

Abstract
Members of the European Society for Immunodeficiencies (ESID) and other colleagues have updated the multi-stage expert-opinion-based diagnostic protocol for non-immunologists incorporating newly defined primary immunodeficiency diseases (PIDs). The protocol presented here aims to increase the awareness of PIDs among doctors working in different fields. Prompt identification of PID is important for prognosis, but this may not be an easy task. The protocol therefore starts from the clinical presentation of the patient. Because PIDs may present at all ages, this protocol is aimed at both adult and paediatric physicians. The multi-stage design allows cost-effective screening for PID of the large number of potential cases in the early phases, with more expensive tests reserved for definitive classification in collaboration with a specialist in the field of immunodeficiency at a later stage.
Keywords: diagnostic protocol, immunological evaluation, primary immunodeficiency, update
Introduction
In 2006, the Clinical Working Party of the European Society for Immunodeficiencies (ESID) published a multi-stage diagnostic protocol suitable for all doctors [1]. The protocol started from the clinical presentation of both paediatric and adult patients. Many primary immunodeficiency diseases (PIDs) present in childhood, but the most common clinically significant PID, ‘common variable immunodeficiency disorders’ (CVID), has a peak onset in the second and third decades of life. The multi-stage design allowed timely identification of potential PID by all doctors, while more costly elaborate tests were reserved for definitive classification at a later stage, in collaboration with an immunologist specialized in the field of immunodeficiency and a specialized laboratory.
Since 2006, many new PIDs have been identified; the International Union of Immunological Societies (IUIS) Expert Committee on Primary Immunodeficiencies published updates of their classification of PIDs, the latest in 2009 [2]. We have therefore updated the 2006 diagnostic protocol, using the IUIS 2009 paper and its references as the basis for clinical disease entities of PIDs. Additionally, a PubMed search was performed from 2007 onwards; several papers discussing the recognition of potential PID in everyday practice were found [3–13], and all were based mainly on expert opinion. All ESID members received an invitation to participate in this effort. [Searchstrategy, papers selected for algorithms designed for identification of potential PID patients in everyday clinical practice published in English in international papers: 1. ‘Related citations’ for the original paper [1] (three relevant hits, references [3–5]); ‘Immunologic Deficiency Syndromes/*classification[MeSH] NOT HIV NOT AIDS NOT HTLV NOT Simian’ (no additional relevant hits); ‘Immunologic Deficiency Syndromes/*diagnosis[MeSH] NOT HIV NOT AIDS NOT HTLV NOT Simian’ (eight additional relevant hits, including the original ESID paper, references [1,4,6–11]); two additional papers suggested by contributors (references [12,13]).]
While the general outline of the diagnostic protocol has remained the same, novel PIDs have been incorporated. The body of knowledge concerning PIDs has expanded considerably; therefore, possible diagnoses are now presented separately from the clinical protocols. Because evidence supporting diagnostic decisions is still limited, the protocols are based largely on consensus of expert opinions.
Do not forget PID and pick up the signs; it is life-saving
Considering the possibility of a PID is the key to the diagnosis. Unfortunately, the awareness of PIDs among professionals is low, as PIDs are considered rare and complex diseases. However, the incidence of PIDs ranges – depending on the disease – from 1:500 for often asymptomatic immunoglobulin (Ig)A deficiency to 1:500 000 [14,15]; all PIDs taken together may be as frequent as 1:2000 [16]. Like any other diagnostic process, symptoms from the history (Table 1a), signs on physical examination (Table 1b) and baseline blood tests (Table 1c) should alert any physician to the possibility of PID in children and adults, even though they are unfamiliar with the precise possible diagnosis.
Table 1.
