

Abstract
The non-catalytic region of tyrosine kinase (Nck) is proposed to play an essential role in T cell activation. However, evidence based on functional and biochemical studies has brought into question the critical function of Nck. Therefore, the aim of the present work was to investigate the role of Nck in T cell activation. To study this, the human Jurkat T cell line was used as a model for human T lymphocytes. The short interfering (si) RNA targeting Nck1 gene was used with electroporation to knock-down Nck1 protein expression in Jurkat T cells. Primary human CD4 T cells were also transfected with the siRNA of Nck1. The results showed that decreased Nck1 protein expression did not affect the apoptosis of the transfected Jurkat T cells compared with control siRNA-transfected cells and non-transfected cells. Upon CD3ε/CD28 stimulation, knock-down of Nck1 in Jurkat T cells caused a decrease in CD69 expression and in interleukin (IL)-2 secretion. Similarly, knock-down of Nck1 in primary CD4 T cells also caused decreased CD69 expression. However, no significant alterations of CD69 and IL-2 expression were found upon phytohaemagglutinin (PHA)/phorbol myristate acetate (PMA) stimulation. Knock-down of Nck1 had no effect on the proliferation of Jurkat T cells stimulated with either PHA or anti-T cell receptor (TCR) monoclonal antibody (C305). The reduced Nck1 expression in Jurkat cells was also associated with a reduced phosphorylation of extracellular regulated kinase (Erk)1 and Erk2 proteins upon CD3ε/CD28 stimulation. In conclusion, the decreased Nck1 protein in Jurkat T cells resulted in an impairment of TCR–CD3-mediated activation involving a defective Erk phosphorylation pathway.
Keywords: CD3, Jurkat T cells, Nck1, siRNA, T cell activation
Introduction
The central event of the adaptive immune system is when the T cell receptor (TCR)–CD3 complex recognizes and binds to a foreign peptide presented by a major histocompatibility complex (MHC) molecule, initiating a specific immune response. Numerous proteins and protein complexes constitute the T cell signalling pathway. Nck is a multi-functional adaptor protein of 47 kDa, containing three N-terminal Src homology 3 (SH3) domains and one C-terminal SH2 domain [1,2]. There are two types of human Nck proteins, namely Nck1/Nckα and Nck2/Nckβ (also known as Grb4). Nck1 and Nck2 exhibit a high degree of sequence homology in the interaction modules at 68% identical and 79% similar over their entire amino acid sequences [1,2]. Mice with deleted Nck1 and Nck2 die in the early embryonic stage, whereas single Nck1 or Nck2 deletion has no apparent phenotype [3]. Although this indicates some functional redundancy of the two isoforms, there are several reports that have indicated non-overlapping functions [4–6].
The modular architecture of the Nck provides a scaffold for a variety of protein–protein interactions, and more than 60 interacting partners have been identified [7]. To date, Nck1- or Nck2-specific downstream targets have rarely been reported, and in many instances the T cell activation pathways have not been attributed clearly to Nck1 or Nck2. Therefore, Nck1 and Nck2 are generally termed Nck in the literature. In many cell types, the SH2 domain binds to the phosphotyrosine residues of receptors or cytosolic proteins and the SH3 domains bind to proline-rich sequences (PRS) in corresponding target proteins, leading to the formation of multi-protein complexes. In T cells, Nck has been reported to act as a link between the TCR–CD3 complex and regulatory molecules that mediate signalling and reorganization of actin cytoskeletons [8–10].
Nck may be involved in intracellular T cell signalling both in an immunoreceptor tyrosine-based activation motif (ITAM)-requiring mechanism [11–14] and in a non-ITAM-requiring mechanism [15,16]. Nck–CD3ε interaction induced both interleukin (IL)-2 release and CD69 expression upon anti-CD3ε antibody (OKT3) stimulation in Jurkat T cells, suggesting that recruitment of Nck to the TCR–CD3 complex may be important for TCR signalling and immunological synapse formation. Nck-defective murine T cells fail to proliferate and to produce IL-2 upon stimulation with anti-CD3ε antibody, but do with phorbol 12-myristate 13-acetate (PMA) and ionomycin [17]. Nck deletion also impairs TCR-mediated calcium mobilization and ERK phosphorylation in activated T cells. Even these this data suggest the importance of Nck in optimum TCR-mediated activation, Szymczak et al. showed that interaction between the CD3ε PRS and Nck is not required for T cell development and function [18]. They observed no differences in CD69 expression after staphylococcal enterotoxin B (SEB) stimulation of T cells from mice expressing wild-type or mutant CD3ε PRS. Moreover, the Nck-CD3ε is also capable of down-regulating T cell activation by inhibiting CD3ε ITAM phosphorylation and/or by reducing TCR cell surface expression [19].
Because the function of Nck1 in T cell receptor signalling is still controversial, the aim of the present study was to assess the role of Nck1 in T cell activation and function. Nck1 knock-down Jurkat T cells were assessed for CD69 and IL-2 expression after stimulation with anti-CD3/anti-CD28 antibodies, anti-TCR antibody or phytohaemagglutinin (PHA)/phorbol myristate acetate (PMA). CD69 expression in Nck1 knock-down human CD4 T cells was assessed. In addition, the role of Nck1 in TCR activation involving the phosphorylation of ERK protein was assessed.
