

Abstract
The ready access to commercially available multiplex assays and the importance of inflammation in disease pathogenesis has resulted in an abundance of studies aimed at identifying surrogate biomarkers for different clinically important questions. Establishing a link between a biomarker and disease pathogenesis, however, is quite complex, and in some instances this complexity is compounded by post-translational modifications and the use of immunoassays that do not always discriminate between the different forms of the same protein. Herein, we provide a detailed description of an assay system that has been established to discriminate the agonist form of CXCL10 from the NH2-terminal truncated form of the molecule generated by dipeptidylpeptidase IV (DPP4) cleavage. We demonstrate the utility of this assay system for monitoring agonist and antagonist forms of CXCL10 in culture supernatant, patient plasma and urine samples. Given the important role of CXCL10 in chronic inflammatory diseases and its suggested role as a predictive marker in managing patients with chronic hepatitis C, asthma, atopic dermatitis, transplantation, tuberculosis, kidney injury, cancer and other diseases, we believe that our method will be of general interest to the research and medical community.
Keywords: BCG, bladder cancer, chemokines/monokines, hepatitis C virus
Introduction
CXCL10 (also known as interferon-induced protein-10 or IP-10) is a member of the CXC chemokine family with proinflammatory and anti-angiogenic properties [1,2]. It has also been demonstrated to suppress bone marrow colony formation, regulate T cell maturation and modulate expression of various adhesion molecules [3–5]. It is an inducible chemokine, and expression is stimulated at the transcription level by interferons (IFN types I, II and III) acting on IFN-stimulated response element (ISRE) or gamma-activated site (GAS) promoter elements; or by tumour necrosis factor (TNF)-α and related nuclear factor (NF)-κB activating pathways [1]. In addition, engagement of host pattern recognition receptors [e.g. Toll-like receptor (TLR) or retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs)] may directly activate CXCL10 transcription via phosphorylation of IFN regulatory factor 3 (IRF3). Many cell types have been reported to secrete CXCL10, including endothelial cells, hepatocytes, keratinocytes, fibroblasts, mesangial cells, astrocytes and immune cells [6–12].
Chemokine signalling is an important component of the regulatory circuit governing the host immune response to infection, stress or tissue damage. Indeed, many studies have evaluated a role for CXCL10 and it has been reported to be induced in many viral infections [e.g. hepatitis C virus (HCV), HBV, herpes simplex virus 1 (HSV)-1, Chikungunya, enterovirus, human rhinovirus, Japanese encephalitis][13–15]); bacterial and parasite infections (e.g. shigella, tuberculosis, leshmania, malaria) [16,17]; allergy and autoimmune diseases (e.g. asthma, systemic lupus erythemytosus, autoimmune arthropathies, dermatitis) [18]; and cancer (e.g. melanoma, renal, cervical) [1,4,19–21]. In a subset of these diseases, CXCL10 has been reported to be a prognostic or diagnostic marker with potential use in the management of patients. For example, several independent studies have demonstrated that baseline levels of CXCL10 are predictive of the failure to respond to HCV treatment [22,23]. It is also an important component of predictive algorithms that are being validated for use in monitoring acute kidney injury and lung inflammation [24–27].
CXCR3 is the receptor for CXCL10, and is shared by two other alpha-chemokines: CXCL9 [also known as monokine induced by IFN-γ (MIG)] and CXCL11 [also known as IFN-inducible T cell chemoattractant (ITAC)][28,29]. CXC-chemokines bind to their G-protein-coupled receptors and mobilize intracellular Ca++, which results in receptor internalization and the initiation of signalling pathways that facilitate chemotaxis as well as other defined biological activities. Binding to, and activation of, the receptor is thought to be a two-step process. First, the core of the ligand binds the outer surface of the receptor; a second step involves the reorientation of the flexible N-terminal tail of the protein, triggering its binding to a distinct domain within the receptor [30,31]. Post-secretion modification of CXCL10 has been described, including C-terminal cleavage by metal metalloproteinase 9 (MMP9 or gelatinase B) and citruillination by peptidylarginine deiminase (PAD), both of which leave the protein in an agonist state [32–34]. Also reported is the N-terminal cleavage of two amino acids by members of the X-prolyl dipeptidyl peptidase (DPP) family, the most characterized being dipeptidylpeptidase IV (DPP4 or CD26) [35,36]. DPP4 has been shown to cleave several chemokines, including members of the α-chemokine family (CXCL4, CXCL10, CXCL11) [37,38]. Importantly, DPP4 truncation of CXCL10 generates a dominant negative form of the protein, which is capable of binding CXCR3 but does not induce signalling [22,38].
Given the importance of chemokines and, in particular, CXCL10, in inflammatory processes, it is surprising how little information is available concerning the different biologically relevant forms of the molecular. One major challenge has been the development of quantitative assays that detect chemokines in biological fluids at physiologically and pathologically relevant concentrations. Currently available assays do not discriminate between the active and NH2-terminus cleaved forms of CXCL10. We generated and validated a multiplex immunoassay that employs specific antibodies to differentiate the native form of CXCL10 (agonist) and the NH2-truncated form generated by DPP4 cleavage (antagonist). We also provide new data relevant for the study of HCV patients, monitoring CXCL10 in culture supernatants and plasma; and for the monitoring of bladder cancer patients receiving BCG therapy, monitoring CXCL10 in urine samples.
Patients and methods
Samples and patients
HCV-infected patients
Sustained virological responders (SVR) were defined as individuals absent of HCV RNA for longer than 6 months after termination of therapy; those who remained persistently infected are referred to as chronically infected HCV patients. Plasma from patients was collected in BD P700 tubes, specialized vacutainers that are preloaded with ethylenediamine tetraacetic acid (EDTA) and DPP4 inhibitor, thus preventing extra-corporeal cleavage of CXCL10. Samples were obtained as part of the study protocol C07-11 approved by the INSERM clinical investigation department with ethical approval from the ‘Comité de Protection des Personnes Ile-de-France II’ (45 rue des Saints-Pères 75006 Paris). All HCV patients were tested for human immunodeficiency virus (HIV), HBV and human T cell lymphotrophic virus (HTLV) and found to be negative. Additional patient data are presented in Table 1.
Table 1.
Hepatitis C virus (HCV) patient characteristics
| Age, years median (range) | Male gender, n (%) | Extensive fibrosis or cirrhosis, n (%) | GT 1 or 4†, n (%) | ALT × ULN,‡ median (range) | |
|---|---|---|---|---|---|
| Chronic (n = 15) | 55 (40–80) | 8 (53) | 7 (47) | 15 (100) | 1·6 (0·9–5) |
| SVR (n = 15) | 53 (43–65) | 7 (47) | 4† (29) | 14† (100) | 0·46 (0·3–1·1) |
Missing data for one patient in each group.
ALT levels reported as fold change compared to upper limit of normal (ULN). SVR: sustained virological responders.
Bladder cancer patients
Urine samples were obtained from patients with a histologically confirmed diagnosis of superficial bladder cancer who were recommended to receive bacilli Calmette–Guérin (BCG) therapy according to existing guidelines. All patients studied were enrolled into an observational study (2007/40) approved by the Institut Pasteur clinical investigation department with ethical approval from the ‘Comité de Protection des Personnes Ile-de-France II’. As part of the standard of care, all patients received six weekly intravesical instillations of 81 mg live BCG (Connaught strain Immucyst®). During the fifth installation, urine was collected before BCG and 4, 8 and 24 h after BCG installation. Urine was centrifuged, filtered and stored at −80°C until testing was performed. Both study protocols conformed to the ethical guidelines of the Declaration of Helsinki. All patients provided informed consent.