Symptoms and signs that could point to potential PID
| (a)History |
|---|
| The hallmark of PID: infection history |
| Recurrent (probably) bacterial infections (more frequent than expected at the patient's age) |
| More than one severe infection (e.g. meningitis, osteomyelitis, pneumonia, sepsis) |
| Infections that present atypically, are unusually severe or chronic or fail regular treatment (especially if i.v. antibiotics are needed) |
| Abscess of internal organ |
| Recurrent subcutaneous abscesses (especially in children) |
| Prolonged or recurrent diarrhoea |
| Any infection caused by an unexpected or opportunistic pathogen (e.g. pneumocystis) |
| Severe or long-lasting warts, generalized mollusca contagiosa |
| Extensive candidiasis, recurrent oral thrush in children >1 year |
| Complications of vaccination (disseminated BCG or varicella infection, paralytic polio, rotavirus infection) |
| Remember the family history! |
| PID in the family; familial occurrence of similar symptoms (affected males related by the female line, or another clear pattern of inheritance) |
| Unexplained early infant deaths, deaths due to infection |
| Consanguinity in the (grand) parents (known or suspected) |
| Autoimmune disease or haematological malignancy in several family members |
| Other*(could point to PID, but may not) |
| Aplasia or hypoplasia of thymus (X-ray) |
| Angioedema |
| Auto-immune disease (especially auto-immune cytopenias, SLE) |
| Bleeding tendency |
| Congenital cardiac anomalies (mainly conotruncal defects) |
| Chronic diarrhoea, malabsorption, pancreatic insufficiency |
| Delayed separation of umbilical cord (>4 weeks) |
| Delayed shedding of primary teeth |
| Developmental delay (progressive) |
| Difficult-to-treat obstructive lung disease |
| Eczema, dermatitis (severe, atypical) |
| Failure to thrive (child) or wasting (adult) |
| Graft-versus-host reaction after blood transfusion, or mother-to-child (infant) engraftment |
| Granulomas |
| Haemolysis |
| Hypersensitivity to sunlight |
| Hypocalcaemic seizures |
| Inflammatory bowel disease (atypical) |
| Malignancy (mainly lymphoma) |
| Non-allergic oedema |
| Poor wound healing; scarring |
| Recurrent fever |
| Rib or other skeletal anomalies (X-ray) |
| Thymoma |
| Unexplained bronchiectasis, pneumatoceles, interstitial lung disease |
| Vasculitis |
| (b)Physical examination | |
|---|---|
| Skin and appendages | Abnormal hair or teeth. Eczema. Neonatal erythroderma. (Partial) albinism. Pale skin. Incontinentia pigmenti. Nail dystrophy. Extensive warts or molluscae. Congenital alopecia. Vitiligo. Petechiae (early onset, chronic). Cold abscesses. Telangiectasia. Absence of sweating |
| Oral cavity | Gingivostomatitis (severe). Periodontitis. Aphthae (recurrent). Giant oral ulcers. Thrush. Dental crowding. Conical incisors. Enamel hypoplasia. Persistent deciduous teeth |
| Eyes | Retinal lesions. Telangiectasia |
| Lymphoid tissue | Absence of lymph nodes and tonsils. Lymphadenopathy (excessive). Asplenia. Organomegaly (liver, spleen) |
| Neurological | Ataxia. Microcephaly. Macrocephaly |
| Other | Angioedema (without urticaria). Digital clubbing. Dysmorphism. Stunted growth or disproportional growth |
| (c) Baseline blood tests | |
|---|---|
| Haematology | Granulocytopenia, lymphocytopenia, or neutrophilia. Eosinophilia. Giant or absent granules in phagocytes. Howell-Jolly bodies |
| Thrombocytopenia. Small platelets | Anaemia (aplastic, haemolytic) |
| Chemistry | Hypocalcaemia. Hypofibrinogenaemia. Hypertriglyceridaemia. Hyperferritinaemia. Low CRP and other inflammatory parameters during infections |
In alphabetical order. BCG: bacille Calmette–Guérin; CRP: C-reactive protein; i.v.: intravenous; PID: primary immunodeficiency; SLE: systemic lupus erythematosus.
This is important, as successful treatment of a child with severe PID such as severe combined immunodeficiency (SCID) is dependent upon rapid recognition [17]. Non-immunologists such as general paediatricians play a vital role. Leucocyte differential and immunoglobulin isotype levels enable detection in most cases; these can be performed in many hospitals. Less urgent, but still important if future organ damage and decreased quality of life and life-span are to be prevented, is the timely recognition of late-onset as well as less pronounced forms of PID in older children and adults [18]. Non-immunologists such as primary care physicians, general paediatricians, pulmonologists and ear, nose and throat (ENT) specialists play an important role here. Common examples are antibody deficiencies such as CVID and specific anti-polysaccharide antibody deficiency (SPAD) [19,20]. These generally present with recurrent respiratory infections, by far the most common clinical presentation of PID. Confusingly, this clinical presentation is often encountered in everyday practice, especially in young children, but also in older children and adults in any pulmonology or ENT service. Most of these patients do not have PID. However, when more than one pneumonia occurs, bronchiectasis is present, the infections fail to clear with conventional treatment or continue to occur when a young child grows older, immunological investigations are needed, and consultation of an immunologist is highly recommended.