Materials and methods
Cell culture
Jurkat T cells (clone E6-1; American Type Culture Collection, Rockville, MD, USA) were grown in the RPMI-1640 medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco), 100 IU/ml penicillin/streptomycin (JRH Biosciences, Victoria, Australia), and 2 mm l-glutamine (JRH Biosciences) at 37°C in the humidified atmosphere with 5% carbon dioxide.
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats obtained from normal blood donors at the Blood Bank Centre of Naresuan University Hospital. The use of buffy coats was approved by the Ethical Committee of Naresuan University. Primary CD4 T cells were isolated from the PBMC using a magnetic affinity cell sorting (MACS) CD4+ isolation kit II (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Purity of the CD4 T cells was >90% as measured by flow cytometric analysis. CD4 T cells were maintained in the same culture medium as Jurkat T cells pretransfection.
Nck short interfering (si)RNA transfection
In order to knock-down Nck, Jurkat T cells were transiently transfected with control siRNA or siRNA specific for Nck1 mRNA (Invitrogen, Carlsbad, CA, USA) by electroporation. Approximately 2 × 105 cells per electroporation were collected, washed with RPMI-1640 and resuspended in solution R plus stealth RNAi™ siRNA duplexes specific for Nck1 (no. 983; Invitrogen). Negative controls were performed by using negative stealth siRNA low GC content (Invitrogen). Positive controls of transfection were performed using the green fluorescent protein reporter plasmid (pEGFP) (Invitrogen) 10 µl according to the manufacturer's instructions. To determine the concentration of siRNA, the first three most efficient conditions were used with 100 pmol or 50 pmol of Nck1 stealth siRNA and with the non-specific siRNA negative control. Cells were electroporated in a microporator pipette at various voltages (V), pulse widths (msec) and pulse numbers using the MicroPorator (Digital Bio Technology, Seoul, Korea). Primary CD4 T cells were transfected using double pulses of 15 ms at 2100 V. The transfected Jurkat T cells and primary CD4 T cells were subsequently cultured in RPMI-1640 plus FBS for 48 h. Transfection efficiency was determined by measuring the green fluorescence mean of each sample using fluorescence activated sell sorter (FACS)caliber flow cytometry and CellQuestPro software (Becton Dickinson, San Jose, CA, USA).
Western blotting
Normal and transfected Jurkat cells as well as primary CD4 T cells were lysed using M-PER mammalian protein extraction reagent (Pierce Biotechnology, Rockford, IL, USA). Protein concentration was determined by using a bicinchoninic acid (BCA) kit (Pierce Biotechnology), according to the manufacturer's instructions.
Samples (5 µg each) were fractionated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to an Immobilon-P membrane (Millipore, Bedford, MA, USA). After washing three times with Tris-buffered saline (TBS) and 0·1% Tween 20, the membrane was blocked with 25 ml of blocking solution (TBS, 1% bovine serum albumin (BSA), 10% FBS and 0·1%Tween 20) for 1 h. The membrane was incubated with 10 µl rabbit monoclonal antibodies raised against Nck1, Nck2 or β-actin (Cell Signaling Technology, Danver, MA, USA). Then, horseradish peroxidase-conjugated goat anti-rabbit antibody diluted 1:1000 was added for 1 h. Protein–antibody interaction was then visualized by chromogenic detection using a Western breeze chromogenic immunodetection system (Invitrogen). Molecular weights were determined by comparing with SeeBlueR Plus2 Pre-stained Standard (Invitrogen) and an equal loading was determined by the presence of β-actin.
Extracellular regulated kinase (ERK) phosphorylation
In order to determine the effect of Nck1 knock-down on ERK phosphorylation, normal and transfected Jurkat cells were stimulated with PHA plus PMA or anti-CD3ε/anti-CD28 monoclonal antibody (mAb). Briefly, the cells were washed in ice-cold PBS and resuspended in ice-cold RPMI-1640 containing 6 µg/ml PHA plus 1 ng/ml PMA or 10 µg/ml anti-CD3ε mAb plus 10 µg/ml anti-CD28 mAb before incubation at 37°C in a water bath to activate signalling for 10 min [20]. At the end of the incubation time, ice-cold RPMI-1640 was added rapidly into the cells and they were centrifuged quickly in a refrigerated microcentrifuge for 30 s [21]. Cell pellets were lysed in buffer containing 20 mm Tris-HCl (pH 8), 137 mm NaCl, 2 mm ethylenediamine tetraacetic acid (EDTA), 10% glycerol, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 1 mm phenylmethylsulphonyfluoride (PMSF), 500 µm sodium orthovanadate, 1 mm sodium fluoride (NaF) and 1% Triton X-100. The supernatant containing cytoplasmic proteins was collected after centrifugation at 15 300 g for 15 min at 4°C. Equal protein amounts were resolved to 8% SDS-PAGE and were then transferred onto polyvinylidene fluoride (PVDF) membrane. The membrane was probed with anti-phospho-ERK (The202/Tyr204; Upstate Biotechnology, Lake Placid, NY, USA), developed with the superSignal West Pico chemiluminescence kit (Pierce Biotechnology), and then observed under a CCD camera (ImageQuant LAS 4000; GE Healthcare Life Sciences, Pittsburgh, PA, USA). The same membrane was stripped and reprobed for β-actin.
