

Abstract
Immune responses to human leucocyte antigen (HLA) and self-antigen collagen V (Col-V) have been proposed in the pathogenesis of chronic rejection (bronchiolitis obliterans syndrome, BOS) following human lung transplantation (LTx). In this study, we defined the role for the shift in immunodominant epitopes of Col-V in inducing T helper phenotype switch leading to immunity to Col-V and BOS. Sera and lavage from BOS+ LTx recipients with antibodies to Col-V were analysed. Two years prior to BOS, patients developed antibodies to both Col-V,α1(V) and α2(V) chains. However, at clinical diagnosis of BOS, antibodies became restricted to α1(V). Further, lung biopsy from BOS(+) patients bound to antibodies to α1(V), indicating that these epitopes are exposed. Fourteen Col-V peptides [pep1–14, pep1–4 specific to α1(V), pep5–8 to α1,2(V) and pep9–14 to α2(V)] which bind to HLA-DR4 and -DR7, demonstrated that prior to BOS, pep 6, 7, 9, 11 and 14 were immunodominant and induced interleukin (IL)-10. However, at BOS, the response switched to pep1, 4 and 5 and induced interferon (IFN)-γ and IL-17 responses, but not IL-10. The T helper (Th) phenotype switch is accompanied by decreased frequency of regulatory T cells (Tregs) in the lavage. LTx recipients with antibodies to α1(V) also demonstrated increased matrix metalloproteinase (MMP) activation with decreased MMP inhibitor, tissue inhibitor of metalloproteinase (TIMP), suggesting that MMP activation may play a role in the exposure of new Col-V antigenic epitopes. We conclude that a shift in immunodominance of self-antigenic determinants of Col-V results in induction of IFN-γ and IL-17 with loss of tolerance leading to autoimmunity to Col-V, which leads to chronic lung allograft rejection.
Keywords: antibody, autoimmunity, epitopes, HLA, lung transplant
Introduction
Lung transplantation (LTx) is a therapeutic option for patients with end-stage pulmonary disorders [1]. However, the success of LTx has been limited by chronic rejection, which is diagnosed clinically as bronchiolitis obliterans syndrome (BOS). Progression of BOS can be slowed, but is unresponsive to the current immunosuppressive therapies [2,3]. Lung allografts sustain injuries due to ischaemia–reperfusion [4], alloimmunity [5], external pathogens [6] and gastro-oesophageal reflux [7], with subsequent release of immunological mediators and growth factors, leading to luminal occlusion and fibrous scarring of small airways [8]. Such an inflammatory milieu is conducive for the development of not only alloimmune responses but also immune responses to self-antigens. Immune response to self-antigens such as collagen V (Col-V) and K-α1-tubulin (K-α1T) have been demonstrated in both animal models of obliterative airway disease (OAD) and human subjects following LTx [9,10] and are proposed to be involved in the immunopathogenesis of chronic rejection of the transplanted organ [11].
Evidence that autoimmune responses are dynamic with evolving specificities to self-antigens have been demonstrated in experimental and spontaneous animal models of autoimmunity [12]. Recent evidence indicates that disease progression may be due to the activation and recruitment of autoreactive lymphocytes [13,14]. The autoreactive lymphocytes have been reported to be specific for epitopes that are distinct from the disease-inducing epitope, which causes tissue damage [12]. Cellular immune responses against an alloantigen have also been shown to spread to additional epitopes within the parent or other self-proteins, a phenomenon termed as intramolecular and intermolecular epitope spreading, respectively [15,16]. Furthermore, inflammation within the allograft and the resulting damage can lead to unmasking of the previously ‘cryptic’ self-antigenic determinants which can trigger autoimmunity. During inflammation, up-regulation of major histocompatibility complex (MHC) and co-stimulatory molecules can also result in infiltration by antigen-presenting cells (APCs), which can lead to lowering of the T cell activation threshold, thereby priming autoreactive T cells with ‘low-affinity’ T cell receptors (TCRs) [17,18].
Previous studies from our laboratory and others have shown that at the onset of BOS there is a significant increase of both serum cytokine levels and the frequency of CD4+ T cells secreting interferon (IFN)-γ and interleukin (IL)-17, along with reduction in the frequency of IL-10-secreting T cells [17,19]. We and others have also noted a decrease in regulatory T cells (Tregs) prior to BOS [14,17,20]. However, the mechanisms leading to a switch in the T cell phenotypes and resulting cytokines remain unknown. In this report, we present evidence for a primary role for self-antigen epitope shift causing a switch in Th-phenotype and cytokines leading to immune responses to self-antigens and chronic rejection following human lung transplantation.