Family history is a vital clue to the diagnosis of PID, as although patients with recurrent infections do not often have PID, this becomes much more likely when it ‘runs in the family’. This also holds true for adult patients who can present with late-onset forms of disease.
Pattern recognition is the key to identification
PIDs tend to present in one of eight different clinical presentations (Table 2, column 1), determined by the underlying pathology of the disease (Table 3). Either initially or during follow-up some patients may show features of more than one clinical presentation, which can be confusing. Encountered pathogens (Table 2, column 2) can help to clarify the pattern, because specific immunological defects will lead to particular patterns of infection [21]. Associated features (Table 2, column 3) and age of presentation can also help. Most PIDs present in childhood but due to, for example, hypomorphic mutation, typical paediatric disease may present later [22]. CVID is the most common PID presenting in adulthood [5].
Table 2.
Pattern recognition gives direction to the diagnostic process
 |
Table 3.
In-depth differential diagnosis of the clinical presentations
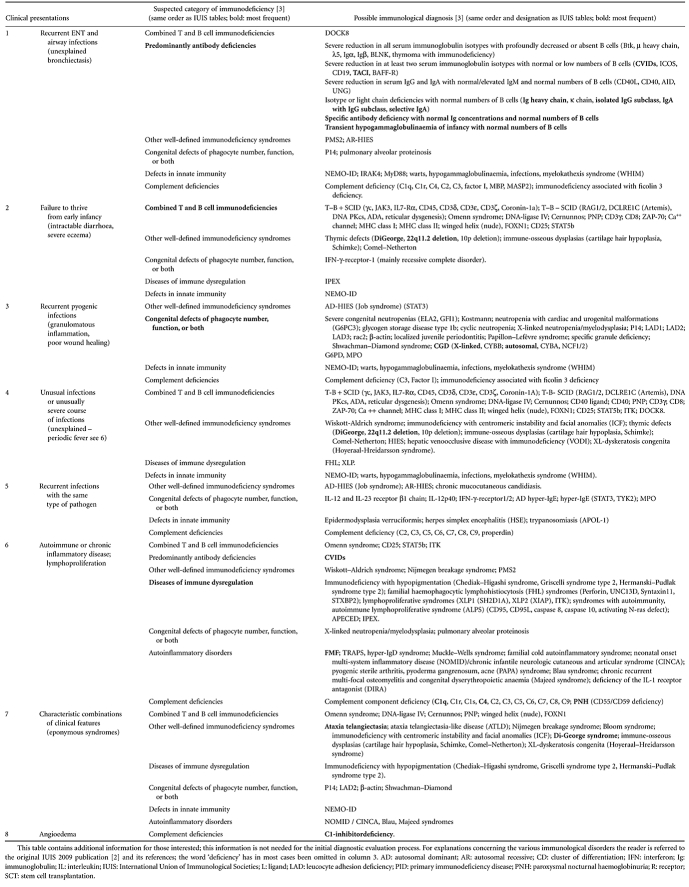 |
In column 5 of Table 2, directions towards the appropriate multi-stage diagnostic protocol for suspected immunodeficiency (Figs 1–3; Tables 4 and 5) are given, using the clinical presentation as the starting-point. In the protocols, severe defects are ruled out first with widely available screening tests (step 1; Figs 1–3). Less severe forms of PID can be diagnosed later (steps 2–4; Figs 1–3), after more frequent non-immunological diseases have been ruled out (Table 2, column 4). It is essential to use age-matched reference values [23–25] to avoid misinterpreting test results, especially in young infants who normally have a relative lymphocytosis and a high level of maternal immunoglobulins in their blood. Beyond the first step of each protocol, and in all cases where a severe PID such as SCID is suspected, timely collaboration with an immunologist to decide on further diagnostic steps and to aid with the interpretation of the results is highly recommended.