Apoptosis assay
The level of apoptosis was determined using FITC Annexin-V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA), following the manufacturer's instructions. Normal and transfected Jurkat T cells were either not stimulated or stimulated with 300 ng/ml anti-TCR antibody (clone C305; Millipore, Temecula, CA, USA) for 24 h. Cells were examined using FACScalibur flow cytometer (Becton Dickinson) and CellQuestPro software.
Cell proliferation assays
A colorimetric 5-bromo-2′-deoxyuridine (BrdU) ELISA kit (Roche Diagnostics, Mannheim, Germany) was used to measure cell proliferation using the manufacturer's instructions. Normal and transfected Jurkat T cells (2 × 104 cells/well) were cultured in the presence or absence of 5 µg/ml PMA, 300 ng/ml anti-TCR monoclonal antibody (C305) or 100 U/ml IL-2 (Pierce Biotechnology) for 24 h. Absorbance was measured at 450 nm on a microplate reader (PerkinElmer Life Sciences, Downers Grove, IL, USA). All proliferation assays were performed in triplicate. Culture medium alone and cells incubated with peroxidase-labelled anti-BrdU in the absence of BrdU were used as controls for non-specific binding.
Detection of CD69
Untransfected and transfected Jurkat T cells and primary CD4 T cells were stimulated with 1 µg/ml PHA plus 10 ng/ml PMA or 10 µg/ml anti-CD3ε (mAb) (OKT3) plus 10 µg/ml anti-CD28 mAb (eBioscience, San Diego, CA, USA) for 24 h at 37°C before treatment with 20 mm ethylenediamine tetraacetic acid (EDTA; BD Biosciences, San Jose, CA, USA) for 15 min at room temperature. The cells were washed twice with phosphate-buffered saline (PBS) containing 0·5% FBS, fixed in 1% paraformaldehyde, washed with PBS containing 2·0% FBS and then incubated with phycoerythrin (PE)-conjugated mouse anti-human CD69 mAb or isotype control for 30 min at 4°C protected from light. Finally, cells were washed, resuspended in a staining buffer (PBS containing 0·5% BSA) and CD69 expression analysed by a FACScalibur using CellQuestPro software.
Measurement of IL-2 production
Normal and transfected Jurkat T cells (1 × 105 cells/ml) were incubated with 6 µg/ml PHA plus 1 ng/ml PMA as described previously [22] or with 10 µg/ml anti-CD3ε mAb plus 10 µg/ml anti-CD28 mAb at 37°C for 24 h; 100 µl cell-culture supernatants were collected, centrifuged and stored at −80°C until assayed. IL-2 levels were determined using a commercial enzyme-linked immunosorbent assay (ELISA) kit (R&D, Minneapolis, MN, USA) following the manufacturer's instructions. The optical density at 450 nm was read using a microplate reader (Perkin Elmer).
CD3 expression
Transfected Jurkat T cells were harvested, washed in ice-cold PBS, and then stained with fluorescein isothiocyanate (FITC)-conjugated anti-human CD3 antibody (OKT3; BioLegend, San Diego, CA, USA) or isotype control for 30 min at 4°C in the dark. Then, the cells were washed, resuspended in a staining buffer (PBS containing 0·5% BSA) and CD3 expression analysed using a FACScalibur (Becton-Dickinson).
Statistical analysis
All experiments were performed in duplicate, as stated otherwise, and were repeated on three different occasions. Statistical analyses were performed using spss software. All data were expressed as mean ± standard deviation (s.d.). Differences between experimental groups were analysed with analysis of variance (anova) followed by Dunnett's comparison test. The differences were considered to be significant when P < 0·05.
Results
Nck1 siRNA transfection in Jurkat T cells and primary CD4 T cells inhibited Nck1 protein expression
siRNA has been used successfully for targeting Nck in T cell blasts [23]. To determine the proper condition for transfection of Jurkat T cells, three electroporation parameters, which were voltage, pulse width and pulse number (1700 V, 20 ms, 1; 1600 V, 10 ms, 3; or 1400 V, 20 ms, 2) and two concentrations (100 pmol or 50 pmol) of siRNA against Nck1 and non-specific siRNA negative control were used. As shown in Fig. 1a, the Nck1 protein expression was reduced in Nck1 siRNA-transfected groups at 48 h after transfection. The greatest reduction of Nck1 protein level appeared when Jurkat T cells were electroporated at 1600 V, 10 ms, and 3 with 50 pmol of Nck1 siRNA compared with blank control and non-specific siRNA negative control. The Nck1 siRNA knock-down was specific because it did not affect expression of the Nck2 and unrelated protein β-actin (Fig. 1a). A similar reduction of Nck1 protein level was also seen in Nck1 knock-down primary CD4 T cells (Fig. 1b).
Fig. 1.
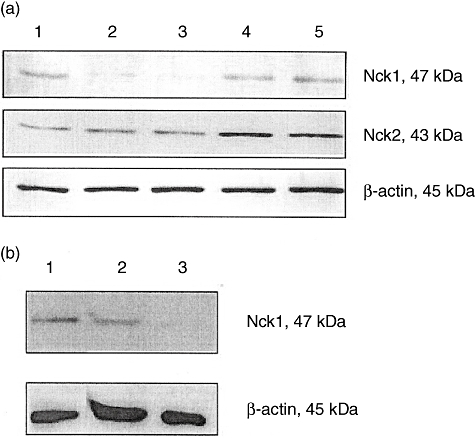
Expression of Nck1 protein was reduced after Nck1-specific short interfering (si)RNA transfection. E6-1 Jurkat T cells (a) and primary CD4 T cells (b) were transfected with Nck1-specific or negative control siRNA for 48 h. Cell extracts were subjected to Western blot analysis with anti-Nck1 monoclonal antibody (mAb) and anti-β-actin mAb. (a) Lane 1, blank control; lane 2, transfection with 100 pmol Nck1 siRNA; lane 3, transfection with 50 pmol Nck1 siRNA; lane 4, transfection with 100 pmol non-specific siRNA negative control; lane 5, transfection with 50 pmol non-specific siRNA negative control. (b) Lane 1, untransfected CD4 T cells; lane 2, negative control siRNA; lane 3, si RNA Nck1.
