cells in T1D patients [54–57], subjects at risk of developing T1D and, more recently, phenotypic T2D patients [58–60]. We have also demonstrated that T cell reactivity to islet proteins in phenotypic T2D patients correlates more strongly with impaired β-cell function compared to autoantibody positivity (Fig. 1), thus demonstrating not only the presence of islet autoimmune responses in T2D patients but autoimmune disease [60]. More recently, we have also observed that the diabetes drug (rosiglitazone), which suppresses the islet reactive T cell responses (anti-inflammatory) in phenotypic T2D patients, can improve β cell function (Reichow et al., unpublished data). Furthermore, rosiglitazone has also been shown to be able to reduce both T cell and macrophage infiltration into the adipose tissue, improving insulin resistance and glucose intolerance [61]. These results support the concept that subsequent improvement in T2D disease can be accomplished by targeting immune responses and most importantly diminishing tissue (islet)-specific T cell responses.
Fig. 1.
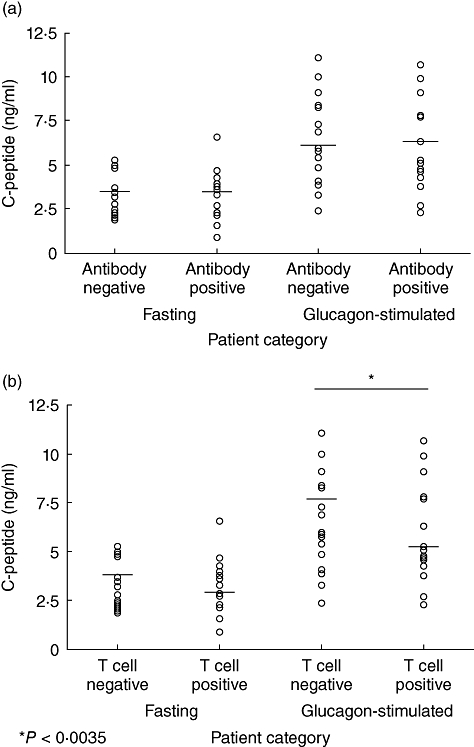
Fasting and glucagon-stimulated C-peptide in phenotypic type 2 diabetes patients separated by antibody-negative (n = 17) and antibody-positive (n = 19) independent of T cell reactivity (a) or separated by T cell responses to islet proteins irrespective of autoantibody responses (b). T cell– (n = 13) and T cell+ (n = 23). Horizontal bars represent means [53].
The importance of quantitating islet autoimmunity though the measurement of the islet-reactive T cells is emphasized by the reports estimating that up to 15–20% of newly diagnosed autoimmune T1D patients are autoantibody-negative [62]. Furthermore, approximately 9% of autoantibody-negative T1D patients carry the highest-risk human leucocyte antigen (HLA) genotype DR3–DQ2/DR4–DQ8, suggesting strongly that these patients had autoimmune diabetes but were undetected with autoantibody testing alone [62]. Similarly, a subgroup of Japanese autoimmune diabetes patients, known as fulminant type 1 diabetes, have been reported to be autoantibody-negative but demonstrate islet-specific T cell responses [63]. In phenotypic T2D patients, we identified the presence of a subgroup of phenotypic T2D patients who are autoantibody-negative, but demonstrate islet-specific autoimmunity with islet-reactive T cells similar to classic T1D patients [60]. These T cell islet-reactive positive phenotypic T2D patients also demonstrated a more severe β cell lesion than the patients who had not yet developed islet-reactive T cell responses [60], thus implicating the islet-reactive T cells in T2D patients in the β cell functional demise associated with T2D pathogenesis. Moreover, these studies demonstrate further the importance of assaying for islet autoimmune T cell responses when determining the presence of islet autoimmunity in T2D patients.