Antibody generation
Production of monoclonal single-chain antibodies specific for the agonist form and also the neo-epitope created by DPP4 cleavage of CXCL10 were generated using the Human Combinatorial Antibody Library Technology (AbD Serotec; http://www.morphosys.com/technologies/morphosys-technologies). The diversity of the antibody repertoire is established through the use of one of seven heavy-chain genetically fused to one of six light-chain variable region genes, giving rise to 42 frameworks in the master library. Highly variable complementarity determining regions (CDRs) are inserted into these genetic frameworks to mimic the antibody repertoire and allowing for the generation of 45 × 1012 functional human antibody specificities in Fab format. Serial panning was performed, alternating between selection of the captured antibodies bound to the peptide or protein of interest; and depletion of those antibodies that bind to the alternate form of the NH2-terminal peptide or CXCL10 protein. Peptides corresponding to the NH2-terminal 7 amino acids of DPP4 cleaved CXCL10 (LSRTVRC) and peptide corresponding to the native protein (VPLSRTVRC) were conjugated to bovine serum albumin (BSA) and transferrin (TRF) for use. DPP4 cleaved and native protein was also employed in the screen in order to ensure that the antibody recognized the epitope within the folded protein conformation. The primary screens for CXCL10 antibodies were carried out in 384 plates. Briefly, 384-well Maxisorp plates (Nalge Nunc International, Rochester, NY, USA) were coated with 20 µl/well of antigen at 5 µg/ml in phosphate-buffered saline (PBS) overnight at 4°C. After PBS washes, the wells were saturated with 100 µl 5% non-fat dry milk in PBS–Tween 0·05% for 1–2 h at room temperature. Twenty µl of the tested antibodies were added to each well, diluted at 2 µg/ml in PBS–Tween for 1 h; 20 µl/well of anti-Fab2-AP conjugate (AbD Serotec, Raleigh, NC, USA) was used as secondary antibody. After the last washing step, 20 µl of AttoPhos (Roche Applied Science, Indianapolis, IN, USA) was added to each well for detection. Values obtained with unspecific antigens were used for the calculation of background (data not shown).
Direct enzyme-linked immunosorbent assay (ELISA) for secondary screening was performed in 96-well Maxisorp plates (Nunc) coated with 50 µl of a peptide solution at 10 µg/ml, or with 50 µl of a CXCL10 protein solution at 0·4 µg/ml, in PBS overnight at 4°C. After PBS washes, the wells were saturated with 200 µl 0·05% Tween-20, 1% BSA in PBS for 1–2 h at room temperature. One hundred µl of the tested antibodies were added to each well, diluted at 2 µg/ml in 1% BSA blocking buffer. Revelation was performed with anti-human Fab horseradish peroxidise (HRP)-conjugated antibody (AbD Serotec). After the last washing step, 100 µl of tetramethylbenzidine (TMB) substrate (Sigma-Aldrich, St Louis, MO, USA) was added to each well and 100 µl of HCl 1 N to stop the reaction. Plates were read in a Labsystems Multiskan MS device (Thermofisher Scientific, Waltham, MA, USA).
Generation of NH2-truncated CXCL10
To generate CXCL10 (3–77), recombinant human CXCL10 at a final concentration of 0·5 mg/ml (Peprotech, Rocky Hill, NY, USA) was incubated with recombinant DPP4, at a final concentration of 1·25 U/ml (Sigma) in a 100 mm Tris-HCL pH 8 solution for 2 h at 37°C. Cleavage was monitored by Western blot analysis (Fig. 1a). To resolve proteins with a two amino acid difference, we utilized a 15% bis-acryalmide gel, 48 cm plates with a 1-mm spacer, and ran the gel in Tris-Tricine buffer. Hybond-P membrane (GE Healthcare, formerly Amersham Biosciences, Piscataway, USA) and a semi-dry Western blot transfer procedure (EBU-4000 CBS Scientific, Del Mar, CA, USA) was used. The membrane was saturated with a 1% Tween-20, 1% BSA Tris-buffered saline (TBS) solution. To detect CXCL10, a mouse anti-human monoclonal antibody (clone 33036; R&D Systems Inc, Minneapolis, MN, USA) was used with an anti-mouse immunoglobulin (Ig)G (H + L) coupled to HRP (Vector Laboratories, Burlingame, CA, USA) and enhanced chemiluminescence (ECL)-plus detection system (Amersham). Confirmatory analysis was performed using surface-enhanced desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry (Fig. 1b). One µl of the digested product was spotted onto an H4 protein chip. Arrays were incubated and washed as per the manufacturer's instructions and analysed using the ProteinChip System Series 4000 (Biorad, Hercules, CA, USA). The range of molecular weights (MWs) investigated was from 0 to 20 000 Da. The focus mass was set at 8·5 kDa. Data were analysed using Ciphergen Express Software.
Fig. 1.
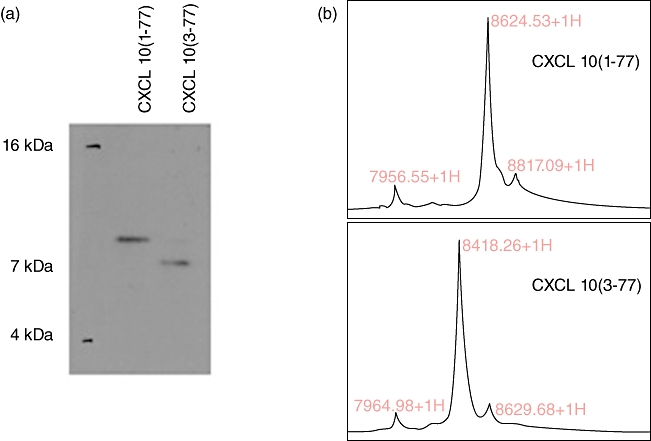
NH2 truncation of recombinant CXCL10 by dipeptidylpeptidase IV (DPP4). Human recombinant CXCL10 (1–77) was incubated in vitro with recombinant DPP4 to obtain CXCL10 (3–77). (a) Native or digested protein (150 ng) was loaded onto a 15% bis-acrylamide gel for Western blot analysis; (b) 0·5 µg was spotted on an H4 column for mass spectrometry analysis. The difference in molecular weight between CXCL10 (1–77) and CXCL10 (3–77) is consistent with a two amino acid truncation.
Assays for discriminating agonist CXCL10 and NH2-truncated CXCL10
Upon identification of candidate antibodies [clone 12010 for CXCL10 (1–77) and 9852 for CXCL10 (3–77)], sandwich ELISAs were established. Maxisorp plates (Nunc) were coated with 50 µl of a 2 µg/ml solution of the capture antibody in PBS overnight at 4°C. The wells were saturated with 200 µl 0·05% Tween-20, 1% BSA in PBS for 1 to 2 h at room temperature. A standard curve was obtained by diluting recombinant CXCL10 (1–77) or CXCL10 (3–77) in saturation buffer and incubated at room temperature for 2 h. Biotinylated goat polyclonal anti-CXCL10 was used for detection (R&D Systems) (0·25 µg/ml as a final concentration), incubated for 1·5 h min at room temperature. Finally, HRP–streptavidin (BD Biosciences Pharmingen, San Diego, CA, USA) followed by 100 µl of TMB substrate (Sigma) was used to reveal a signal; 100 µl of HCl 1 N was used to stop the reaction. Plates were read in a Labsystems Multiskan MS (Thermofisher Scientific, Waltham, MA, USA) device.