Loss of Nck1 did not induce apoptosis in Jurkat T cells
It has been shown that Nck translocates to the nucleus upon DNA damage. This accumulation is essential for the activation of downstream regulators of the DNA damage cascade and cell-cycle arrest [24]. To test whether or not the loss of Nck1 was associated with apoptosis, annexinV-FITC and PI staining were performed using flow cytometry (Fig. 2).
Fig. 2.
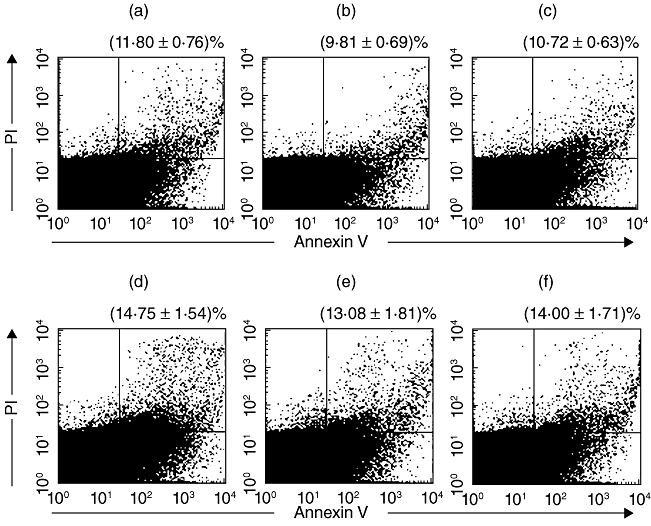
Nck1 knock-down did not induce apoptosis in Jurkat T cells. E6-1 Jurkat T cells were transfected with Nck1-specific short interfering (si)RNA (a), untransfected (b) or transfected with non-specific siRNA negative control (c). Jurkat T cells were also stimulated with anti-T cell receptor (TCR) monoclonal antibody (mAb) and there was no difference in the % apoptosis of T cell receptor (TCR)-stimulated Jurkat cells transfected with Nck1-specific siRNA (d), untransfected (e) or transfected with non-specific siRNA negative control (f). The lower left zone showed the viable cells (annexinV-/PI-), the upper left zone represented necrosis cells (annexinV-/PI+), the lower right zone showed early apoptosis (annexinV+/PI-) and the upper right zone showed the late apoptosis (annexinV+/PI+). The combination of the upper and lower left zones represented the total apoptotic cells. Shown are mean percentage ± standard deviation of apoptotic cells from three experiments.
The percentage of apoptotic cells in the Nck1 siRNA-transfected group was 11·80% ± 0·76%, which did not differ from that of the non-transfected control group (9·81% ± 0·69%) and the non-specific siRNA negative control group (10·72% ± 0·63%) (Fig. 2a–c). Similar, the percentage of apoptotic cells in transfected Jurkat cells stimulated with anti-TCR antibody was 14·75% ± 1·54%, which also differed from the untransfected control (13·08% ± 1·81%) and the negative siRNA control (14·00% ± 1·71%) groups (Fig. 2d–f). This finding indicated that a cytotoxic effect of siRNA treatment was minimal and Jurkat T cell survival was independent of Nck1 expression.
Loss of Nck1 did not impair Jurket T cell proliferation
To determine whether Nck1 was involved in proliferation of T cells, normal and transfected Jurkat T cells were cultured for 24 h in the presence of PHA, anti-TCR antibody and IL-2. The proliferative response in the Nck1 siRNA-transfected cells did not differ significantly from that in non-specific siRNA-transfected cells (Fig. 3). The results suggested that Nck1 was not required for the proliferation of Jurkat T cells stimulated with PHA, anti-TCR antibody or IL-2.
Fig. 3.
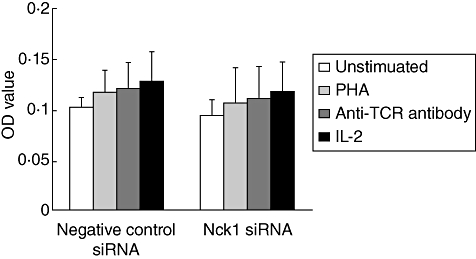
Nck1 was not required for PHA- and IL-2-induced Jurkat T cell proliferation. Nck1 short interfering (si)RNA-transfected or negative control siRNA-transfected Jurkat T cells were unstimulated (□), stimulated with phytohaemagglutinin (PHA) ( ), anti-T cell receptor (TCR) antibody (
), anti-T cell receptor (TCR) antibody ( ) or interleukin (IL)-2 (
) or interleukin (IL)-2 ( ) for 24 h and proliferation measured using 5-bromo-2′-deoxyuridine (BrdU). Values are presented as mean ± standard deviation from three experiments. OD: optical density read at 450 nm.