Therefore, it appears that islet autoimmune disease may be involved in the continued β cell functional demise associated with the progressive nature of T2D disease. However, is the islet autoimmunity that develops in T2D the same as the islet autoimmunity which develops in T1D? Comparing islet autoantibodies associated with T1D and T2D patients suggests potential differences. The most common islet autoantibodies found in childhood T1D patients are islet cell autoantibodies (ICA), glutamate decarboxylase autoantibodies (GADAb), insulinoma-associated antigen-2 autoantibodies (IA-2), zinc transporter autoantibodies (ZnT8) and insulin autoantibodies (IAA), with many patients demonstrating positivity for multiple islet autoantibodies. In fact, positivity for an increasing number of islet autoantibodies is associated with a progressively greater risk of developing T1D [64–67]. In contrast, for phenotypic T2D patients, GADAb and ICA are much more common than IAA, IA-2 and ZnT8 autoantibodies and singular positivity for either ICA or GADAb is more characteristic of autoimmune phenotypic T2D patients [68–73]. One important issue to stress is the islet autoantibodies used to categorize and identify autoimmune T2D patients are islet autoantibodies identified originally in T1D patients. Therefore, there may be other islet autoantibodies specific to autoimmunity in phenotypic T2 diabetes that have not yet been identified which would classify them more accurately. In support of this concept, Seissler et al. [74] demonstrated that GAD and IA-2 could block ICA staining in approximately 60% of sera from T1D patients, but in a much lower percentage of sera from autoimmune T2D patients.
When T cell recognition of islet proteins is compared between T1D and T2D patients (Fig. 2), islet proteins that T cells from both groups of patients recognize are identified, but differences in the islet proteins recognized by the T cells from T1D and T2D patients are also observed [75]. These results demonstrate that the development of islet autoimmunity in T1D and T2D patients appears to follow a slightly different roadmap to islet autoimmune disease. This is not totally surprising, as the autoimmune development in T2D patients appears to arise as a sequela of the chronic inflammatory responses associated with obesity, whereas the autoimmune responses in T1D may have a more specific environmental trigger. Recently, obesity has also been demonstrated to be a potential accelerant of the diabetes disease processes and subsequent complications in classic T1D patients [76–79]. These studies suggest further that islet autoimmune development in both T1D and T2D may be more similar than appreciated previously.
Fig. 2.
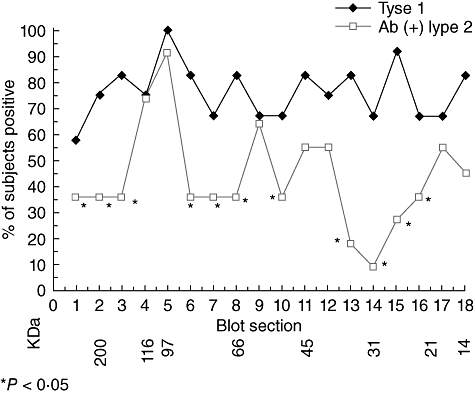
Similarities and differences identified in islet proteins recognized by peripheral blood mononuclear cells from type 1 diabetes and autoantibody-positive phenotypic type 2 diabetes patients. Asterisks identify significant (P < 0·05) differences in percentage of patients responding to a particular molecular weight region containing islet proteins [66].
Accumulating data support the concept that not only are islet autoreactivity and inflammation present in T2D, but also islet autoimmune disease. Moreover, the development of islet autoimmune disease appears to be one of the factors associated with the progressive nature of the T2D disease process. Understanding the islet autoimmune cell-mediated pathogenesis in phenotypic T2D patients may lead to the development of new, more efficacious and safer antigen-based intervention strategies directed at the developing cell-mediated islet autoimmunity both in T1D and T2D.
Disclosure
None.