Whole blood stimulation
TruCulture™ tubes were preloaded with 300 units recombinant IFN-α2 (Intron-A; Schering-Plough, Kirkland, QC, Canada) or media alone. The tubes contained Na heparin and a total volume of 2 ml, prior to blood sampling. The TruCulture syringe-based stimulation system is designed to avoid the introduction of errors inherent in traditional culture-based models in which leucocytes are manipulated ex vivo (e.g. transportation of samples to the laboratory, Ficoll separation). An important feature of the collection system is that they are designed to allow only 1 ml of blood to be drawn into the syringe; thereafter, the syringe locks. The blood/medium mixture was incubated for 20 h at 37°C. At the end of this incubation, the supernatant of the culture tube was separated from the cells by inserting a plunger into the syringe. Subsequently the culture supernatant was aliquoted and stored at −80°C until analysis. IFN-α2 TruCulture stability testing was preformed (data not shown) and all collections were performed within the optimal window for use.
Analysis of DPP4 activity
The activity of DPP4 was measured using a luciferase-based assay (DPP4-Glo protease Assay; Promega), following the supplier's instructions. The DPP4-Glo™ Protease Assay is a homogeneous, luminescent assay that measures DPP4 activity. The DPP4-Glo™ assay provides a proluminescent DPP4 substrate, Gly-Pro-aminoluciferin, in a buffer system optimized for DPP4 and luciferase activities. Following DPP4 cleavage, the substrate for luciferase (aminoluciferin) is released, resulting in the luciferase reaction and the production of light. The signal is proportional to the amount of DPP4 activity present. Human serum was serially diluted between 0·25% and 2·5% in 10 mm Tris–HCL pH 8 with 0·1% prionex stabilizer. Plates were read using the Tristar LB941 device (Berthold Technologies, Oak Ridge, TN, USA).
Data presentation and statistical analyses
Values obtained from our CXCL10 three-plex are presented as pg/ml. Values above the least detectable dose (LDD) possess excellent precision with coefficients of variation (CV) < 10%. For individual assays, there is the possibility of achieving a value that is measurable, but below the LDD. In such instances, we define a per run lower assay limit (LAL), the lowest value that may be calculated from the standard curve. Values below the LDD and above the LAL may be real values whose precision is examined closely. For data analysis and presentation, values below the LAL were replaced with a value that is 50% of the lowest value in the data set. The LDD for the assays are indicated. The Mann–Whitney U-test was used to determine whether a difference existed between two groups of individuals. A two-tailed comparison was performed in all cases. A P-value <0·05 was considered to be statistically significant.
Results
Generation of antibodies specific for CXCL10 (1–77) and CXCL10 (3–77)
Five antibody clones were identified from the primary screen and subjected to testing in a direct ELISA assay. Whole protein and the NH2-terminal peptide of DPP4 cleaved CXCL10 were bound to a plate at titrated doses and the specificity of the clones was evaluated. A rabbit polyclonal antibody (pAb), previously raised against the NH2-terminus of the agonist form of CXCL10, served as a control. Based on these studies, clone 9852 was selected for use (Fig. S1). A similar strategy was utilized to generate monoclonal reagents specific for the agonist form of CXCL10, and resulted in the identification of six clones. The secondary screen revealed that only clone 12010 recognized the epitope within the context of the full-length protein at reasonable affinity (Fig. S2).
Using these unique antibodies, we generated and validated a three-plex assay that permits simultaneous quantification of total (i.e. all forms), CXCL10 (1–77) and CXCL10 (3–77). The assay was tested rigorously for the fundamental parameters of least detectable dose, precision, cross-reactivity, correlation, linearity, spike-recovery, dynamic range, matrix interferences and stability. Details can be found within the Supplementary information. Representative standard curves (Fig. S3) and assay parameters (Table 2) are shown. In addition, we cross-validated the single-plex and multiplex assays, which demonstrated accuracy and precision for our method (all assays showed an R2 > 0·9, data not shown).
Table 2.
Parameters of CXCL10 three-plex assay
| Analyte | Units | LAL† | LDD‡ | LLOQ§ | Dynamic range |
|---|---|---|---|---|---|
| CXCL10 | pg/ml | 46 | 110 | 171 | 110–47 550 |
| Long CXCL10 | pg/ml | 606 | 706 | 971 | 706–250 000 |
| Short CXCL10 | pg/ml | 130 | 533 | 443 | 533–250 000 |
LAL (lower assay limit) is defined as the lowest value that can be calculated from the standard curve. The value shown here is for plasma samples; urine samples (not shown) have a slightly lower LAL.
LDD (least detectable dose) is measured by determining the mean of 20 blank samples and adding 3 standard deviations to the mean.
LLOQ (lower limit of quantitation) is the value at which the assay precision reaches >30%.
HCV patients rapidly catabolize CXCL10 due to higher ex vivo DPP4 activity
There are nearly 170 million HCV infected individuals worldwide [39]. While cure is possible, 20–60% of chronically infected patients (numbers vary depending on viral genotype and IL28B haplotype) who receive pegylated (peg)-IFN-α2/ribavirin (RBV) therapy fail to achieve a sustained viral response (SVR) [40]. To define more clearly the distinct clinical outcomes of HCV infection, many investigators have performed biomarker studies, with CXCL10 proving to be a valuable discriminatory analyte [41–43]. We have reported recently that the dominant form of CXCL10 circulating in chronic HCV patients is an antagonist molecule lacking the two NH2-terminal amino acids [22].
To gain additional insight into the catabolism of CXCL10 we evaluated ex-vivo stimulation of whole blood obtained from HCV patients, examining the forms of CXCL10 present after stimulation with recombinant IFN-α2. In order to ensure standardization of whole blood stimulation, the TruCulture System was employed [44]. Briefly, TruCulture collection syringes were preloaded with buffered medium and 300 U recombinant IFN-α2 (Intron-A; Schering Plough). One ml of blood was drawn up into the TruCulture tubes; thereafter the blood/medium mixture was incubated for 20 h at 37°C and the culture supernatant was separated from the cells and frozen immediately for storage at −80°C until analysis. Of note, TruCulture tubes containing media alone were used as a control.
Samples from 30 HCV patients were obtained; half the donors had previously received treatment, and responded successfully based on their achieving SVR; and half the donors failed treatment and remained persistently infected. After a 24-h ex-vivo stimulation, the majority of the IFN-induced CXCL10 remained in the native form (Fig. 2a). Notably, the concentration of CXCL10 (1–77) was slightly higher than CXCL10 (total). We therefore plotted the results of these two assays against each other and recalibrated the data based on the assumption that the assays should read out equivalent levels of protein (Fig. 2b). The coefficient of determination, R2, was equal to 0·96, supporting this assumption and indicating a close correlation for the two assays. Forcing the y-intercept through the origin, we calculated a slope of 1·67, which was used as a correction for all future results of CXCL10 (1–77) assays.
Fig. 2.
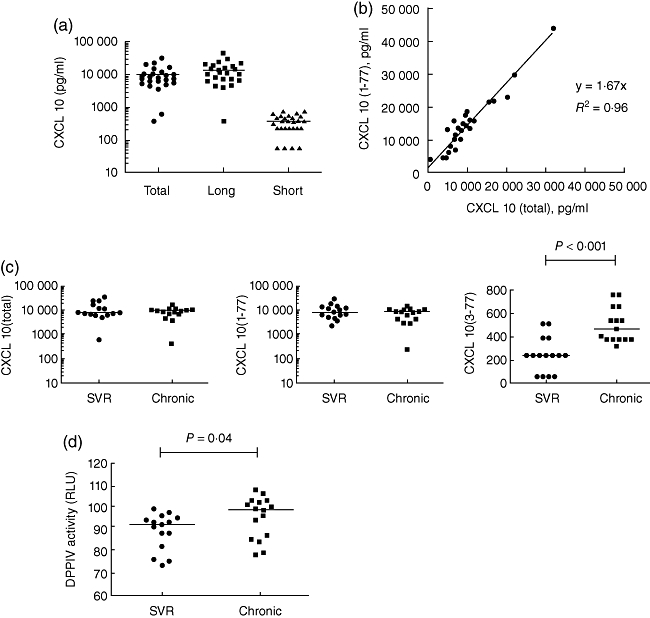
Chronic hepatitis C virus (HCV) patients rapidly catabolize CXCL10 due to higher ex-vivo dipeptidylpeptidase IV (DPP4) activity. (a) Whole blood taken from HCV patients was stimulated with 100 IU/ml interferon (IFN)-α2 (final concentration) using TruCulture collection tubes. Culture supernatants were analysed using the CXCL10 triplex assay. (b) CXCL10 (1–77) concentration was plotted against CXCL10 (total) concentration. The coefficient of determination, R2, and the curve equation are represented. (c) CXCL10 (total), CXCL10 (1–77) and CXCL10 (3–77) concentration in the two patient groups – sustained virological responders (SVR) and those who remained persistently infected after treatment (chronic). (d) DPP activity was measured in SVR and chronic patients. (c,d) A Mann–Whitney U-test was used to compare the two groups and, where significant, P-value is indicated.