) for 24 h and proliferation measured using 5-bromo-2′-deoxyuridine (BrdU). Values are presented as mean ± standard deviation from three experiments. OD: optical density read at 450 nm.
Nck1 is required for TCR–CD3-mediated activation
To assess the influence of Nck1 down-regulation on the expression levels of the CD69, negative control and Nck1 knock-down Jurkat T cells were stimulated with PHA/PMA or anti-CD3ε/anti-CD28 antibodies. In the Nck1 siRNA-transfected group, CD69 was detectable by both types of stimulation. After PHA/PMA stimulation, Nck1 siRNA-transfected cells had similar CD69 expression compared with the negative control siRNA group. However, after anti-CD3ε/anti-CD28 antibody stimulation, Nck1 siRNA-transfected cells had significantly lower CD69 expression compared with the negative control siRNA group (Fig. 4a,b). Similarly, CD69 expression was decreased in Nck1 knock-down primary CD4 T cells (Fig. 4c,d). In addition, knock-down of Nck1 did not affect CD3 expression Jurkat T cells (Fig. 5). These results indicated clearly that CD69 expression upon TCR–CD3-mediated stimulation was affected by Nck1 down-regulation.
Fig. 4.
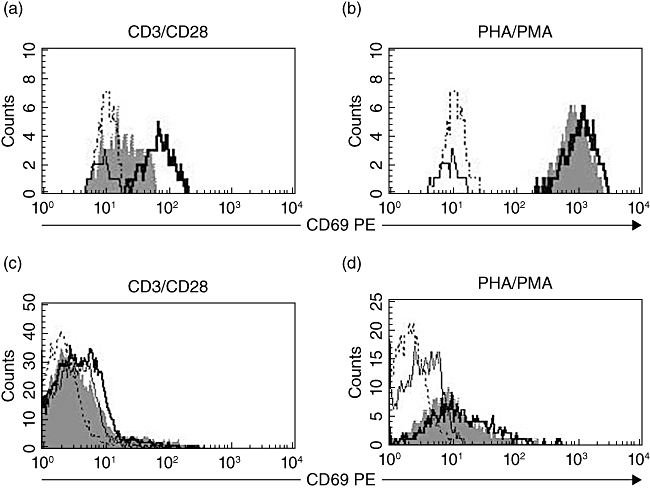
Nck1-regulated expression of CD69 in T cell receptor (TCR)–CD3-mediated activation. Jurkat T cells and primary human CD4 T cells were stimulated with anti-CD3ε/anti-CD28 antibodies (a,c) or phytohaemagglutinin (PHA)/phorbol myristate acetate (PMA) (b,d) for 24 h. Histograms show CD69 expression of untransfected Jurkat T cells and CD4 T cells. Dotted line, unstimulated; bold solid line, negative control short interfering (si)RNA-transfected; grey histogram, Nck1 siRNA-transfected. Each cell population was stained with anti-CD69 phycoerythrin (PE) and isotype control antibodies (solid line) and was analysed by flow cytometry for CD69 expression.
Fig. 5.
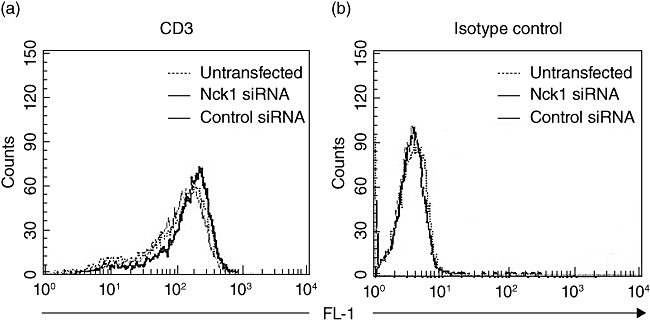
CD3 expression of Nck1 knock-down Jurkat cells. Jurkat T cells were transfected with Nck1 short interfering (si)RNA or negative control siRNA.
Knock-down of Nck1 resulted in the reduction of IL-2 production
The impact of the Nck1 knock-down on IL-2 production induced by PHA/PMA or anti-CD3ε/anti-CD28 mAb was examined. Consistent with the CD69 results, Jurkat T cells expressing reduced levels of Nck1 failed to respond to anti-CD3ε/anti-CD28 antibody stimulation by producing IL-2 when compared with cells transfected with the negative control siRNA (Fig. 6). In contrast, IL-2 production upon PHA/PMA stimulation from Nck1 siRNA-transfected cells was comparable to that from negative control siRNA-transfected cells. Because stimulation with PHA/PMA bypasses the TCR signalling machinery, these results suggested that Nck1 reduction specifically induced an impairment of TCR–CD3-mediated T cell activation and function.
Fig. 6.