Fig. 1.
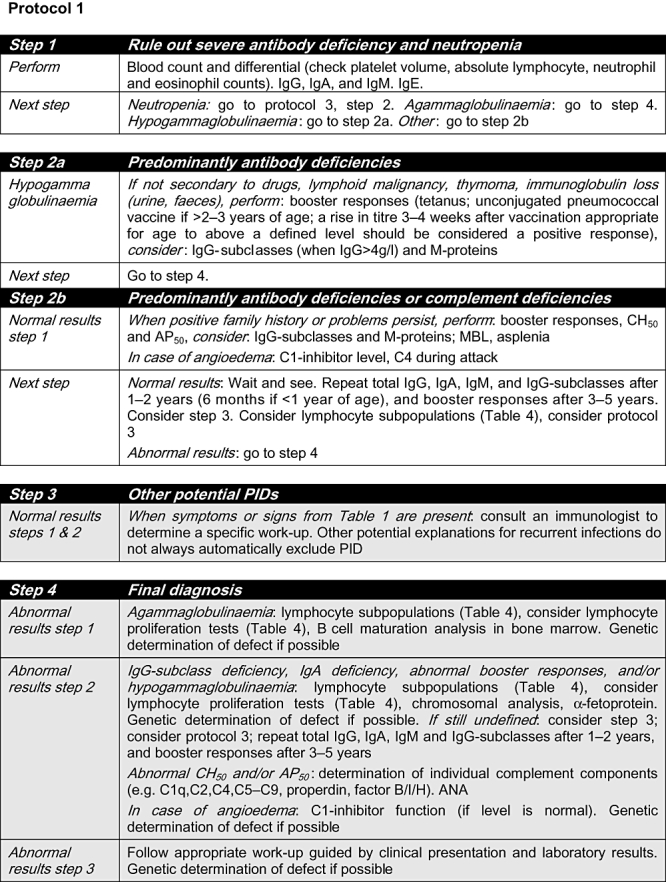
Protocol 1. ANA: anti-nuclear antibody; C: complement; CD: cluster of differentiation; Ig: immunoglobulin; MBL: mannose binding lectin; PID: primary immunodeficiency. Grey shading: consultation with an immunologist is highly recommended.
Fig. 3.
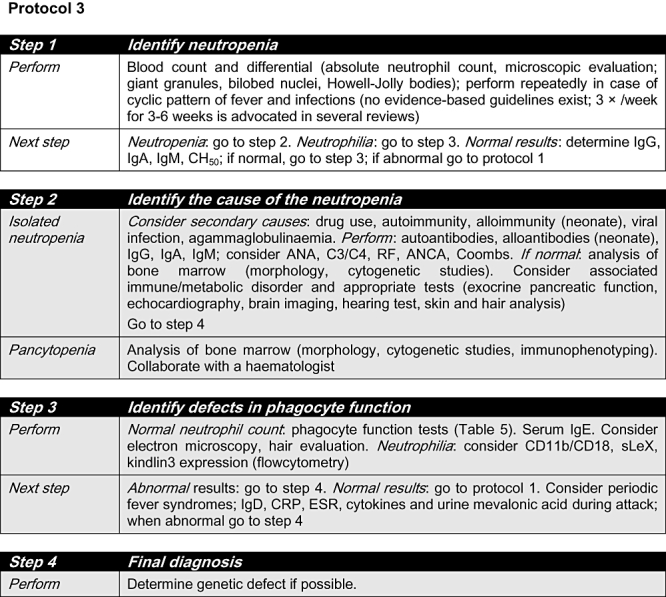
Protocol 3. ANA: anti-nuclear antibody; ANCA: anti-neutrophil cytoplasmic antibodies; C: complement component; CD: cluster of differentiation; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; GCSF: granulocyte–colony-stimulating factor; Ig: immunoglobulin; RF: rheumatoid factor; sLeX: sialyl-Lewis X. Grey shading: consultation with an immunologist is highly recommended.
Table 4.