References
- 1.Jager A, Kuchroo VK. Effector and regulatory T cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72:173–84. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dumitriu IE, Araguas ET, Baboonian C, Kaski JC. CD4+CD28null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. 2009;81:11–19. doi: 10.1093/cvr/cvn248. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamarron BF, Chen WJ. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7:651–8. doi: 10.7150/ijbs.7.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goverman J, Brabb T, Paez A, Harrington C, von Dassow P. Initiation and regulation of CNS autoimmunity. Crit Rev Immunol. 1997;17:469–80. [PubMed] [Google Scholar]
- 6.Burns J, Rosenzwerg A, Zwerman B, Lisak RP. Isolation of myelin basic protein-reactive T cell lines from normal human blood. Cell Immunol. 1983;81:435–40. doi: 10.1016/0008-8749(83)90250-2. [DOI] [PubMed] [Google Scholar]
- 7.Homann D, von Herrath M. Regulatory T cells and type 1 diabetes. Clin Immunol. 2004;112:202–9. doi: 10.1016/j.clim.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 8.Hoyne GF. Mechanisms that regulate peripheral immune responses to control organ-specific autoimmunity. Clin Dev Immunol. 2011 doi: 10.1155/2011/294968. 2011:294968 Epub 2011 28 April. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bubanovic I. Auto-immunity as evolutionary by product of adoptive immunity and source of anti-tumor immunity failure. Int J Cancer Res. 2005;1:81–6. [Google Scholar]
- 10.Brooks-Worrell B, Palmer JPP. Is diabetes mellitus a continuous spectrum? Clin Chem. 2011;57:158–61. doi: 10.1373/clinchem.2010.148270. [DOI] [PubMed] [Google Scholar]
- 11.Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 1997;20:1183–97. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 12.McCance DR, Hanson RL, Pettitt DF, Bennett PH, Hadden DR, Knowler WC. Diagnosing diabetes mellitus: do we need new criteria? Diabetologia. 1997;40:247–55. doi: 10.1007/s001250050671. [DOI] [PubMed] [Google Scholar]
- 13.Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt2+ T cells. J Exp Med. 1987;166:823–32. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–8. [PubMed] [Google Scholar]
- 15.Lampeter EF, Homberg M, Quabeck K, et al. Transfer of insulin-dependent diabetes between HLA-identical siblings by bone marrow transplantation. Lancet. 1993;341:1243–4. doi: 10.1016/0140-6736(93)91148-f. [DOI] [PubMed] [Google Scholar]
- 16.Ehses JA, Ellingsgaard H, Boni-Schnetzler M, Donath MY. Pancreatic islet inflammation in type 2 diabetes: from α and β cell compensation to dysfunction. Arch Physiol Biochem. 2009;115:240–7. doi: 10.1080/13813450903025879. [DOI] [PubMed] [Google Scholar]
- 17.Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes. Diabetes Care. 2008;31:S161–4. doi: 10.2337/dc08-s243. [DOI] [PubMed] [Google Scholar]
- 18.Donath MY, Boni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic β-cell in type 2 diabetes. Physiology. 2009;24:325–31. doi: 10.1152/physiol.00032.2009. [DOI] [PubMed] [Google Scholar]
- 19.Boni-Schnetzler M, Ehses JA, Faulenbach M, Donath MY. Insulitis in type 2 diabetes. Diabetes Obes Metab. 2008;10:S20–S204. doi: 10.1111/j.1463-1326.2008.00950.x. [DOI] [PubMed] [Google Scholar]
- 20.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1304–10. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 21.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 22.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–9. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulation on activation, normal T cell expressed and secreted up-regulation in adipose tissue in obesity. Circulation. 2007;115:1029–38. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 24.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameter. Nat Med. 2009;15:930–40. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in T regulatory cell numbers. PLoS ONE. 2011;6:e16376. doi: 10.1371/journal.pone.0016376. doi: 10.1371/journal.pone.0016376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilan Y, Maron R, Tukpah A-M, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci. 2010;107:9765–70. doi: 10.1073/pnas.0908771107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meijer K, de Vries M, Al-Lahham S, et al. Human primary adipocytes exhibit immune cell function: adipocytes prime inflammation independent of macrophages. PLoS ONE. 2011;6:e17154. doi: 10.1371/journal.pone.0017154. doi: 10.1371/journal.pone.0017154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European prospective investigation into cancer and nutrition (EPIC)-Potsdam study. Diabetes. 2003;52:812–17. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 29.Herder C, Brunner EJ, Rathmann W, et al. Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes: the Whitehall II study. Diabetes Care. 2009;32:421–3. doi: 10.2337/dc08-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pradhan DA, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin-6 and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 31.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 32.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 33.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2010;152:346–57. doi: 10.1059/0003-4819-152-6-201003160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1β-induced insulin resistance in adipocytes through down-relation of insulin receptor substrate-1 expression. Endocrinology. 2007;148:241–51. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weigert C, Hennige AM, Lehmann R, et al. Direct cross-talk of interleukin-6 and insulin signal transduction via insulin receptor substrate-1 in skeletal muscle cells. J Biol Chem. 2006;281:7060–7. doi: 10.1074/jbc.M509782200. [DOI] [PubMed] [Google Scholar]
- 36.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 37.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389:610–14. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 38.Lee MS. Role of innate immunity in diabetes and metabolism: recent progress in the study of inflammasomes. Immune Netw. 2011;11:95–9. doi: 10.4110/in.2011.11.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickup JC. Inflammation and activation innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–23. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 40.Setty AR, Curhan G, Choi HK. Obesity, waist circumference, weight change, an the risk of psoriasis in women: Nurses' Health Study II. Arch Intern Med. 2007;167:1670–5. doi: 10.1001/archinte.167.15.1670. [DOI] [PubMed] [Google Scholar]
- 41.Hass DJ, Brensinger CM, Lewis JD, Lichtenstein GR. The impact of increased body mass index on the clinical course of Crohn's disease. Clin Gasteroenterol Hepatol. 2006;4:482–8. doi: 10.1016/j.cgh.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Deckert M, Sanchez-Ruiz M, Brunn A, Schluter D. Role of CD8 T-cell-mediated autoimmune diseases of the central nervous system. Crit Rev Immunol. 2010;30:311–26. doi: 10.1615/critrevimmunol.v30.i4.10. [DOI] [PubMed] [Google Scholar]
- 43.Mars LT, Saikali P, Liblau RS, Arbour N. Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim Biophys Acta. 2011;1812:151–61. doi: 10.1016/j.bbadis.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zierden M, Kuhnen E, Odenthal M, Dienes HP. Effects and regulation of autoreactive CD8+ T cells in a transgenic mouse model of autoimmune hepatitis. Gasteroenterology. 2010;139:975–86. doi: 10.1053/j.gastro.2010.05.075. [DOI] [PubMed] [Google Scholar]
- 45.Longhi MS, Ma Y, Mieli-Vergani G, Vergani D. Adaptive immunity in autoimmune hepatitis. Dig Dis. 2010;28:63–9. doi: 10.1159/000282066. [DOI] [PubMed] [Google Scholar]
- 46.Faustman DL, Davis M. The primacy of CD8 T lymphocytes in type 1 diabetes and implications for therapies. J Mol Med. 2009;87:1173–8. doi: 10.1007/s00109-009-0516-6. [DOI] [PubMed] [Google Scholar]