Next, we re-analysed the patient data, segregating those patients that had achieved SVR and those who remained chronically infected (Fig. 2c). Interestingly, there was no difference between the levels of CXCL10 induced by ex-vivo stimulation with type I IFN; however, there was evidence of increased catabolism of CXCL10 in patients who remained actively infected by HCV. Statistical analysis revealed a difference for the CXCL10 (3–77) assay with a P-value < 0·001. To account for this finding, we tested the culture supernatants for the X-prolyl DPP activity using a luciferase-based enzymatic assay (Promega). Indeed, we observed higher DPP activity in the culture supernatants of chronically infected HCV patients compared to those who achieved an SVR (Fig. 3d). Notably, the CXCL10 (3–77) concentration present in the null TruCulture tubes (media alone) was <200 pg/ml for all patients (data not shown). Thus, we conclude that the DPP4 present in our TruCulture system acts on newly produced CXCL10.
Fig. 3.
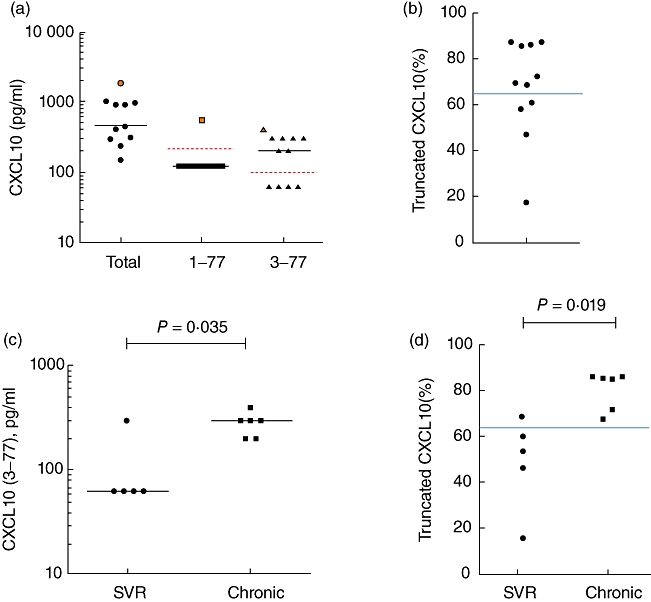
Plasma CXCL10 (3–77) concentrations are higher in chronic patients than in sustained virological responder (SVR) VR patients. (a) Plasma samples were obtained from hepatitis C virus (HCV) patients and concentration of CXCL10 (total), CXCL10 (1–77) and CXCL10 (3–77) were analysed. The donor with the highest level of CXCL10 (total) is marked in orange with corresponding symbols indicated for the respective assays. (b) The calculated fraction of truncated CXCL10 is represented [% truncated CXCL10 = [(CXCL10 (total) – CXCL10 (1–77))/CXCL10 (1–77)]. (c,d) Plasma CXCL10 (3–77) concentration and the percentage of truncated CXCL10 is shown for the two patient groups – chronic versus SVR. A Mann–Whitney U-test was used to compare the two groups and, where significant, P-value is indicated. Dotted red lines indicate the lower assay limit (LAL) for the respective assays. Solid blue line indicates the percentage of CXCL10 (3–77) that is capable of exerting a dominant negative effect on CXCR3.
Evidence for in vivo cleavage of CXCL10 in patients with chronic HCV infection
To demonstrate the utility of this assay for in vivo use, samples were collected from HCV patients post-treatment and the different forms of CXCL10 were assessed utilizing our CXCL10 three-plex. In all patients, the CXCL10 levels were detectable and elevated compared to healthy donors (data not shown). Strikingly, 10 of 11 donors had undetectable levels of the CXCL10 (1–77) (Fig. 3a). Based on the CXCL10 (3–77) assay, seven of 11 donors had detectable levels of the product generated by DPP4 cleavage (Fig. 3a).
Data from the in-vivo analysis suggested that it is not possible to achieve mass conservation: [CXCL10 (1–77) + CXCL10 (3–77) ≠ CXCL10 (total)]. While such an analysis is complicated by the fact that these assays utilize distinct capture antibodies, we believe that the data indicate the presence of additional antagonist forms of CXCL10. One possibility is that following loss of the proline in the penultimate position of the NH2-terminus, other N-terminal aminopeptidases (e.g. APN or CD13) may act on the protein, thus resulting in loss of our neo-epitope. To gain insight into this question, we substituted negative values for all assays with (½ × lower assay limit) and used a simple calculation (total – long/total) to estimate the percentage of truncated CXCL10. Seven of 11 donors had catabolized >66% of their plasma CXCL10 (Fig. 3b). The solid blue line (>66%) indicates the ratio at which we observe the truncated form of CXCL10 demonstrating dominant negative activity, based on in-vitro Ca++ flux studies (data not shown and [22]). Interestingly, and confirming prior results in an independent patient population [22], chronic HCV patients had higher plasma CXCL10 (3–77) compared to patients who had cleared their virus (Fig. 3c). Moreover, the chronic patients had a greater percentage of catabolized CXCL10 based on the comparison of total and CXCL10 (1–77) assays (Fig. 3d).
In addition to monitoring baseline levels of CXCL10 that result from chronic HCV infection and liver inflammation, it is also possible to evaluate the form of CXCL10 after therapeutic treatment with recombinant IFN-α2. Patients were enrolled into a substudy in order to evaluate the CXCL10 induced by the first dose of peg-IFN-α2/RBV therapy. Two representative patients are shown in order to highlight the variable in-vivo response to treatment. In one individual (patient IM-34), CXCL10 peaked at 6 h and a significant fraction of the circulating molecule was detectable using the CXCL10 (1–77)-specific antibodies (Fig. 4a). Based on in-vitro experiments, this fraction of CXCL10 (1–77) is sufficient to achieve CXCR3 activation (data not shown). In contrast, some of the individuals receiving peg-IFN-α2/RBV therapy have a delayed induction of CXCL10, with only a small fraction being present in the intact form (<10%) (Fig. 4b). One caveat is that we may have missed the peak of CXCL10 (1–77), but arguably these data suggest that in-vivo catabolism in some individuals is quite rapid. This also applies to the further truncation of CXCL10 (3–77), as suggested by the low levels of this molecule detected in our patient samples. This method for immune monitoring will be instrumental in our evaluation of treatment trials utilizing DPP4 inhibitors; such studies are currently under way, the hypothesis being that preservation of the agonist form will enhance T and natural killer (NK) cell trafficking to the liver with the hope of increasing response to peg-IFN-α2/RBV therapy.
Fig. 4.