Materials and methods
Human subjects and peripheral blood leucocyte (PBL) isolation
Twelve patients who underwent LTx at Washington University Medical Center/Barnes-Jewish Hospital who developed BOS and developed Col-V antibodies were selected for this study after obtaining informed consent. Chronic lung allograft rejection (BOS) was diagnosed according to standard International Society for Heart and Lung Transplantation guidelines and BOS of grades 3 and 4 were chosen for the study. The standard immunotherapy protocol for all patients consisted of cyclosporine A, azathioprine and prednisone. The LTx recipients (LTxR) with both human leucocyte antigen (HLA)-class II types DR4 and DR7 were chosen in our study (Table 1). The whole blood from these patients was used for PBL isolation. A cohort of time-matched (mean post-LTx duration 45·5 ± 9·3 months) stable LTxR (n = 7) who had antibodies to Col-V were used as controls. The serum was isolated from the whole blood by centrifugation at 200 g for 20 min. The peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood by Ficoll-Hypaque density gradient centrifugation (Pharmacia, Uppsala, Sweden) and stored at −135°C until evaluation [20]. The CD4+ T cells were isolated from PBMCs by positive selection on a magnetic affinity cell sorter (MACS) bead isolation kit (Miltenyi Biotec Inc., Auburn, CA, USA). The samples selected for analysis were obtained at least 1 year post-transplant and the patients were free of any acute rejection and/or infection.
Table 1.
Patient clinicodemographics
| Pt HLA | HLA mismatch | BOS- free survival (months) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Age (years) | Sex | Race | Pathology | Type (R/B) | DR type | A,B | DR | |
| Pt1 | 43 | M | C | COPD | B | 4 | 2 | 0 | 75 |
| Pt2 | 56 | F | C | COPD | B | 7 | 4 | 2 | 96 |
| Pt3 | 55 | F | C | A1AT | B | 4 | 3 | 1 | 68 |
| Pt4 | 48 | M | C | COPD | B | 4 | 3 | 1 | 42 |
| Pt5 | 60 | M | C | COPD | R | 7 | 4 | 2 | 39 |
| Pt6 | 49 | M | C | CF | B | 4 | 0 | 2 | 45 |
| Pt7 | 61 | F | C | COPD | B | 7 | 3 | 1 | 88 |
| Pt8 | 60 | M | C | COPD | R | 4 | 4 | 2 | 56 |
| Pt9 | 48 | M | C | CF | B | 7 | 2 | 1 | 70 |
| Pt10 | 52 | F | C | COPD | B | 4 | 4 | 1 | 48 |
| Pt11 | 55 | M | C | COPD | B | 7 | 3 | 2 | 43 |
| Pt12 | 47 | M | C | COPD | B | 4 | 3 | 1 | 54 |
M: male; F: female; C: Caucasian; COPD: chronic obstructive pulmonary disease; A1AT: alpha1 anti-trypsin deficiency; CF: cystic fibrosis; B: bilateral lung transplant; R: right lung transplant; HLA: human leucocyte antigen; BOS: bronchiolitis obliterans syndrome.
Western blot analysis
The purified proteins, Col-V α1 and α2, were obtained from Dr Brand (University of Tennessee, Memphis, TN, USA). Peptide-epitopes were ordered from Peptide 2·0 Inc. (Chantilly, VA, USA). All purified proteins and peptides were free of endotoxin by limulus amoebocyte lysate (LAL) assay. The proteins were run on 4–12% gradient Bis-Tris IPG Gel (Invitrogen, Carlsbad, CA, USA) electrophoresis [10]. The peptides were run on 10–20% Tris–glycine gels. The proteins were transferred to a nitrocellulose membrane and blocked with Tris-buffered saline (TBS) with 5% skimmed milk protein overnight. After one washing with TBS, the nitrocellulose membrane was cut into 0·5-cm wide strips. Each strip was blotted with a different patient serum or serum from a normal volunteer (negative control) (1:10 dilution) overnight. The serum was washed from the membranes, which were then incubated for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-human serum (1/10 000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Immunoreactivity was detected using the SuperSignal West Pico Chemiluminescent Substrate Western blot detection system (Pierce, Rockford, IL, USA). The blots were read using Bio-Rad Universal Hood II (Hercules, CA, USA). Western blots for the epitopes were run on 10–20% Tris–glycine gel and adopting a similar procedure as described above.
Quantitation of antibodies to whole Col-V, α1(V) and α2(V)
Enzyme-linked immunosorbent assay (ELISA) plate (Nunc) was coated with Col-V (BD Biosciences, San Jose, CA, USA) α1(V) and α2(V) at 1 µg/ml in phosphate-buffered saline (PBS) at 4°C and blocked with 1% bovine serum albumin (BSA) for 2 h. All the collagens were tested and found to be free of endotoxin contamination by chromogenic LAL assay and isoelectric focusing. Patient's serum at 1:750 and 1:1500 dilutions were used for the quantification of collagen antibodies. Detection was performed with anti-mouse HRP or anti-human HRP-labelled antibody followed by development using tetramethylbenzidine substrate and read at 450 nm. A sample was considered positive if the values were over average cut-off values obtained from normal sera from 10 different individuals. Concentration of antibodies was calculated based on a standard curve using the binding of known concentration of anti-Col-V antibodies (Santa Cruz Biotechnology).