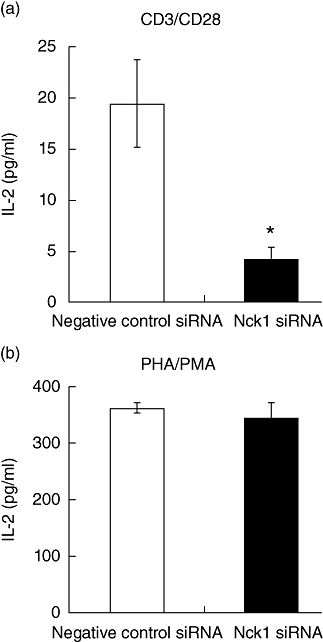
Nck1 was required for interleukin (IL)-2 production by Jurkat T cells. Negative control short interfering (si)RNA-transfected (□) or Nck1 siRNA-transfected ( ) Jurkat T cells were stimulated with anti-CD3ε/anti-CD28 antibodies (a) or phytohaemagglutinin (PHA)/phorbol myristate acetate (PMA) (b) for 24 h. Results represent mean ± standard deviation of three experiments. *P < 0·05.
) Jurkat T cells were stimulated with anti-CD3ε/anti-CD28 antibodies (a) or phytohaemagglutinin (PHA)/phorbol myristate acetate (PMA) (b) for 24 h. Results represent mean ± standard deviation of three experiments. *P < 0·05.
Knock-down of Nck1 resulted in an impaired ERK phosphorylation
The importance of Nck1 on ERK phosphorylation was assessed. The tranfected and untransfected Jurkat cells were stimulated with PHA plus PMA or anti-CD3ε/anti-CD28 mAb. Then, the phosphorylation of ERK was determined and the result clearly showed that knock-down of Nck1 was associated with a defective ERK phosphorylation (Fig. 7).
Fig. 7.
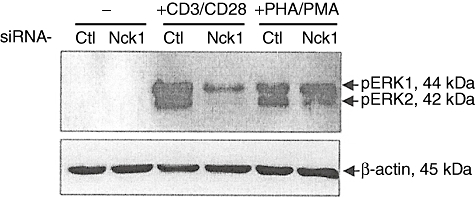
Extracellular regulated kinase (ERK) phosphorylation was decreased in Nck1 knock-down Jurkat cells. Jurkat T cells were treated with specific Nck1 short interfering (si)RNA (Nck1) or control siRNA (Ctl). After 48 h of cultivation, cells were subsequently stimulated with 10 µg/ml anti-CD3ε monoclonal antibody (mAb) plus 10 µg/ml anti-CD28 mAb or with 6 µg/ml phytohaemagglutinin (PHA) plus 1 ng/ml phorbol myristate acetate (PMA) or none (−) for 10 min at 37°C. Cytoplasmic proteins were immunoblotted with anti-phospho-Erk1/2 (Thr202/Tyr204, Thr185/Tyr187) antibody.
Discussion
Nck1 is an important regulator of the cytoskeletal reorganization and the formation of the immune synapse between T cells and antigen-presenting cells (APCs). Two different models for the association of Nck with the TCR–CD3 complex have been proposed. In the ITAM-requiring pathway, Nck is recruited to the membrane proximal site via interaction with the SH-2-domain containing 76 kDa leucocyte protein (SLP-76), forming a trimolecular complex of SLP–76/Vav1/Nck which is involved in the Wiskott–Aldrich syndrome protein (WASP)-dependent actin cytoskeletal rearrangement [9,25]. In the non-ITAM-requiring pathway, the N-terminal SH3 domain of Nck binds to a PRS in CD3ε that is exposed due to a conformational change in the TCR complex after TCR ligation [15].
In this present report, Nck1 siRNA was transfected into Jurkat T cells used as a cell model to knock-down Nck1 protein in order to investigate the role of Nck1 in T cell activation and function. siRNA, 20–25-nucleotide double- stranded RNA, mediates sequence-specific elimination of complementary mRNA [26–28]. For delivering siRNA into the T cell line, an effective and cost-saving method is the capillary electroporation system [29]. A partial knock-down of Nck by electroporated siRNA transfection can yield 50–60% reduction of Nck protein level in PHA-stimulated T cell blasts [23]. The Nck1 protein expression was inhibited after transfection with Nck1-specific siRNA, suggesting that the transfection was successful. CD69 expression represents one of the earliest available markers for T cell activation. CD69 acts as a potent signal-transmitting receptor on lymphocytes. It is involved in cytokine gene regulation and cell migration upon lymphocyte activation [30]. The reduced IL-2 secretion by Nck1 knock-down Jurkat T cells stimulated with anti-CD3ε/anti-CD28 antibodies was accompanied by lower expression of CD69. This is not unexpected, because surface expression of CD69 is a marker of Ras activation, which is critical for proliferation and transcription of the IL-2 gene [31]. Nck is known to interact with Grb2 and SOS GTP exchange factor [10,32,33], leading to enhanced transcription from a Ras-dependent reporter gene. Ras signalling affects many cellular functions, including cell proliferation. This reduced IL-2 secretion was unlikely to result from cell apoptosis, as no difference in the percentage of apoptotic cells was found between the transfected and control groups. However, the reduced CD69 and IL-2 expression in this present study was associated with defective ERK phosphorylation. This finding supports previous studies that Nck deletion causes reduced ERK phosphorylation and cytokine production by T cells [17,34].