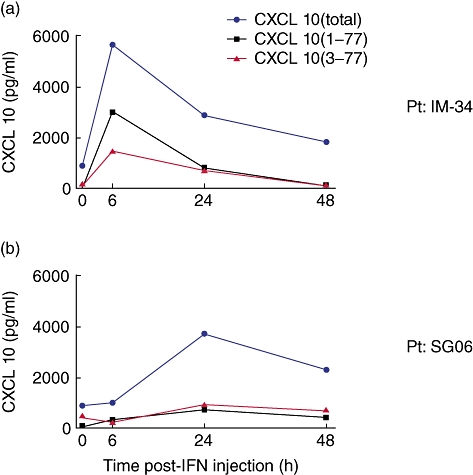
Kinetic analysis of CXCL10 in chronic hepatitis C virus (HCV) patients after treatment with IFN-α2. Plasma concentrations of CXCL10 (total, blue; 1–77, black; and 3–77, red) were measured from two HCV patients at interval time-points after their receiving their first injection of pegylated-interferon.
Inflammation-induced CXCL10 catabolism in the urine of patients with carcinoma of the bladder
We next explored the possibility of monitoring urine CXCL10 concentration utilizing our assay system. We and others have demonstrated that following intravesical BCG therapy, given in the context of treatment for carcinoma of the bladder, there is robust induction of proinflammatory cytokines and chemokines, including CXCL10 [45,46]. In fact, in our prior biomarker studies in which we evaluated 90 inflammatory proteins and plasma metabolites (human map version 1·6, RBM), CXCL10 was the most highly induced molecule, showing an increase of 225-fold at the third instillation of BCG [46]. Of note, the kidney epithelium expresses high levels of DPP4 that is presumably shed (but may be secreted actively) into the urine, accounting for the high DPP activity (data not shown and [47,48]). We therefore hypothesized that although acutely produced as a result of BCG instillation, the CXCL10 may be cleaved rapidly by DPP4, thus partially abrogating its ability to recruit CXCR3+ cells to the bladder.
Using samples that were collected as part of an ongoing observational clinical study in patients with bladder cancer receiving intravesical BCG, we determined the concentration of the different forms of CXCL10. Samples were obtained from 10 patients and analysis focused on the fifth instillation comparing pre- versus 4 h post-BCG treatment. Confirming prior data, we observed a median fold change of >50-fold across the patient group. While extraordinarily high levels of CXCL10 were detected following BCG treatment (range: 0·4– > 1000 ng/ml), the majority was not sensitive to the CXCL10 (1–77) antibody (Fig. 5a). As observed in the plasma of HCV patients, we were able to detect elevated levels of CXCL10 (3–77); however, additional N-terminal truncation of the molecule is likely. Kinetic studies supported this hypothesis, as CXCL10 is rapidly cleaved at its NH2-terminus and cleared, a combination of degradation and micturation (Fig. 5b).
Fig. 5.
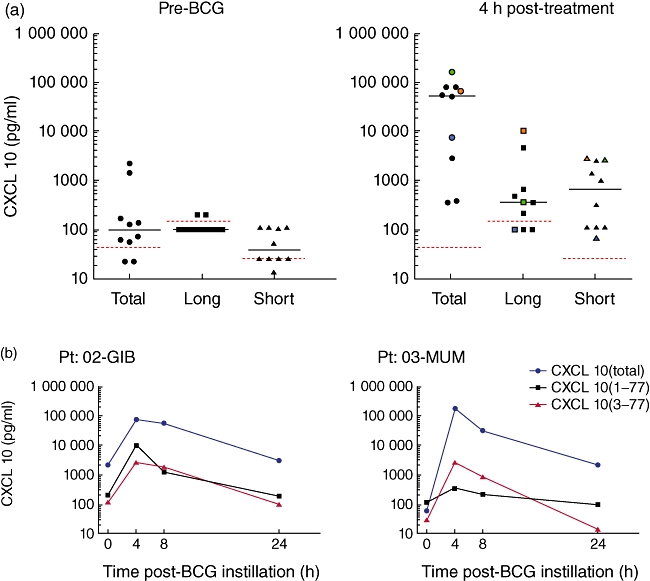
Urinary CXCL10 in bacilli Calmette–Guérin (BCG) treatment patients exists in a truncated form. Samples were obtained from bladder cancer patients during their fifth instillation of BCG. (a) Pre- and 4 h post-BCG urines were collected and CXCL10 three-plex was performed. Dotted red lines indicate the lower assay limit (LAL) for the respective assays, preformed using a proprietary buffer for measurements of urine samples. Note that the LAL for urine assays was lower than for plasma assays. At the 4-h time-point, three representative patients are marked in green, orange or blue to allow tracking of total, long and short CXCL10 concentrations. (b) Kinetic studies are shown for two patients (CXCL10 total, blue; 1–77, black; and 3–77, red).
Discussion
Biomarker discovery is regarded as a priority research area, with the aim to improve diagnosis, guide therapeutic interventions, monitor disease pathogenesis and reduce costs through improved management of patients. Plasma proteins are a bedrock for clinically useful surrogate markers. For example, monitoring liver or cardiac enzymes are indispensible biomarkers for hepatic or myocardial injury [49,50]. The – omics revolution has provided exciting new tools; however, the steps to a validated and commercialized assay are arduous, especially when advancing methods on unknown molecules [51]. As a result, many have chosen to start their discovery process using validated assays based on high-quality antibodies. It is indeed in this way that CXCL10 has emerged as an important biomarker in the prediction of response to therapy in patients with chronic HCV, as well as several other disease states (reviewed above). We have discovered that even with precise assays, the insight offered through biomarker studies may be misleading with respect to a better understanding of disease pathogenesis. Indeed, many studies have highlighted the challenge presented by post-translational modification of proteins that may impact their receptor binding, activity or bioavailability [52].
In the current study, we present the development of new tools that permit discrimination of the intact, agonist form of CXCL10 (1–77) and the NH2-terminal truncated antagonist form of CXCL10 (3–77). Specifically, these unique antibodies have helped to correct the interpretation of elevated plasma concentrations of CXCL10 in the context of chronic HCV infection [22], and provided insight into an intriguing new drug target that may improve response to peg-IFN/RBV therapy. Additionally, we provide new information about the use of ex-vivo whole-blood stimulation systems that may be important for monitoring DPP4 action on IFN-induced substrates. Finally, our manuscript suggests that even under instances of acute inflammation, as is the case for intravesical BCG therapy in patients with bladder cancer, CXCL10 catabolism may be limiting T cell and NK cell recruitment. We discuss briefly the role of CXCL10 in the context of HCV and bladder cancer therapy and highlight the utility, but also the limitations, of our newly developed CXCL10 assays.
NH2-truncated CXCL10 in chronic HCV
It has been noted for some time that chemokines are interesting host factors from the perspective of biomarkers for treatment outcome. This is based on their role as regulators of inflammation; the knowledge that several factors are induced only upon cell stress or cell damage; and the demonstration that many are modulated by therapeutic IFN [53]. Moreover, it has been shown that viral clearance correlates with NK activity and HCV-reactive CD8+ T cell responses [54–57]– cell types that are known to respond to IFN-induced chemokines (e.g. CXCL10) [5,58].
To our knowledge, the methodology presented herein provides the first opportunity to study chemokine antagonism using whole blood culture devices or in vivo using plasma samples, both of which offer the possibility of linking it to disease pathogenesis. Interestingly, when we compared whole blood samples obtained from SVR and chronic patients, stimulated by recombinant IFN-α2, we observed higher levels of CXCL10 (3–77) in chronically infected patients (Fig. 2c). This is due probably to higher plasma concentrations of DPP4 in the plasma of chronically infected patients ([22] and Fig. 2d), but may also reflect higher levels of cell associated DPP4 on circulating lymphocytes [59]. These data illustrate the utility of monitoring the different forms of CXCL10 in ex-vivo culture and demonstrate a potential application, as DPP activity may be regulated differentially during inflammatory and infectious disease situations.