Immunohistochemistry
The deposition of specific antibodies to Col-V in the lungs and binding by antibodies present in the serum was analysed by immunohistological staining. The serum from BOS patients collected at various time-points (−2 years and +6 months) was pooled and biotinylated by EZ-Link® Sulfo-NHS-Biotinylation Kit (Thermo Fisher Scientific, Rockford, IL, USA) using the manufacturer's buffers and protocol [21]. The lung tissue from BOS+ patients was embedded in Freeze Tissue matrix and cut at 5-µm thickness. The sections were fixed in cold alcohol for 2 min (−20°C), air-dried and treated with 3% H2O2 and blocked with biotin/avidin system components (Avidin/Biotin Blocking Kit, Vector Laboratories, Burlingame, CA, USA). The sections were incubated overnight with biotinylated serum (1:5). Washed sections were blocked and treated with streptavidin–fluorescein isothiocyanate (FITC) (Santa Cruz Biotechnologies). The tissues sections were viewed with Zeiss Axiovert 200 with an AxioCam MRm camera.
Epitope identification and peptide synthesis
Col-V peptides that bind to common HLA class II (DR4 and DR7) were identified using the HLA class II-peptide binding computer algorithm from the Bioinformatics and Molecular Analysis Section of the National Institutes of Health at http://www.syfpeithi.de/Scripts/MHCServer.dll/EpitopePrediction.htm. Peptides were synthesized by Peptide 2·0 Inc. (Chantilly). The purity of peptides was determined by high-performance liquid chromatography and mass spectrometry. Sequences with overlapping consensus were clubbed together in the final peptide synthesis. Of the initial 42 peptides (Supplementary Fig. S1) identified and screened, 14 peptides (Table 2) were selected which individually showed specific stimulation of CD4 T cells.
Table 2.
Peptides used for cytokine induction assay
| Peptide # | ColV# | Sequence | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pep1 | Col5A1.74 | A | Y | R | V | T | K | D | A | Q | L | S | A | P | T | K | |
| Pep2 | Col5A1.135 | S | P | V | F | L | Y | E | D | H | T | G | K | P | G | P | |
| Pep3 | Col5A1.283 | P | E | D | L | G | K | E | P | T | P | S | K | K | P | V | |
| Pep4 | Col5A1.337 | A | D | D | L | E | G | E | F | T | E | E | T | I | R | N | |
| Pep5 | Col5A1.712 | G | P | Q | G | E | P | G | P | P | G | Q | Q | G | N | P | Col5A2.9 (60%) |
| Pep6 | Col5A1.1486 | G | L | I | G | L | I | G | P | P | G | E | Q | G | E | K | Col5A2.790 (67%) |
| Pep7 | Col5A1.1727 | Q | M | T | F | L | R | L | L | S | A | S | A | H | Q | N | Col5A2.1026 (80%) |
| Pep8 | Col5A1.1796 | K | T | V | L | E | I | D | T | P | K | V | E | Q | V | P | Col5A2.1099 (47%) |
| Pep9 | Col5A2.275 | P | G | E | V | G | F | A | G | S | P | F | A | R | G | F | |
| Pep10 | Col5A2.365 | P | G | P | M | G | P | L | G | I | P | G | S | S | G | F | |
| Pep11 | Col5A2.419 | P | G | A | I | G | T | D | G | T | P | G | A | K | G | P | |
| Pep12 | Col5A2.948 | S | G | E | Y | W | I | D | P | N | Q | G | S | V | E | D | |
| Pep13 | Col5A2.999 | P | V | W | Y | G | L | D | M | N | R | G | S | Q | F | A | |
| Pep14 | Col5A2.1078 | N | I | R | F | R | Y | I | V | L | Q | D | T | C | S | K | |
Col: collagen.
Enzyme-linked immunospot (ELISPOT) assay
ELISPOT assays were performed as described previously [17]. Briefly, MultiScreen 96-well filtration plates (Millipore, Billerica, MA, USA) were coated overnight at 4°C with 5·0 µg/ml capture human cytokine-specific monoclonal antibody (mAb) (BD Biosciences) in 0·05 m carbonate–bicarbonate buffer (pH 9·6). The plates were blocked with 1% BSA for 2 h and washed three times with PBS. Subsequently, 3 × 105 CD4+ T cells (isolated from PBLs by MACS beads) were cultured in triplicate in the presence of Col-V epitopes (1–14) (40 µg/ml) and irradiated feeder autologous PBMCs (APCs) (1:1 ratio). After 5 days (for IL-10 and IFN-γ) and 3 weeks (for IL-17), along with changing the supernatant, the plates were washed three times with PBS and three times with 0·05% PBS/Tween 20. Later, 2·0 µg/ml biotinylated human cytokine-specific mAb (BD Biosciences) in PBS/BSA/Tween 20 was added to the wells. After an overnight incubation at 4°C, the plates were washed three times and HRP-labelled streptavidin (BD Biosciences) diluted 1:2000 in PBS/BSA/Tween 20 was added to the wells. After 2 h, the assay was developed by 3-amino-9-ethylcarbazole substrate reagent (BD Biosciences) for 5–10 min. The plates were washed with tap water to stop the reaction and air-dried. The spots were analysed in an ImmunoSpot Series I Analyzer (Cellular Technology, Shaker Heights, OH, USA), and the results were expressed as spots per million cells (s.p.m.). Spots obtained by culturing T cell lines with APCs alone were subtracted from the number of spots in the experimental cultures.