Basic protocol for in vitro determination of lymphocyte subpopulations and function
| (a) Determine the absolute count of the following lymphocyte subpopulations, and compare the results with age-matched reference values | |
| CD3+ | T lymphocytes |
| CD3+/CD4+ | Helper-T lymphocytes |
| CD3+/CD4+/CD27+/CD45RA+ | Naive helper-T lymphocytes |
| CD3+/CD8+ | Cytotoxic T lymphocytes |
| CD3+/HLA-DR+ | Activated T lymphocytes |
| CD3+/TCR-αβ+/CD4-/CD8- | ‘Double-negative’ TCR-αβ+ T cells |
| CD3+/TCR-γδ+ | TCR-γδ+ subset of T lymphocytes |
| CD19+ or CD20+ | B lymphocytes |
| CD19+/CD27+/IgM-/IgD- | Switched memory-B lymphocytes |
| CD3-/CD16+ and/or CD56+ | NK cells |
| (b) Determine the uptake of [3H]-thymidine (or CFSE or activation markers) and compare the results with, preferably, age-matched controls after stimulation with: | |
| Mitogens (e.g. PHA, PMA + ionomycin, PWM) | |
| Consider monoclonal antibodies (e.g. CD2 ± CD28, CD3 ± CD28) | |
| Antigens (e.g. tetanus, after booster vaccination; PPD, candida) | |
| Consider allogeneic cells | |
Part (a) can be performed in many hospitals, part (b) is performed in specialized laboratories only. For correct interpretation of the results, collaboration with an immunologist specialized in immunodeficiency and/or a specialized laboratory is highly recommended. CD: cluster of differentiation; CFSE: carboxyfluorescein succinimidyl ester; HLA: human leucocyte antigen; NK: natural killer; PHA: phytohaemagglutinin; PMA: phorbol myristate acetate; PWM: pokeweed mitogen; TCR: T cell receptor.
Table 5.
Protocol for determination of granulocyte function
| (a) Oxidative burst and flow cytometry |
| Flow cytometric analysis using dihydrorhodamine (DHR) |
| Nitroblue tetrazolium test (NBT) to a stimulant (PMA, LPS) |
| Chemoluminescence test |
| Immunophenotyping (CD18, CD11b, sLeX, kindlin3) |
| (b) Chemotaxis, granule contents, bacterial killing, phagocytosis |
| Migration to a chemoattractant (e.g. fMLP) |
| Immunohistochemistry of granule contents, electron microscopy |
| Bacterial killing (e.g. of Staphylococcus aureus) |
| Phagocytosis (e.g. zymosan uptake, FITC-conjugated latex beads) |
Part (a) can be performed in many hospitals, part (b) is performed in specialized laboratories only. For correct interpretation of the results, collaboration with an immunologist specialized in immunodeficiency and/or a specialized laboratory is highly recommended. CD: cluster of differentiation; FITC: fluorescein isothiocyanate; FMLP: formyl-met-leu-phe, a bacterial peptide; LPS: lipopolysaccharide; PMA: phorbol myristate acetate.
Fig. 2.
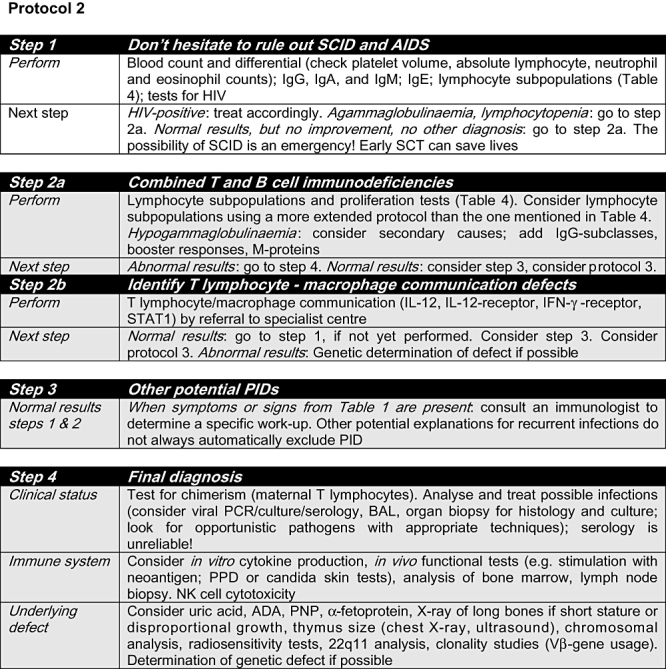
Protocol 2. ADA: adenosine deaminase; AIDS: acquired immunodeficiency syndrome; BAL: bronchoalveolar lavage; CD: cluster of differentiation; HIV: human immunodeficiency virus; Ig: immunoglobulin; IFN: interferon; IL: interleukin; NK: natural killer; PID: primary immunodeficiency; PNP: purine nucleoside phosphorylase; PPD: purified protein derivative; SCID: severe combined immunodeficiency; SCT: stem cell transplantation; STAT: signal transducers and activators of transcription. Grey shading: consultation with an immunologist is highly recommended.