Regarding in-vivo assessment of CXCL10, these assays have provided an important proof-of-principle of how these new reagents may impact our understanding of existing biomarkers for treatment responsiveness in patients with chronic HCV (Figs 3 and 4). Additionally, the demonstration of NH2-truncation of CXCL10 has informed us about mechanisms of immune regulation and provides insight into novel drug targets that may help to improve response to therapy. It is interesting to note that many of the pieces to this puzzle have been known; however, a synthesis of disparate observations from different fields required the development of novel assay systems such as the one described in this study. Several independent cohorts have shown CXCL10 to be a negative predictive marker for response to therapy in chronic HCV patients [23,41,60,61]. This observation has held for genotypes 1 and 4 patients [23], including difficult-to-treat patients with high levels of liver fibrosis, co-infected HIV/HCV-infected patients, and individuals with extrahepatic manifestations of HCV chronic infection (e.g. mixed cryoglobulinaemia and thyroiditis). Regarding the role of DPP4 in modulating chemokine function, it was known that CXCL10 may be truncated in vitro to generate a dominant negative form, CXCL10 (3–77 aa), capable of binding CXCR3 without signalling and competitively inhibiting binding by the agonist form [35,38]. DPP4 is expressed constitutively by a wide range of cell types, including hepatocytes, and healthy donors harbour physiological plasma activity in the range of 12–25 U/l. Published kinetic studies suggest that this should be sufficient to cleave even high concentrations of CXCL10 [62]. Moreover, there have been two clinical reports that have made a link between DPPs and chronic HCV infection [63,64]. Importantly, through the application of novel tools and a critical examination of the available data, it has been possible to provide an important and unique perspective regarding the inflammatory state of chronic HCV patients. Not only is the identification of in-vivo antagonist CXCL10 important from the perspective of biomarker discovery, the possibility of protecting agonist CXCL10 by inhibiting DPP4 may prove valuable for enhancing response to treatment in chronic HCV patients.
DPP4 activity in urine may limit inflammation during BCG therapy
Non-muscle invasive bladder carcinoma is treated by the intravesical instillation of BCG [65]. While the mechanism of action of BCG is still being elucidated, it is clear that intravesical therapy triggers a profound innate immune response. We have demonstrated recently that following BCG treatment, the bladder urothelium, as well as infiltrating immune cells, both release CXCL10 into the bladder parenchyma. Notably, CXCL10 leaks into the urine and may be quantified for use as a potential biomarker [66]. As a result of chemokine production, the expectation would be the creation of an inflammatory microenvironment. Specifically, a CXCL10 gradient might be capable of attracting CXCR3-expressing T and NK cells. Indeed, these cell populations traffic into the inflamed bladder ([46] and unpublished data, Biot & Albert).
Following from our studies in HCV patients, we queried as to whether DPP4 activity may also impact the functional status of CXCL10 produced within the bladder mucosa. Interestingly, high DPP4 concentrations are found in urine, and enzymatic activity has been show to be a useful biomarker in several clinical situations (e.g. marker of cholestasis, glomerulopathies) [67–69]. Our findings provide the first evidence for in-situ truncation of urinary CXCL10 (Fig. 5). Interestingly, these data suggest that chemokine antagonism may limit the inflammatory response to BCG during intravesical therapy. This was somewhat surprising, as the induction of CXCL10 in the therapeutic setting is similar to the ex-vivo stimulation of whole blood, and in the latter instance CXCL10 was principally intact. Following the in-vivo results from bladder cancer patients, we suggest that DPP4 may provide a mechanism for limiting migration of CXCR3-positive cells into the bladder. While this may provide a protective mechanism for the bladder mucosa, it also has the potential to limit the efficacy of BCG therapy. Along these lines, it suggests that inhibition of DPP4 in patients receiving BCG therapy may provide a mechanism for enhancing anti-tumour responses and/or possibly allow for shortening of therapy.
One additional consideration concerns the role of CXCL10 in limiting angiogenesis. In fact, when CXCL10 was first identified in the urine of patients receiving BCG therapy, Poppas et al. suggested that the cytokine-mediated anti-angiogenic environment may contribute to the inhibition of future tumour growth and progression [45]. Interestingly, while NH2-terminal truncation of CXCL10 affects trafficking of CXCR3 expressing cells, it does not impact its anti-angiogenic properties [38]. This supports a model in which DPP4 participates in restoring tissue integrity following the onset of inflammation.
Technical considerations and limitations of the assay
While the detection of circulating CXCL10 (3–77) supports a role for in-vivo DPP4 activity, we could not account for the total amount of CXCL10 detected. In other words, total CXCL10 > CXCL10 (1–77) + CXCL10 (3–77). As the assays utilize different capture antibodies, it is difficult to compare directly the amount of CXCL10 detected in the different ELISAs. That said, we favour the hypothesis that once cleaved by DPP4 (or possibly other members of the DPP family), the truncated form of CXCL10 becomes a substrate for other aminopeptidases (e.g. APN or CD13) [70]. Several candidate enzymes were tested, including APN and several MMPs; however, it remains unclear as to which N-terminal proteases are responsible for the subsequent catabolism of CXCL10 (3–77). To overcome this limitation, we were able to calculate the percentage of NH2-truncated CXCL10 by establishing a ratio of total and CXCL10 (1–77) concentrations (Fig 3b,d).
It is also worth mentioning that the lower assay limit for CXCL10 (1–77) and CXCL10 (3–77) is relatively high: 606 pg/ml and 130 pg/ml, respectively. As such, the assays do not permit direct assessment of CXCL10 concentrations that might be found in healthy individuals. Improved antibodies and/or new assay systems will be required in order to ascertain the functional state of CXCL10 in the resting state.
In summary, our paper demonstrates the utility of a new panel of CXCL10 assays that permit discrimination of agonist (CXCL10, 1–77) and antagonist (CXCL10, 3–77) forms of CXCL10 in culture supernatant, patient plasma and urine samples. Given the important role of CXCL10 in chronic inflammatory diseases and its suggested role as a predictive marker in managing patients with chronic hepatitis C, asthma, atopic dermatitis, transplantation, tuberculosis, kidney injury, cancer and others diseases, we believe that our method will be of general interest to the research and medical community.
Acknowledgments
The authors would like to thank members of the Centre d'Immunology Humaine (CIH) and the Pôle Integré de Recherche Clinique (PIRC) Institut Pasteur; and the members of the Liver Unit, Hopital Cochin for support of this work. The work was funded by grants obtained from L'Institut National du Cancer (INCA), the European Research Council (ERC) and a private donation from Caisse de Retraite et de Prévoyance des Clercs et Employés de Notaires (CRPCEN).
Disclosure
Laurie Stephen, James Mapes and Manfred Schmolz are employees of Rules Based Medicine, the sole owners of the TruCulture stimulation system; and the commercial party licensed to develop the CXCL10 3-plex assay.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Development of antibodies specific for CXCL10 (3–77). (a) As a secondary screen following selection of clones, direct enzyme-linked immunosorbent assay (ELISA) was performed using plate-bound recombinant CXCL10 (1–77, black; 3–77, red) or peptide corresponding to the NH2-sequence of the truncated form of CXCL10 (LSRTVR peptide, blue). (b) A sandwich ELISA was developed by coating the selected clone (#9852). A standard curve is shown, using recombinant CXCL10 (1–77, black; and 3–77, red).
{kind=link}
Fig. S2. Development of antibodies specific for CXCL10 (1–77). (a) As a secondary screen following selection of clones, direct enzyme-linked immunosorbent assay (ELISA) was performed using plate-bound recombinant CXCL10 (1–77, black; 3–77, red) or peptide corresponding to the NH2-sequence of the intact and truncated form of CXCL10 (VPLSRTVR peptide, grey; LSRTVR peptide, blue). (b) A sandwich ELISA was developed by coating the selected clone (#12010). A standard curve is shown, using recombinant CXCL10 (1–77, black; and 3–77, red).