Forkhead box P3 (FoxP3) DNA methylation assay
Baron et al. have originally described the DNA methylation assay as a reliable criterion to determine the natural Treg frequency in peripheral blood [22,23]. We adopted a similar strategy to determine the Treg frequency in peripheral blood of BOS(+) lung transplant recipients [24]. Genomic DNA was isolated using the DNeasy blood and tissue kit (Qiagen). Bisulphite treatment of genomic DNA was performed on the extracted DNA. A methyl-sensitive polymerase chain reaction (PCR) was used to analyse the methylation status of the cytosine–guanine dinucleotide (CpG) island of the FoxP3 promoter. Genomic DNA was prepared using a blood DNA extraction kit (Qiagen). The FoxP3 CpG island was PCR amplified using the following specific primers: FoxP3 enhancer CpG island from −5782 to −5558 was PCR amplified using 5′-AATGTGGGTATTAGGTAAAATTTTT-3′ (forward) and 5′-AAACCCTAAAACTACC TCTAAC-3′ (reverse) primers. FoxP3 CpG island from −5252 to −5030 was PCR amplified using 5′-TAGGTGATTGATAAGTAGGAGAAGTTAGTA-3′ (forward) and 5′-TACCCCCATTACTTATAACCATTTC-3′ (reverse) primers. Real-time PCR was performed in a final reaction volume of 20 µl using iCycler 480 Probes Master (Roche Diagnostics, Indianapolis, IN, USA) containing 15 pmol each of methylation or non-methylation-specific forward and reverse primers for Treg-specific demethylated region (TSDR), and 30 ng of bisulphite-treated genomic DNA. Each sample was analysed in triplicate. Cycling conditions consisted of an initial denaturation of 95°C for 15 min, followed by 50 cycles of 95°C for 30 s, followed by 61°C for 1 min.
Matrix metalloproteinase (MMP) zymography
The MMP2 and 9 activity in the bronchoalveolar lavage (BAL) fluid collected from the patients was determined by gelatin zymography [25,26]. The protein concentration of the supernatants was determined using the bicinchoninic acid (BCA) protein assay (Pierce), and 5 µg of supernatant proteins were resolved by non-reducing 10% sodium dodecyl sulphate-polymerase chain reaction (SDS-PAGE) through Novex Tris-Glycine gels containing 0·1% gelatin (Invitrogen). The gels were then washed for 30 min in Novex zymogram denaturing buffer (Invitrogen) to remove the SDS, and then incubated in Novex zymogram developing buffer (Invitrogen) for 24 h at 37°C. Areas of protease activity were visualized after staining the gels with 0·5% Coomassie blue R-250 (Invitrogen). The gelatinolytic activity of the MMPs were analysed quantitatively by the optical density of the bands using the Kodak image analysis system (Gel Logic 100 System; Kodak, Inc., Rochester, NY, USA).
RT-PCR analysis
mRNA gene expression profile of MMP2 and 9, and TIMP 1 and 2 in the BAL fluid was analysed using the FAm-labelled RT–PCR primers for MMP2,9 and TIMP1, 2 (Applied Biosystems, Foster City, CA, USA) as per the manufacturer's recommendation. Briefly, total RNA was extracted from 106 cells using TRIzol reagent (Sigma-Aldrich, St. Louis, MO, USA). The RNA was reverse-transcribed and real-time PCR was performed in a final reaction volume of 20 µl using iCycler 480 Probes Master (Roche Diagnostics, Indianapolis, IN, USA). Each sample was analysed in triplicate. Cycling conditions consisted of an initial denaturation of 95°C for 15 min, followed by 40 cycles of 95°C for 30 s, followed by 61°C for 1 min.
Statistical analysis
Statistical analysis was performed using Microcal Origin 6·0 and GraphPad Prism, version 4·02 (GraphPad Software, LaJolla, CA, USA). Paired or unpaired two-tailed Student's and Fisher's exact t-tests were used as appropriate to compare the individual antibody responses to Col-V, α1(V) and α2(V). Statistical significance was defined at P < 0·05. In the FoxP3 methylation assay, female patients were accounted for by a multiplication factor of 1·8 due to X-inactivation.
Results
Antibodies to Col-V restricted to α1(V) chain at the onset of BOS
Studies from our laboratory and others have reported development of antibodies to Col-V in human LTxR diagnosed with chronic rejection [9,17]. Col-V in lung is a heterotrimer consisting of two chains of α1 and one chain of α2 moulded as a helix [27]. To determine the specificity of antibodies to individual chains of Col-V developed during post-transplant, we used sera from 12 LTxR who have developed BOS following LTx and had antibodies to Col-V. Western blot analysis was carried out to determine the specificity of the antibodies to specific chains of Col-V [α1(V) and α2(V)]. We determined the specificity to individual chains of Col-V using sera drawn 6 months after diagnosis with BOS. The results demonstrate that (Fig. 1a) all 12 patients developed antibodies for Col α1(V) and only two of 12 had antibodies for α2(V) at the time of clinical diagnosis of BOS. Even the two patients who had antibodies to both chains eventually lost antibodies to α2(V), but retained antibodies to α1(V). These results suggest that LTxR suffering from BOS have antibody specificity restricted to α 1(V).