Secondary immunodeficiencies present in a similar fashion to PIDs. Human immunodeficiency virus (HIV) infection occurs much more frequently in some parts of the world. Also, drugs, malignancies and diseases which cause protein and/or lymphocyte loss may cause secondary immunodeficiency; this is more common than unrecognized PID in adults [5]. It is important to eliminate these possibilities before making a definitive diagnosis of PID.
Many new PIDs have been identified in the past decades, and more are likely in the near future, so this multi-stage diagnostic protocol will need to be revised from time to time.
Take-home messages
The key to detect a PID is to consider the possibility.
PIDs almost always present with one or more of eight clinical presentations; these can be used as the starting-point to enter the appropriate diagnostic protocol.
SCID is an emergency.
Timely recognition of antibody deficiency prevents future organ damage.
If PID is suspected or runs in the family, delay live-attenuated vaccinations and do not postpone immunological investigations.
Use age-matched reference values to avoid misinterpretation of immunological test results.
Acknowledgments
This work was supported in part by the NIHR Biomedical Research Centres funding scheme (K. Gilmour) and BMBF PIDNET (C. Klein), which enabled them to spend time on the multi-stage diagnostic protocol for suspected immunodeficiency. P. Soler Palacín gratefully acknowledges Fabiola Caracseghi for her useful help in reviewing the manuscript.
Contributors to the study
E. de Vries, Department of Paediatrics, Jeroen Bosch Hospital 's-Hertogenbosch, the Netherlands; A. Alvarez Cardona, Primary Immunodeficiency Investigation Unit, Instituto Nacional de Pediatría, Universidad Autónoma de México, Ciudad de Mexico, Mexico; A. H. Abdul Latiff, Division of Clinical Immunology and Paediatrics School of Medicine and Health Sciences, Monash University, Sunway Campus, Malaysia; R. Badolato, Clinica Pediatrica dell'Università di Brescia c/o Spedali Civili, Brescia, Italy; N. Brodszki, Department of Paediatric Immunology, Lund University Hospital, Lund, Sweden; A. J. Cant, Great North Children's Hospital, Newcastle upon Tyne, UK; J. Carbone, Department of Immunology, Gregorio Marañon Hospital, Madrid, Spain; J. T. Casper, Medical College of Wisconsin, Department of Paediatrics, Immunology/BMT, MACC Fund Research Center, Milwaukee, USA; P. Čižnár, 1st Paediatric Department, Comenius University Medical School, Children' University Hospital, Bratislava, Slovakia; A. V. Cochino, Department of Paediatrics, University of Medicine and Pharmacy ‘Carol Davila’, Bucharest, Romania; B. Derfalvi, 2nd Department of Paediatrics, Immunology–Rheumatology–Nephrology Unit, Semmelweis University Budapest, Budapest, Hungary; G. J. Driessen, Department of Paediatric Infectious Disease and Immunology, Erasmus MC, University Medical Center Rotterdam, Rotterdam, the Netherlands; R. Elfeky, Department of Pediatrics,Ain Shams University, Cairo, Egypt; D. El-Ghoneimy, Department of Paediatric Allergy & Immunology, Faculty of Medicine, Ain Shams University, Cairo, Egypt; T. Espanol, Immunology