{kind=link}
Fig. S3. Standard curves for CXCL10 3-plex. Standard curves for the CXCL10 multiplex are shown for the (a) CXCL10 (total), (b) CXCL10 (1–77) and (c) CXCL10 (3–77) assays. Biological CXCL10 was produced by culturing whole blood with recombinant interferon (IFN)-a2 (Intron-A; Schering Plough) for 24 h. The supernatant of these cultures was used as a biological standard for measurement of total and CXCL10 (1–77). Recombinant protein cleaved by DPP4 was used as standard for the measurement of CXCL10 (3–77) assay.
{kind=link}
References
- 1.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–15. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu M, Guo S, Hibbert JM, et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011;22:121–30. doi: 10.1016/j.cytogfr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taub DD, Longo DL, Murphy WJ. Human interferon-inducible protein-10 induces mononuclear cell infiltration in mice and promotes the migration of human T lymphocytes into the peripheral tissues and human peripheral blood lymphocytes–SCID mice. Blood. 1996;87:1423–31. [PubMed] [Google Scholar]
- 4.Angiolillo AL, Sgadari C, Taub DD, et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182:155–62. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen JE, de Lemos C, Moos T, Christensen JP, Thomsen AR. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol. 2006;176:4235–43. doi: 10.4049/jimmunol.176.7.4235. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan G, Luster AD, Hancock G, Cohn ZA. The expression of a gamma interferon-induced protein (IP-10) in delayed immune responses in human skin. J Exp Med. 1987;166:1098–108. doi: 10.1084/jem.166.4.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J Exp Med. 1987;166:1084–97. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apolinario A, Majano PL, Lorente R, Nunez O, Clemente G, Garcia-Monzon C. Gene expression profile of T-cell-specific chemokines in human hepatocyte-derived cells: evidence for a synergistic inducer effect of cytokines and hepatitis C virus proteins. J Viral Hepatol. 2005;12:27–37. doi: 10.1111/j.1365-2893.2005.00540.x. [DOI] [PubMed] [Google Scholar]
- 9.Harvey CE, Post JJ, Palladinetti P, et al. Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J Leukoc Biol. 2003;74:360–9. doi: 10.1189/jlb.0303093. [DOI] [PubMed] [Google Scholar]
- 10.Cardozo AK, Proost P, Gysemans C, Chen MC, Mathieu C, Eizirik DL. IL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia. 2003;46:255–66. doi: 10.1007/s00125-002-1017-0. [DOI] [PubMed] [Google Scholar]
- 11.Hensbergen PJ, van der Raaij-Helmer EM, Dijkman R, et al. Processing of natural and recombinant CXCR3-targeting chemokines and implications for biological activity. Eur J Biochem. 2001;268:4992–9. doi: 10.1046/j.0014-2956.2001.02433.x. [DOI] [PubMed] [Google Scholar]
- 12.van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi–Goutieres syndrome. Glia. 2008;56:568–78. doi: 10.1002/glia.20639. [DOI] [PubMed] [Google Scholar]
- 13.Quint JK, Donaldson GC, Goldring JJ, Baghai-Ravary R, Hurst JR, Wedzicha JA. Serum IP-10 as a biomarker of human rhinovirus infection at exacerbation of COPD. Chest. 2010;137:812–22. doi: 10.1378/chest.09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh MF, Lai SL, Chen JP, et al. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J Immunol. 2006;177:1855–63. doi: 10.4049/jimmunol.177.3.1855. [DOI] [PubMed] [Google Scholar]
- 15.Chen JP, Lu HL, Lai SL, et al. Dengue virus induces expression of CXC chemokine ligand 10/IFN-gamma-inducible protein 10, which competitively inhibits viral binding to cell surface heparan sulfate. J Immunol. 2006;177:3185–92. doi: 10.4049/jimmunol.177.5.3185. [DOI] [PubMed] [Google Scholar]
- 16.Campanella GS, Tager AM, Khoury JKE, et al. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci USA. 2008;105:4814–19. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain V, Armah HB, Tongren JE, et al. Plasma IP-10, apoptotic and angiogenic factors associated with fatal cerebral malaria in India. Malaria J. 2008;7:83–98. doi: 10.1186/1475-2875-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smoller BR, McNutt NS, Gray MH, Krueger J, Hsu A, Gottlieb AB. Detection of the interferon-gamma-induced protein 10 in psoriasiform dermatitis of acquired immunodeficiency syndrome. Arch Dermatol. 1990;126:1457–61. [PubMed] [Google Scholar]
- 19.Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo T, Nakazawa H, Ito F, et al. Favorable prognosis of renal cell carcinoma with increased expression of chemokines associated with a Th1-type immune response. Cancer Sci. 2006;97:780–6. doi: 10.1111/j.1349-7006.2006.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arenberg DA, Kunkel SL, Polverini PJ, et al. Interferon-gamma-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med. 1996;184:981–92. doi: 10.1084/jem.184.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casrouge A, Decalf J, Ahloulay M, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121:308–17. doi: 10.1172/JCI40594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romero AI, Lagging M, Westin J, et al. Interferon (IFN)-gamma-inducible protein-10: association with histological results, viral kinetics, and outcome during treatment with pegylated IFN-alpha 2a and ribavirin for chronic hepatitis C virus infection. J Infect Dis. 2006;194:895–903. doi: 10.1086/507307. [DOI] [PubMed] [Google Scholar]
- 24.Nakaya I, Wada T, Furuichi K, et al. Blockade of IP-10/CXCR3 promotes progressive renal fibrosis. Nephron Exp Nephrol. 2007;107:e12–21. doi: 10.1159/000106505. [DOI] [PubMed] [Google Scholar]
- 25.Lazzeri E, Rotondi M, Mazzinghi B, et al. High CXCL10 expression in rejected kidneys and predictive role of pretransplant serum CXCL10 for acute rejection and chronic allograft nephropathy. Transplantation. 2005;79:1215–20. doi: 10.1097/01.tp.0000160759.85080.2e. [DOI] [PubMed] [Google Scholar]
- 26.Heresi GA, Aytekin M, Newman J, Dweik RA. CXC-chemokine ligand 10 in idiopathic pulmonary arterial hypertension: marker of improved survival. Lung. 2010;188:191–7. doi: 10.1007/s00408-010-9232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nance S, Cross R, Fitzpatrick E. Chemokine production during hypersensitivity pneumonitis. Eur J Immunol. 2004;34:677–85. doi: 10.1002/eji.200324634. [DOI] [PubMed] [Google Scholar]
- 28.Muller M, Carter S, Hofer MJ, Campbell IL. Review: the chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity – a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36:368–87. doi: 10.1111/j.1365-2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- 29.Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann NY Acad Sci. 2009;1173:310–17. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation – a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- 31.Rosenkilde MM, Andersen MB, Nygaard R, Frimurer TM, Schwartz TW. Activation of the CXCR3 chemokine receptor through anchoring of a small molecule chelator ligand between TM-III, -IV, and -VI. Mol Pharmacol. 2007;71:930–41. doi: 10.1124/mol.106.030031. [DOI] [PubMed] [Google Scholar]
- 32.Van den Steen PE, Husson SJ, Proost P, Damme JV, Opdenakker G. Carboxyterminal cleavage of the chemokines MIG and IP-10 by gelatinase B and neutrophil collagenase. Biochem Biophys Res Commun. 2003;310:889–96. doi: 10.1016/j.bbrc.2003.09.098. [DOI] [PubMed] [Google Scholar]
- 33.Loos T, Mortier A, Gouwy M, et al. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: a naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood. 2008;112:2648–56. doi: 10.1182/blood-2008-04-149039. [DOI] [PubMed] [Google Scholar]