Fig. 1.
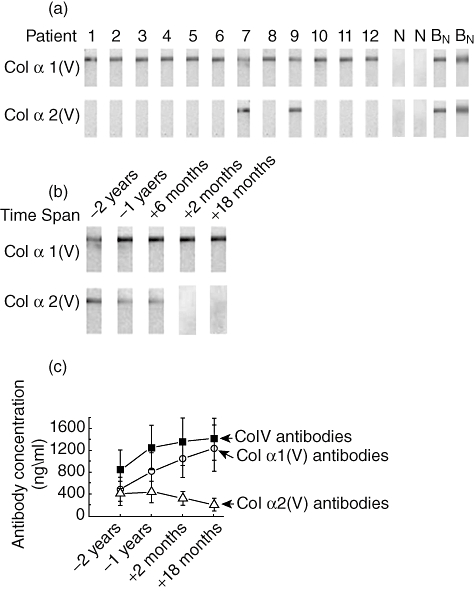
Collagen-V (Col-V) antibody characterization for individual fragments α1(V) and α2(V) by Western blot analysis on bronchiolitis obliterans syndrome [BOS(+)] patients. (a) Western blot analysis of membrane strips coated by α1(V) and α2(V) individually. Patients 1–12 refer to the 12 BOS(+) lung transplant (LTx) recipients, ‘N’ refers to a representative of two (of five tested) normal healthy adults, and ‘BN’ refers to the representive of two (of seven) time-matched stable LTx recipients. (B) Serial analysis for the development of antibodies to α1(V) and α2(V) from 2 years before onset of BOS to 18 months after the onset of BOS; and (c) concentrations of antibodies to whole Col-V, α1(V) and α2(V). The presented numbers of mean ± standard error of the mean for 12 patients.
To determine the temporal relationship between the development of antibodies and BOS, we analysed sera from all 12 LTxR collected at 2 years (−2 years), 1 year (−1 year) and 6 months before (−6 months) as well as 2 months (+2 months) and 18 months after (+18 months) the development of BOS. As shown in Fig. 1b, LTxR developed antibodies to both α1(V) and α2(V), 2 years prior to BOS. However, at BOS and later (18 months after diagnosis) only antibodies to Col α1(V) persisted. To verify that this is not due to a loss of antibodies to α2(V) over time differences, a cohort matched for the time-period (mean post-LTx duration 45·5 ± 9·3 months) and have stable lung function (n = 7) were tested. All these patients had antibodies to both chains of Col-V (Fig. 1a). This suggests strongly that the loss of antibodies to α2(V) is not due to a mere loss of antibody titres caused by the temporal-effect of duration following LTx. Furthermore, quantitative analysis of the presence of specific Col-V antibodies by ELISA (Fig. 1c) demonstrated that there was an increase in the α1(V) (12 of 12 patients) over this time-period from 490 ± 220 ng/ml (−2 years) to 1240 ± 420 ng/ml (+18 months) after BOS (P-value < 0·001). At the same time, there was a drop in the α2(V) antibodies from 420 ± 170 ng/ml (−2 years) to 210 ± 50 ng/ml (+18 months) after BOS (P-value = 0·04). These results suggest strongly that α1(V) fragment of Col-V is the immunodominant portion and immune response to this may be involved in the immunopathogenesis of BOS.
Immunodominance of Col α1(V) during BOS
To demonstrate further the significance of Col α1(V)-specific antibody responses in BOS patients, we performed immunohistochemical staining of the lung biopsy collected 6 months after the development of BOS. To this end, patient lung biopsy sections were incubated with autologous biotinylated serum immunoglobulins and antibody deposition was imaged with streptavidin–FITC. As shown in Fig. 2a, there is specific deposition of the antibodies to α1(V) chain, indicating that Col α1(V) is the predominantly deposited extracellular matrix protein in the BOS lung. However, when biotinylated patient sera were preincubated overnight at 4°C were pre-adsorbed to α1(V) protein before being incubated with lung biopsy tissue there was a significant decrease in the antibody binding (Fig. 2b). In contrast, antibody pre-adsorbed to α2(V) protein did not show any significant inhibition (Fig. 2c), indicating BOS patient antibody specificity to α 1(V) of Col-V.
Fig. 2.