Unit, University Hospital Vall d'Hebron, Barcelona, Spain; A. Etzioni, Meyer's Children Hospital, Faculty of Medicine, Technion, Haifa, Israel; E. Gambineri, Department of Sciences for Woman and Child's Health, University of Florence, ‘Anna Meyer’ Children's Hospital, Florence, Italy; K. Gilmour, Camelia Botnar Laboratories, Great Ormond Street for Children NHS Trust, London, UK; L. I. Gonzalez-Granado, Immunodeficiencies Unit, Department of Paediatrics, Hospital 12 octubre, Madrid, Spain; M. N. Guseva, Consulting Center of Saint-Petersburg Pediatric Medical Academy, Saint-Petersburg, Russia; M. H. Haverkamp, Department of Infectious Diseases, Leiden University Medical Center, Leiden, the Netherlands; M. Helminen, Department of Paediatric Infectious Diseases, University Hospital of Tampere, Tampere, Finland; M. Hönig, Department of Paediatrics, University Hospital Ulm, Ulm, Germany; M. G. Kanariou, Specific Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, ‘Aghia Sophia’ Children's Hospital, Athens, Greece; M. Kirschfink, Institute of Immunology, University of Heidelberg, Heidelberg, Germany; C. Klein, University Children's Hospital, Dr von Haunersches Kinderspital, Munich, Germany; T.W. Kuijpers, Division of Paediatric Hematology, Immunology and Infectious diseases, Emma Children's Hospital, Academic Medical Center, Amsterdam, the Netherlands; N. Kutukculer, Department of Pediatrics, Division of Pediatric Immunology, Ege University, Izmir, Turkey; B. Martire, Dipartimento di Biomedicina dell'Eta′ Evolutiva, Policlinico Università di Bari, Bari, Italy; I. Meyts, Department of Paediatrics, University Hospitals Leuven, Leuven, Belgium; T. Niehues, Helios Klinikum Krefeld; Krefeld Immunodeficiency Centre KIDZ, Krefeld, Germany; C. Pignata, Department of Paediatrics, ‘Federico II’ University, Naples, Italy; S. M. Reda, Department of Paediatric Allergy and Immunology, Faculty of Medicine, Ain Shams University, Cairo, Egypt; E. D. Renner, University Children's Hospital, Ludwig Maximilians Universität, München, Germany; N. Rezaei, Molecular Immunology Research Centre and Research Group for Immunodeficiencies, Children's Medical Center, Tehran University of Medical Sciences, Tehran, Iran; M. Rizzi, Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany; M. A. Sampalo Lainz, Department of Immunology, Puerta del Mar Universitary Hospital, Cadiz, Spain; R. B. Sargur, Department of Immunology, Northern General Hospital, Sheffield, UK; A. Sediva, Institute of Immunology, University Hospital Motol, Prague, Czech Republic; M. G. Seidel, Paediatric Immunology Outpatient Clinic, St Anna Children's Hospital, Vienna, Austria; S. L. Seneviratne, Department of Clinical Immunology, St Mary's Hospital and Imperial College, London, UK; P. Soler-Palacín, Pediatric Infectious Diseases and Immunodeficiencies Unit, Vall d'Hebron University Hospital, Barcelona, Spain; A. Tommasini, Laboratory of Immunopathology, Institute for Maternal and Child Health, IRCCS Burlo Garofolo, Trieste, Italy; K. Warnatz, Centre of Chronic Immunodeficiency, University Hospital of Freiburg, Freiburg, Germany.
Disclosure
None.