- 47.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 48.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winer S, Paltser G, Chan Y, et al. Obesity predisposes to TH17 bias. Eur J Immunol. 2009;39:2629–35. doi: 10.1002/eji.200838893. [DOI] [PubMed] [Google Scholar]
- 50.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Auto-immune inflammation from the Th17 perspective. Autoimmun Rev. 2007;6:169–75. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Cnop M, Welsh N, Jonas J-C, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes. Diabetes. 2005;545(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 52.Mandrup-Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol. 2003;66:1433–40. doi: 10.1016/s0006-2952(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 53.DeFronzo RA, Tipathy D, Schwenke DC, et al. for the ACT NOW study Pioglitazone for Diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011;364:1104–15. doi: 10.1056/NEJMoa1010949. [DOI] [PubMed] [Google Scholar]
- 54.Seyfert-Margolis V, Gisler TD, Asare AL, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes. 2006;55:2588–94. doi: 10.2337/db05-1378. [DOI] [PubMed] [Google Scholar]
- 55.Herold KC, Brooks-Worrell B, Palmer JPP, et al. The type 1 diabetes TrailNet research group: validity and reproducibility of measurement of islet autoreactivity by T-cell assays in subjects with early type 1 diabetes. Diabetes. 2009;58:2588–95. doi: 10.2337/db09-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brooks-Worrell BM, Starkebaum GA, Greenbaum C, Palmer JPP. Peripheral blood mononuclear cells of insulin-dependent diabetic patients: respond to multiple islet cell proteins. J Immunol. 1996;157:5668–74. [PubMed] [Google Scholar]
- 57.Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JPP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol. 2001;166:5265–70. doi: 10.4049/jimmunol.166.8.5265. [DOI] [PubMed] [Google Scholar]
- 58.Brooks-Worrell BM, Juneja R, Minokadeh A, Greenbaum CJ, Palmer JPP. Cellular immune response to human islet proteins in antibody-positive type 2 diabetic patients. Diabetes. 1999;48:983–8. doi: 10.2337/diabetes.48.5.983. [DOI] [PubMed] [Google Scholar]
- 59.Brooks-Worrell BM, Reichow JL, Goel A, Ismail H, Palmer JPP. Identification of autoantibody-negative autoimmune type 2 diabetes patients. Diabetes Care. 2011;34:168–73. doi: 10.2337/dc10-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goel A, Chiu H, Felton J, Palmer JPP, Brooks-Worrell B. T cell responses to islet antigens improves detection of autoimmune diabetes and identifies patients with more severe β-cell lesions in phenotypic type 2 diabetes. Diabetes. 2007;56:2110–15. doi: 10.2337/db06-0552. [DOI] [PubMed] [Google Scholar]
- 61.Foryst-Ludwig A, Hertge M, Clemenz M, et al. PPARgamma activation attenuates T-lymphocyte-dependent inflammation of adipose tissue and development of insulin resistance in obese mice. Cardiovasc Diabetol. 2010;9:64–72. doi: 10.1186/1475-2840-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Miao D, Babu S, et al. Prevalence of autoantibody-negative diabetes is not rare at all ages and increases with older age and obesity. J Clin Endocrinol Metab. 2007;92:88–92. doi: 10.1210/jc.2006-1494. [DOI] [PubMed] [Google Scholar]
- 63.Nagata M, Moriyama H, Kotani R, et al. Immunological aspects of ‘fulminant type 1’ diabetes. Diabetes Res Clin Pract. 2007;77:1–5. doi: 10.1016/j.diabres.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 64.Verge CF, Gianani R, Kawasake I, et al. Number of autoantibodies (against insulin, GAD or ICA512/IA2) rather than particular autoantibody specificities determines risk of type 1 diabetes. J Autoimmun. 1996;9:370–83. doi: 10.1006/jaut.1996.0051. [DOI] [PubMed] [Google Scholar]