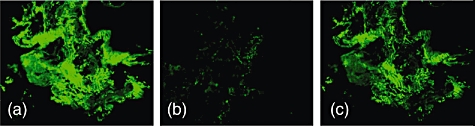
Immunohistochemical staining of the frozen sections taken from lung biopsy specimens of patients at 6 months after the clinical diagnosis of bronchiolitis obliterans syndrome (BOS). (a) Biopsy tissue treated with 6 months post-BOS(+) serum; (b) biopsy tissue treated with 6 months post-BOS(+) serum pre-incubated with collagen (Col) α1(V); and (c) biopsy tissue treated with 6 months post-BOS(+) serum pre-incubated with Col α2(V).
The switch in the Col-V peptide specificities of peripheral blood Th subsets of BOS patients correlate with antibody reactivity to immunodominant epitopes of Col α1(V) and α2(V). To determine the immunodominant epitopes of Col α1(V) and Col α2(V), we identified peptides of Col-V using an HLA class-II binding prediction program (detailed in Materials and methods). Initially, 42 peptides (Supplementary Fig. S1) were synthesized based on prediction probability and tested for cytokine responses. Of these 42 peptides, 14 peptides (pep1–14) (Table 2) which demonstrated significant cytokine response to ELISPOT assay were chosen for further studies.
We have demonstrated previously that immediately after LTx there is a high frequency of IL-10-producing T cells specific for self-antigens, including Col-V, and prior to development of BOS there is a significant reduction in IL-10 with concomitant increases in IFN-γ and IL-17 [17]. To determine the immune mechanisms, ELISPOT analysis was performed using serial samples obtained following LTx. We determined the ability of individual peptides derived from Col-V to stimulate IL-10 and calculated the precursor frequency of antigen-specific CD4+ T cells at (–)2years, (–)1 year and (+)6 months) following LTx (Fig. 3a). Two years before the onset of BOS, pep6, 7, 9, 11 and 14 induced high IL-10 responses (precursor frequency of 180 ± 21, 210 ± 24, 212 ± 21, 196 ± 23 and 182 ± 32, respectively). In contrast, pep5 resulted in minimal IL-10 response (78 ± 16) and all other peptides did not induce any IL-10 responses. More significant is that at (–)1 year and (+)6 months there was significant reduction of IL-10 responses to all peptides, with concomitant increases in IFN-γ and IL-17. Pep1, 4 and 5 stimulated high IFN-γ and IL-17 responses, respectively, at 6 months after the development of BOS (Fig. 3b,c). The individual Th responses to peptides by all LTxR are shown in the supplementary data Fig. S2a–c). Th responses in stable LTxR and normal smokers were predominantly IL-10 with minimal or no IFN-γ and IL-17 response (data not shown).
Fig. 3.
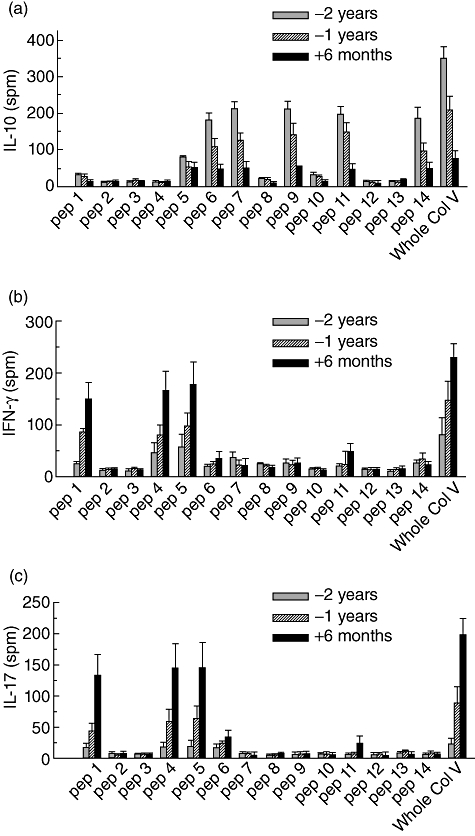
T helper (Th)-precursor frequency upon stimulation with individual peptides and whole collagen-V (Col-V). (a) Col-V epitope-induced interleukin (IL)-10 precursor frequency; (b) Col-V epitope-induced interferon (IFN)-γ precursor frequency; and (c) Col-V epitope-induced IL-17 precursor frequency. The data are representative of experiments performed in triplicate, and the mean ± standard error of the mean bars for the 12 patients were illustrated. The results are expressed as spots/million cells (s.p.m.). The specific Th-precursor frequency for the individual patient and respective peptide are shown in supplementary Fig. S4.
We also determined serially the serum cytokine levels of IL-10, IFN-γ and IL-17 in patients who developed BOS using the Luminex multiplex kit. Similar to ELISPOT results, there was a higher concentration of IL-10 at 2 years before the development of BOS. However, 6 months after BOS, there were increased levels of IFN-γ and IL-17 levels (Fig. 4). Decreases in Treg frequency and a decline in IL-10 cytokine has been proposed to play an important role in the development of autoimmunity [28]. IL-10 has also been shown to be a dominant cytokine by Tregs[29]. Due to our finding that a change in IL-10 was associated with increases in IFN-γ- and IL-17-secreting T cells upon stimulation with collagen peptides, we determined the change in the frequency of peripheral Tregs in LTxR who have developed BOS using the DNA methylation assay and a significant drop from 7 to 0·2% at BOS was noted (Fig. 4d). This finding supports that increases in IFN-γ and IL-17 production may be due to a decrease in the peripheral Tregs, favouring a role for Tregs in the cytokine switch noted in LTxR with BOS.