An early study has indicated that recruitment of Nck to the PRS of CD3ε is essential for optimal T cell activation and synapse formation [15]. Nck-CD3ε interaction-inducing antibodies was a better inducer for both IL-2 release and CD69 expression than non-inducing antibodies for Jurkat T cells. A recent study employing a highly sophisticated mouse model has shown that Nck-defective T cells fail to proliferate upon stimulation with anti-CD3ε antibody but not with PMA and ionomycin [17]. Furthermore, culture supernatants of T cells from Nck-deficient mice exhibited a reduction in IL-2 content compared to untreated mice after CD3ε/CD28 stimulation. This suggested that the unmodified response of Nck-deficient T cells to PMA and ionomycin stimulation was due to the signalling pathway that bypassed the TCR signalling apparatus, indicating the involvement of Nck in proximal TCR signalling. The present study is in line with those results, as CD3ε/CD28 stimulation was unable to induce CD69 expression and IL-2 secretion in Nck1 knock-down cells. However, the proliferation and CD69 expression of Nck1 knock-down cells stimulated with PHA and PHA/PMA, respectively, were not changed. The stimulation with PHA and PMA bypassed TCR signals by directly triggering lymphocyte-specific protein tyrosine kinase (Lck) and protein kinase C (PKC) activity [35]. Thus, Nck1 impairs TCR–CD3-mediated CD69 expression.
Functional analysis has shown that there are no differences observed in proliferation and CD69 expression after treatment with strong stimulators in T cells from mice expressing wild-type or CD3ε PRS mutation [18]. The data suggest that interaction of CD3ε PRS with Nck might not be essential for T cell development and function. However, in addition to CD3ε, interaction of Nck with SLP-76 or linker for activation of T cells (LAT) [9,25] induces WASP-mediated actin cytoskeleton rearrangement and T cell activation [36–38]. Nck1 may cause recruitment of these regulatory molecules to the plasma membrane for enhancing proximal TCR signal potency. Conflicting data have indicated that the Nck-CD3ε is also capable of down-regulating T cell activation by inhibiting CD3ε ITAM phosphorylation and/or reducing TCR cell surface expression [19]. In this context, the SH 3-1 domain of Nck2 binding to an uncommon PxxDY motif of CD3ε blocks the tyrosine phosphorylation of the ITAM of CD3ε and affects internalization signal of TCR. This discrepancy may be due to differences in the isoforms of Nck being tested. Perhaps Nck1 could bind to PxxDY motif to a lesser degree and subsequent ITAM phosphorylation may not be affected.
In summary, Nck1-specific siRNA transfection did not induce Jurkat T cell apoptosis. Nck1 appears to be important in TCR–CD3-mediated activation, as evidenced by inhibition of T cell function when its expression is down-regulated. Nck1 could bridge TCR activation to downstream signalling mechanism, at least in part, through the ERK phosphorylation cascade. In this scenario, formation of Nck-mediated signalling complex may result in the propagation of proximal TCR signals. The reduced Nck1 expression in our present study led to an impairment CD69 expression and IL-2 production by T cells. Taken together, these data suggest that the Nck1 adaptor protein is a crucial regulator of TCR–CD3-mediated activation and function.
Acknowledgments
The authors thank Miss Kwansuda Supalap and Mr Sang Sri-ampai, Biomedical Research Unit, Office of Research, Faculty of Medicine, Naresuan University, for their technical assistance. This work was supported by Naresuan University Research Grant (fiscal year 2011) and the Faculties of Dentistry and Medicine, Naresuan University (research grants from income of fiscal year 2009). The authors thank Professor Wolfgang W. Schamel, University of Freiburg and Max-Planck Institute for Immunobiology, Germany, for his useful suggestions on the manuscript. We also thank Dr Roger F. Searle, School of Medical Sciences Education Development, Newcastle University, UK, for kindly editing the English language. Mr Jatuporn Ngoenkam and Miss Pussadee Paensuwan are currently supported by the Royal Golden Jubilee PhD programme of the Thailand Research Fund.
Disclosure
The authors declare no conflict of interest.
References
- 1.Lehmann JM, Riethmuller G, Johnson JP. Nck, a melanoma cDNA encoding a cytoplasmic protein consisting of the src homology units SH2 and SH3. Nucleic Acids Res. 1990;18:1048. doi: 10.1093/nar/18.4.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park D. Cloning, sequencing, and overexpression of SH2/SH3 adaptor protein Nck from mouse thymus. Mol Cells. 1997;7:231–36. [PubMed] [Google Scholar]
- 3.Bladt F, Aippersbach E, Gelkop S, et al. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm-derived embryonic structures and for regulating the cellular actin network. Mol Cell Biol. 2003;23:4586–597. doi: 10.1128/MCB.23.13.4586-4597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen M, She H, Kim A, Woodley DT, Li W. Nckbeta adapter regulates actin polymerization in NIH 3T3 fibroblasts in response to platelet-derived growth factor bb. Mol Cell Biol. 2000;20:7867–880. doi: 10.1128/mcb.20.21.7867-7880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan S, Chen M, Woodley D, Li W. Nckbeta adapter controls neuritogenesis by maintaining the cellular paxillin level. Mol Cell Biol. 2007;27:6001–11. doi: 10.1128/MCB.01807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pramatarova A, Ochalski PG, Chen K, et al. Nck beta interacts with tyrosine-phosphorylated disabled 1 and redistributes in Reelin-stimulated neurons. Mol Cell Biol. 2007;23:7210–221. doi: 10.1128/MCB.23.20.7210-7221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lettau M, Pieper J, Janssen O. Nck adapter proteins: functional versatility in T cells. Cell Commun Signal. 2009;7:1. doi: 10.1186/1478-811X-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunnell SC, Hong DI, Kardon JR, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–75. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott–Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15:5725–31. doi: 10.1128/mcb.15.10.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buday L, Wunderlich L, Tamas P. The Nck family of adapter proteins of actin cytoskeleton. Cell Signal. 2002;14:723–31. doi: 10.1016/s0898-6568(02)00027-x. [DOI] [PubMed] [Google Scholar]