- 34.Van den Steen PE, Deroost K, Van Aelst I, et al. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur J Immunol. 2008;38:1082–95. doi: 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- 35.Oravecz T, Pall M, Roderiquez G, et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186:1865–72. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci (Lond) 2005;108:277–92. doi: 10.1042/CS20040302. [DOI] [PubMed] [Google Scholar]
- 37.Ludwig A, Schiemann F, Mentlein R, Lindner B, Brandt E. Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I-TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J Leukoc Biol. 2002;72:183–91. [PubMed] [Google Scholar]
- 38.Proost P, Schutyser E, Menten P, et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood. 2001;98:3554–61. doi: 10.1182/blood.v98.13.3554. [DOI] [PubMed] [Google Scholar]
- 39.Houghton M, Abrignani S. Prospects for a vaccine against the hepatitis C virus. Nature. 2005;436:961–6. doi: 10.1038/nature04081. [DOI] [PubMed] [Google Scholar]
- 40.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–72. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 41.Butera D, Marukian S, Iwamaye AE, et al. Plasma chemokine levels correlate with the outcome of antiviral therapy in patients with hepatitis C. Blood. 2005;106:1175–82. doi: 10.1182/blood-2005-01-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeremski M, Markatou M, Brown QB, Dorante G, Cunningham-Rundles S, Talal AH. Interferon gamma-inducible protein 10: a predictive marker of successful treatment response in hepatitis C virus/HIV-coinfected patients. J Acquir Immune Defic Syndr. 2007;45:262–8. doi: 10.1097/QAI.0b013e3180559219. [DOI] [PubMed] [Google Scholar]
- 43.Antonelli A, Ferri C, Fallahi P, et al. Alpha-chemokine CXCL10 and beta-chemokine CCL2 serum levels in patients with hepatitis C-associated cryoglobulinemia in the presence or absence of autoimmune thyroiditis. Metabolism. 2008;57:1270–7. doi: 10.1016/j.metabol.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Schmolz M, Hurst TL, Bailey DM, et al. Validation of a new highly standardised, lab-independent whole-blood leukocyte function assay for clinical trials (ILCS) Exp Gerontol. 2004;39:667–71. doi: 10.1016/j.exger.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 45.Poppas DP, Pavlovich CP, Folkman J, et al. Intravesical bacille Calmette–Guerin induces the antiangiogenic chemokine interferon-inducible protein 10. Urology. 1998;52:268–75. discussion 75-6. [PubMed] [Google Scholar]
- 46.Bisiaux A, Thiounn N, Timsit MO, et al. Molecular analyte profiling of the early events and tissue conditioning following intravesical bacillus Calmette–Guerin therapy in patients with superficial bladder cancer. J Urol. 2009;181:1571–80. doi: 10.1016/j.juro.2008.11.124. [DOI] [PubMed] [Google Scholar]
- 47.Slimane TA, Lenoir C, Sapin C, Maurice M, Trugnan G. Apical secretion and sialylation of soluble dipeptidyl peptidase IV are two related events. Exp Cell Res. 2000;258:184–94. doi: 10.1006/excr.2000.4894. [DOI] [PubMed] [Google Scholar]
- 48.Stefanovic V, Ardaillou N, Vlahovic P, Placier S, Ronco P, Ardaillou R. Interferon-gamma induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology. 1993;80:465–70. [PMC free article] [PubMed] [Google Scholar]
- 49.Dolci A, Panteghini M. The exciting story of cardiac biomarkers: from retrospective detection to gold diagnostic standard for acute myocardial infarction and more. Clin Chim Acta. 2006;369:179–87. doi: 10.1016/j.cca.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 50.Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. The current state of serum biomarkers of hepatotoxicity. Toxicology. 2008;245:194–205. doi: 10.1016/j.tox.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 51.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 52.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3:509–19. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 53.Zeremski M, Petrovic LM, Talal AH. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J Viral Hepat. 2007;14:675–87. doi: 10.1111/j.1365-2893.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 54.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–54. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 56.Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–52. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 58.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–5. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 59.Mattern T, Scholz W, Feller AC, Flad HD, Ulmer AJ. Expression of CD26 (dipeptidyl peptidase IV) on resting and activated human T-lymphocytes. Scand J Immunol. 1991;33:737–48. doi: 10.1111/j.1365-3083.1991.tb02548.x. [DOI] [PubMed] [Google Scholar]
- 60.Darling JM, Aerssens J, Fanning G, et al. Quantitation of pretreatment serum interferon-gamma-inducible protein-10 improves the predictive value of an IL28B gene polymorphism for hepatitis C treatment response. Hepatology. 2011;53:14–22. doi: 10.1002/hep.24056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lagging M, Romero AI, Westin J, et al. IP-10 predicts viral response and therapeutic outcome in difficult-to-treat patients with HCV genotype 1 infection. Hepatology. 2006;44:1617–25. doi: 10.1002/hep.21407. [DOI] [PubMed] [Google Scholar]
- 62.Lambeir AM, Proost P, Durinx C, et al. Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase IV reveals a striking selectivity within the chemokine family. J Biol Chem. 2001;276:29839–45. doi: 10.1074/jbc.M103106200. [DOI] [PubMed] [Google Scholar]
- 63.Firneisz G, Lakatos PL, Szalay F. Serum dipeptidyl peptidase IV (DPP IV, CD26) activity in chronic hepatitis C. Scand J Gastroenterol. 2001;36:877–80. doi: 10.1080/003655201750313423. [DOI] [PubMed] [Google Scholar]
- 64.Yang SS, Fu LS, Chang CS, Yeh HZ, Chen GH, Kao JH. Changes of soluble CD26 and CD30 levels correlate with response to interferon plus ribavirin therapy in patients with chronic hepatitis C. J Gastroenterol Hepatol. 2006;21:1789–93. doi: 10.1111/j.1440-1746.2006.04677.x. [DOI] [PubMed] [Google Scholar]
- 65.Morales A, Eidinger D, Bruce AW. Intracavitary bacillus Calmette–Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116:180–3. doi: 10.1016/s0022-5347(17)58737-6. [DOI] [PubMed] [Google Scholar]
- 66.Luo Y, Chen X, O'Donnell MA. Mycobacterium bovis bacillus Calmette–Guerin (BCG) induces human CC- and CXC-chemokines in vitro and in vivo. Clin Exp Immunol. 2007;147:370–8. doi: 10.1111/j.1365-2249.2006.03288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rakoczi G, Takacs L, Jakabfi P, et al. Increased urinary dipeptidyl peptidase IV activity in extrahepatic biliary atresia. Lancet. 1995;345:864–5. doi: 10.1016/s0140-6736(95)93003-5. [DOI] [PubMed] [Google Scholar]
- 68.Mitic B, Lazarevic G, Vlahovic P, Rajic M, Stefanovic V. Diagnostic value of the aminopeptidase N, N-acetyl-beta-D-glucosaminidase and dipeptidylpeptidase IV in evaluating tubular dysfunction in patients with glomerulopathies. Ren Fail. 2008;30:896–903. doi: 10.1080/08860220802359048. [DOI] [PubMed] [Google Scholar]
- 69.Perner F, Gyuris T, Rakoczy G, et al. Dipeptidyl peptidase activity of CD26 in serum and urine as a marker of cholestasis: experimental and clinical evidence. J Lab Clin Med. 1999;134:56–67. doi: 10.1016/s0022-2143(99)90054-9. [DOI] [PubMed] [Google Scholar]
- 70.Proost P, Mortier A, Loos T, et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood. 2007;110:37–44. doi: 10.1182/blood-2006-10-049072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.