Fig. 4.
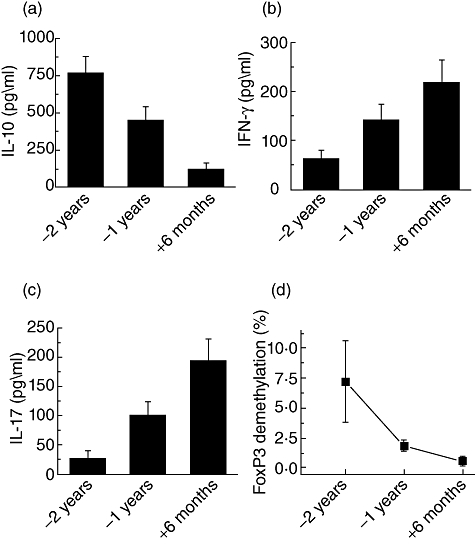
Serum cytokine levels of (a) interleukin (IL)-10; (b) interferon (IFN)-γ; (c) IL-17; and (d) bronchoalveolar lavage (BAL) fluid analysis of forkhead box P3 (FoxP3) DNA methylation assay for the regulatory T cell (Treg) frequency in peripheral blood. The data are representative of experiments performed in triplicate, and the mean ± standard error of the mean bars for the 12 patients are illustrated (P < 0·01).
Enhanced MMP2 and 9 activation in BOS patients
Activation of MMP2 and 9 have been shown to be associated with the development of BOS [30]. MMP2 and 9 are involved in the specific cleavage of Col-V [31]. To determine whether the MMPs activation will correlate with the shift in the Col-V epitopes which we have noted during BOS, bronchoalveolar lavage (BAL) was analysed by gelatin zymography. Preferential activation of MMP2 and 9 was seen in LTxR who developed BOS (Fig. 5a). Quantitative densitometric analysis of MMP activation as shown by the ratio of activated over inactive also demonstrated a 16-fold activation of MMP2 (8·6 ± 2·1) at BOS(−2 years) and 0·5 ± 0·3 at BOS(–) (Fig. 5b) and eightfold activation of MMP9 (0·82 ± 0·12) at BOS(−2 years) and 0·1 ± 0·05 at BOS(–) (Fig. 5c). These results are in agreement with the MMP analysis performed by RT–PCR (Fig. 5d). Analysis for MMP activation in the BAL from patients who had stable lung function (post-LTx 45·5 ± 9·3 months) (n = 7) did not demonstrate enhanced MMP2 and 9 activation. MMP7 did not show much change in expression (data not shown). Furthermore, RT–PCR for tissue inhibitors of MMP2 and MMP9, TIMP-2 and TIMP-1, respectively [32], also demonstrated significantly decreased expression (P < 0·01) prior to BOS (Fig. 5e). Taken together, these results indicate that there is specific activation of MMPs prior to BOS which can result in the exposure of otherwise cryptic Col-V determinants.
Fig. 5.
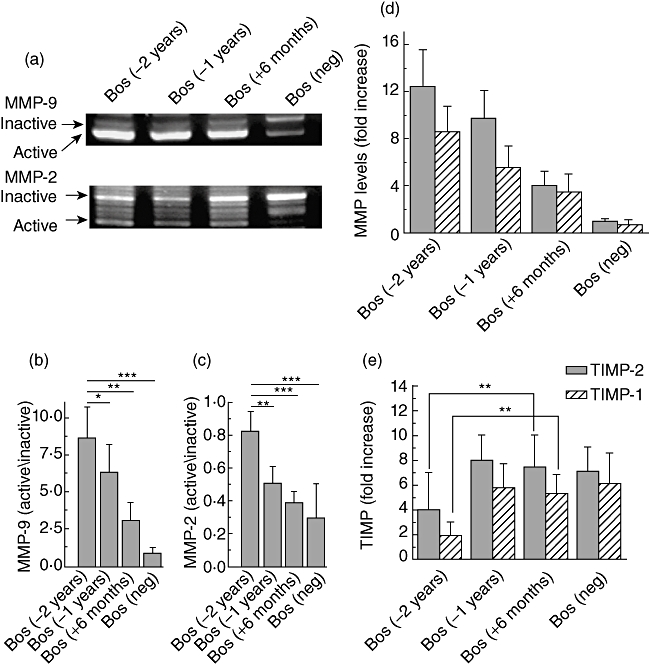
Matrix metalloproteinases (MMP)2 and 9 assays by gelatin zymography. (a) Activity of MMP2 (66 kDa) and MMP9 (97 kDa); (b,c) densitometric analysis of the ratio of activated over inactivated MMP9 and 2, respectively; (d) reverse transcription–polymerase chain reaction (RT–PCR) analysis of the expression of MMP2 and 9; and (e) RT–PCR analysis for the expression of tissue inhibitors of MMPs, tissue inhibitor of metalloproteinase (TIMP)-2 and 1. bronchiolitis obliterans syndrome [BOS(neg)] refers to time-matched stable lung transplant (LTx) recipients. (**) P < 0·01 and (***) P < 0·001).