- 11.Alberola-Ila J, Takaki S, Kerner JD, Perlmutter RM. Differential signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1997;15:125–54. doi: 10.1146/annurev.immunol.15.1.125. [DOI] [PubMed] [Google Scholar]
- 12.Bubeck Wardenburg J, Pappu R, Bu JY, et al. Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity. 1998;9:607–16. doi: 10.1016/s1074-7613(00)80658-5. [DOI] [PubMed] [Google Scholar]
- 13.Galisteo ML, Chernoff J, Su YC, Skolnik EY, Schlessinger J. The adaptor protein Nck links receptor tyrosine kinases with the serine-threonine kinase Pak1. J Biol Chem. 1996;271:20997–1000. doi: 10.1074/jbc.271.35.20997. [DOI] [PubMed] [Google Scholar]
- 14.Lin J, Weiss A. T cell receptor signaling. J Cell Sci. 2001;114:243–44. doi: 10.1242/jcs.114.2.243. [DOI] [PubMed] [Google Scholar]
- 15.Gil D, Schamel WW, Montoya M, Sanchez-Madrid F, Alarcon B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–12. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- 16.Gil D, Schrum AG, Alarcon B, Palmer E. T cell receptor engagement by peptide–MHC ligands induces a conformational change in the CD3 complex of thymocytes. J Exp Med. 2005;201:517–22. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy E, Togbe D, Holdorf AD, et al. Nck adaptors are positive regulators of the size and sensitivity of the T-cell repertoire. Proc Natl Acad Sci USA. 2010;107:15529–34. doi: 10.1073/pnas.1009743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szymczak AL, Workman CJ, Gil D, et al. The CD3epsilon proline-rich sequence, and its interaction with Nck, is not required for T cell development and function. J Immunol. 2005;175:270–75. doi: 10.4049/jimmunol.175.1.270. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi K, Yang H, Ng E, et al. Structural and functional evidence that Nck interaction with CD3epsilon regulates T-cell receptor activity. J Mol Biol. 2008;380:704–16. doi: 10.1016/j.jmb.2008.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tailor P, Tsai S, Shameli A, et al. The proline-rich sequence of CD3ε as an amplifier of low-avidity TCR signaling. J Immunol. 2008;181:243–55. doi: 10.4049/jimmunol.181.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawasdikosol S. Detecting tyrosine-phosphorylated proteins by Western blot analysis. Curr Protoc Immunol. 2010;89:11.3.1–11. doi: 10.1002/0471142735.im1103s89. [DOI] [PubMed] [Google Scholar]
- 22.Sasagawa M, Cech NB, Gray DE, Elmer GW, Wenner CA. Echinacea alkylamides inhibit interleukin-2 production by Jurkat T cells. Int Immunopharmacol. 2006;6:1214–21. doi: 10.1016/j.intimp.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Lettau M, Qian J, Linkermann A, et al. The adaptor protein Nck interacts with Fas ligand: guiding the death factor to the cytotoxic immunological synapse. Proc Natl Acad Sci USA. 2006;103:5911–16. doi: 10.1073/pnas.0508562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kremer BE, Adang LA, Macara IG. Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell. 2007;130:837–50. doi: 10.1016/j.cell.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol. 2005;6:80–9. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- 26.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 27.Hammond SM, Bernstein E, Beach D, Hannon GJ An RN. A-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–6. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 28.Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 29.Kim JA, Cho K, Shin MS, et al. A novel electroporation method using a capillary and wire-type electrode. Biosens Bioelectron. 2008;23:1353–60. doi: 10.1016/j.bios.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Shiow LR, Rosen DB, Brdickova N, et al. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 31.Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity. 1998;9:617–26. doi: 10.1016/s1074-7613(00)80659-7. [DOI] [PubMed] [Google Scholar]
- 32.Hu Q, Milfay D, Williams LT. Binding of NCK to SOS and activation of ras-dependent gene expression. Mol Cell Biol. 1995;15:1169–74. doi: 10.1128/mcb.15.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada S, Pessin JE. Interaction between Src homology (SH) 2/SH3 adaptor proteins and the guanylnucleotide exchange factor SOS are differently regulated by insulin and epidermal growth factor. J Biol Chem. 1996;271:25533–8. doi: 10.1074/jbc.271.41.25533. [DOI] [PubMed] [Google Scholar]
- 34.Roy E, Togbe D, Holdorf A, et al. Fine tuning of the threshold of T cell selection by the Nck adaptors. J Immunol. 2010;185:7518–26. doi: 10.4049/jimmunol.1000008. [DOI] [PubMed] [Google Scholar]
- 35.Smith-Gavin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badour K, Zhang J, Siminovitch KA. Involvement of the Wiskott–Aldrich syndrome protein and other actin regulatory adaptors in T cell activation. Semin Immunol. 2004;16:395–407. doi: 10.1016/j.smim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Snapper SB, Rosen FS, Mizoguchi E, et al. Wiskott–Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9:81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Shehabeldin A, da Cruz LAG, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott–Aldrich syndrome protein-deficient lymphocytes. J Exp Med. 1999;190:1329–42. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]