References
- 1.De Vries E, Clinical Working Party of the European Society for Immunodeficiencies (ESID) Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. 2006;145:204–14. doi: 10.1111/j.1365-2249.2006.03138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies. Notarangelo LD, Fischer A, Geha RS, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samarghitean C, Ortutay C, Vihinen M. Systematic classification of primary immunodeficiencies based on clinical, pathological, and laboratory parameters. J Immunol. 2009;183:7569–75. doi: 10.4049/jimmunol.0901837. [DOI] [PubMed] [Google Scholar]
- 4.Ballow M. Approach to the patient with recurrent infections. Clin Rev Allergy Immunol. 2008;34:129–40. doi: 10.1007/s12016-007-8041-2. [DOI] [PubMed] [Google Scholar]
- 5.Azar AE, Ballas ZK. Evaluation of the adult with suspected immunodeficiency. Am J Med. 2007;120:764–8. doi: 10.1016/j.amjmed.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Wood P, UK Primary Immunodeficiency Network. Primary antibody deficiencies: recognition, clinical diagnosis and referral of patients. Clin Med. 2009;9:595–9. doi: 10.7861/clinmedicine.9-6-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliveira JB, Fleisher TA. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(Suppl 2):S297–305. doi: 10.1016/j.jaci.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roxo Júnior P. Primary immunodeficiency diseases: relevant aspects for pulmonologists. J Bras Pneumol. 2009;35:1008–17. doi: 10.1590/s1806-37132009001000010. [DOI] [PubMed] [Google Scholar]
- 9.Samarghitean C, Vihinen M. Bioinformatics services related to diagnosis of primary immunodeficiencies. Curr Opin Allergy Clin Immunol. 2009;9:531–6. doi: 10.1097/ACI.0b013e3283327dc1. [DOI] [PubMed] [Google Scholar]
- 10.Johnston SL. Clinical immunology review series: an approach to the patient with recurrent superficial abscesses. Clin Exp Immunol. 2008;152:397–405. doi: 10.1111/j.1365-2249.2008.03640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleisher TA. Evaluation of suspected immunodeficiency. Adv Exp Med Biol. 2007;601:291–300. doi: 10.1007/978-0-387-72005-0_31. [DOI] [PubMed] [Google Scholar]
- 12.Slatter MA, Gennery AR. Clinical immunology review series: an approach to the patient with recurrent infections in childhood. Clin Exp Immunol. 2008;152:389–96. doi: 10.1111/j.1365-2249.2008.03641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cassimos DC, Liatsis M, Stogiannidou A, Kanariou MG. Children with frequent infections: a proposal for a stepwise assessment and investigation of the immune system. Pediatr Allergy Immunol. 2010;21:463–73. doi: 10.1111/j.1399-3038.2009.00964.x. [DOI] [PubMed] [Google Scholar]
- 14.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–19. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 15.Fischer A. Human primary immunodeficiency diseases. Immunity. 2007;27:835–45. doi: 10.1016/j.immuni.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497–502. doi: 10.1007/s10875-007-9103-1. [DOI] [PubMed] [Google Scholar]
- 17.Rosen FS. Severe combined immunodeficiency: a pediatric emergency. J Pediatr. 1997;130:345–6. [PubMed] [Google Scholar]
- 18.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–60. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 19.Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev. 2009;22:396–414. doi: 10.1128/CMR.00001-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood PM. Primary antibody deficiency syndromes. Curr Opin Hematol. 2010;17:356–61. doi: 10.1097/MOH.0b013e328338f69e. [DOI] [PubMed] [Google Scholar]
- 21.Stiehm ER, Chin TW, Haas A, Peerless AG. Infectious complications of the primary immunodeficiencies. Clin Immunol Immunopathol. 1986;40:69–86. doi: 10.1016/0090-1229(86)90070-x. [DOI] [PubMed] [Google Scholar]
- 22.Nelson KS, Lewis DB. Adult-onset presentations of genetic immunodeficiencies: genes can throw slow curves. Curr Opin Infect Dis. 2010;23:359–64. doi: 10.1097/QCO.0b013e32833bc1b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanariou M, Petridou E, Liatsis M, Revinthi K, Mandalenaki-Lambrou K, Trichopoulos D. Age patterns of immunoglobulins G, A & M in healthy children and the influence of breast feeding and vaccination status. Pediatr Allergy Immunol. 1995;6:24–9. doi: 10.1111/j.1399-3038.1995.tb00253.x. [DOI] [PubMed] [Google Scholar]
- 24.Comans-Bitter WM, de Groot R, van den Beemd R, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997;30:388–93. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 25.Huck K, Feyen O, Ghosh S, Beltz K, Bellert S, Niehues T. Memory B-cells in healthy and antibody-deficient children. Clin Immunol. 2009;131:50–9. doi: 10.1016/j.clim.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Ming JE, Stiehm ER, Graham JM., Jr Syndromic immunodeficiencies: genetic syndromes associated with immune abnormalities. Crit Rev Clin Lab Sci. 2003;40:587–642. doi: 10.1080/714037692. [DOI] [PubMed] [Google Scholar]