- 65.Naserke HE, Ziegler A-G, Lampasona V, Bonifacio E. Early development and spreading of autoantibodies to epitopes of IA-2 and their association with progression to type 1 diabetes. J Immunol. 1998;161:6963–9. [PubMed] [Google Scholar]
- 66.Kawasaki E, Yu L, Rewers MJ, Hutton JC, Eisenbarth GS. Definition of multiple ICA 512/phogrin autoantibody epitopes and detection of intramolecular epitope spreading in relatives of patients with type 1 diabetes. Diabetes. 1998;47:733–42. doi: 10.2337/diabetes.47.5.733. [DOI] [PubMed] [Google Scholar]
- 67.Greenbaum CJ, Brooks-Worrell BM, Palmer JPP, Lernmark A. Autoimmunity and prediction of insulin dependent diabetes mellitus. In: Marshal SM, Home PD, editors. The diabetes annual/8. Amsterdam: Elsevier; 1994. pp. 21–9. [Google Scholar]
- 68.Juneja R, Hirsch IB, Naik RG, Brooks-Worrell BM, Greenbaum CJ, Palmer JPP. Islet cell antibodies and glutamic acid decarboxylase antibodies but not the clinical phenotype help to identify type 1 1/2 diabetes in patients presenting with type 2 diabetes. Metabolism. 2001;50:1008–13. doi: 10.1053/meta.2001.25654. [DOI] [PubMed] [Google Scholar]
- 69.Mayer A, Fabien N, Gutowski MC, et al. Contrasting cellular and humoral autoimmunity associated with latent autoimmune diabetes in adults. Eur J Endocrinol. 2007;157:53–61. doi: 10.1530/EJE-07-0060. [DOI] [PubMed] [Google Scholar]
- 70.Zavala AV, Fabiano de Bruno LE, Cardoso AI, et al. Cellular and humoral autoimmunity markers in type 2 (non-insulin-dependent) diabetic patients with secondary drug failure. Diabetologia. 1992;35:1159–64. doi: 10.1007/BF00401370. [DOI] [PubMed] [Google Scholar]
- 71.Hosszufalusi N, Yatay A, Rajczy K, et al. Similar genetic features and different islet cell autoantibody pattern of latent autoimmune diabetes in adults (LADA) compared with adult-onset type 1 diabetes with rapid progression. Diabetes Care. 2003;26:452–7. doi: 10.2337/diacare.26.2.452. [DOI] [PubMed] [Google Scholar]
- 72.Murao S, Kondo S, Ohashi J, et al. Anti-thyroid peroxidase antibody, IA-2 antibody, and fasting C-peptide levels predict beta cell failure in patients with latent autoimmune diabetes in adults (LADA) – a 5 year follow-up of the Ehime study. Diabetes Res Clin Pract. 2008;38:114–21. doi: 10.1016/j.diabres.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 73.Wenzlau JM, Moua O, Sarkar SA, et al. SIC30A8 is a major target of humoral autoimmunity in Type 1 diabetes and a predictive marker in prediabetes. Immunology of diabetes V. Ann NY Acad Sci. 2008;1150:256–9. doi: 10.1196/annals.1447.029. [DOI] [PubMed] [Google Scholar]
- 74.Seissler J, DeSonnaville JJJ, Morgenthaler NG, et al. Immunological heterogeneity in type 1 diabetes: presence of distinct autoantibody patterns in patients with acute onset and slowly progressive disease. Diabetologia. 1998;41:891–7. doi: 10.1007/s001250051004. [DOI] [PubMed] [Google Scholar]
- 75.Palmer JPP, Hampe CS, Chiu H, Goel A, Brooks-Worrell BM. Is latent autoimmune diabetes in adults distinct from type 1 diabetes or just type 1 diabetes at an older age? Diabetes. 2005;54:S62–67. doi: 10.2337/diabetes.54.suppl_2.s62. [DOI] [PubMed] [Google Scholar]
- 76.Abraham A, Donaghue KC, Chan AK, Benitez-Aguirre P, Lloyd M, Craig ME. Adiposity at diagnosis of childhood type 1 diabetes – have we reached a plateau? Diabetes. 2011;60:A43–44. [Google Scholar]
- 77.Klingensmith GJ, Kaminski BM, Diabetes P. Consortium: obesity and DKA are common in youth at T1D onset: findings of the pediatric diabetes consortium (PDC) Diabetes. 2011;60:A340. [Google Scholar]
- 78.Libman I, Hughan K, Giwa A, Lee S, Beker D, Arslanian S. Are overweight children with type 1 diabetes mellitus (T1DM) at increased risk of cardiovascular disease (CVD)? Diabetes. 2011;60:A333. [Google Scholar]
- 79.Faulkner MS, Michaliszyn SF, Going S, Wheeler MD. Body mass index and waist circumference are associated with C-reactive protein and heart rate variability in adolescents with Type 1 diabetes. Diabetes. 2011;60:A341. [Google Scholar]