Discussion
The development of immune responses to self-antigens and its possible role in the pathogenesis of chronic rejection (BOS) following human LTx has been reported recently both by our group [10] and others [9]. Following human LTx several immune mediated and non-immune mediated risk factors [donor and recipient age, graft ischaemic time, gastro-oesophageal reflux disease (GERD), metalloproteinase degradation and bacterial/fungal/non-cytomegalovirus (CMV) viral infection] have been associated with development of inflammatory milieu within the graft [1,8]. This inflammatory milieu predisposes to the release of self-antigens or their determinants leading to immune response and development of antibodies to self-antigens (Col-V and K-α1T) [9,10,14]. It is well recognized that T cells play and important role towards distinguishing self and non-self [33]. Activated CD4+ T cells are known to differentiate into either Th1, Th2 or Th17 phenotypes, characterized by production of specific cytokines [9,17,20,28,34]. It is of interest to note that polarization of the T cells into Th phenotypes is often mutually antagonistic and Th2 inhibits the differentiation of Th1 and Th17 [35].
As with LTx, the presence of autoimmune responses to self-antigens in chronic allograft rejection following other solid organ transplantation is well documented [9,20]. The presence of antibodies to self-antigens, myosin and vimentin have been documented in human cardiac transplants and T cells specific for cardiac myosin and vimentin has been shown to be associated with chronic cardiac allograft rejection, i.e. cardiac allograft vasculopathy (CAV) [36–38]. Studies have also suggested that antibodies against vimentin, a cytoskeleton protein, are an independent predictor of coronary atherosclerosis and may contribute to the accelerated onset of transplant associated coronary vasculopathy [37,39]. Similarly, transplant glomerulopathy following renal transplantation is associated with the presence of antibodies directed to the second extracellular loop of the angiotensin II type 1 (AT1) receptor and has been suggested to be a marker for refractory allograft rejection [40,41].
Various mechanisms including molecular mimicry, bystander activation, release of self-antigens and epitope spreading have been hypothesized to play a role in the development of immune responses to self-antigens [42]. In our study, we identified a novel ‘restrictive epitope shift’ mechanism, wherein we demonstrate that a shift of immunodominant epitopes of Col-V plays a crucial role in defining the Th phenotype switch, and the ensuing induction of cytokines specific to Col-V leads to immune responses to Col-V and development of BOS following human LTx.
The presence of antibodies to Col-V in LTxR who develop BOS has been demonstrated recently [43]. Col-V is a heterotrimer of two fragments α1(V), and one fragment of α2(V). In normal lung Col-V is sequestered within the fibrils of Col-I and Col-III. Sequential analysis for antibody development against Col-V in the patients' sera at various time-points following LTx (Figs 1 and 2) demonstrated that patients initially develop (2 years prior to BOS) antibodies to both α1(V) and α2(V), and at diagnosis of BOS the antibodies restrict to α1(V). This is not a time-dependent loss of antibodies to α2(V), as a similar cohort of time-matched (mean post-LTx duration 45·5 ± 9·3 months) stable LTx recipients (n = 7) demonstrated the persistence of antibodies to both chains of Col-V (Fig. 1a). Furthermore, there was no significant immunoglobulin (Ig)G-isotype change in the Col-V antibodies in the serum of the patients prior to and following the development of BOS (data not shown). The mechanism for the shift in the antibody specificity and restriction to 1(V) of Col-V is currently unknown, and in this report we propose that a restrictive shift in the immunodominant portion of Col-V occurs prior to BOS development; this is associated strongly with increased activation of MMPs. This also suggests that antibodies to α1(V) may play a role in the pathogenesis of BOS.
Epitope spreading is a well-recognized phenomenon in solid organ transplantation [44], and it has been proposed that epitope spreading against alloantigens may play an important role in allograft rejection and also in other autoimmune diseases, including multiple sclerosis, type 1 diabetes and myasthenia gravis [42]. However, contrary to the published results, we observed a restriction and ‘shifting’ of immune response to α1(V) from initial responses to both α1(V) and α2(V). To determine the mechanism which leads to the restrictive epitope shifting during chronic rejection following LTx, and whether it correlates with T cell reactivity including Th phenotype switch, we performed ELISPOT assays to determine induction of cytokine synthesis following stimulation with collagen peptides. Our results, using 14 epitopes (1–8 from α1(V), and 9–14 from α2(V) chains), demonstrated that the dominant epitopes 2 years before BOS (pep6, 7, 9, 11 and 14) stimulated IL-10 preferentially, whereas the same epitopes at 6 months after the development of BOS failed to induce IL-10. This indicates that there is a restrictive shift leading to a decrease in the frequency of T cells responsive to these epitopes (pep6, 7, 9, 11 and 14) which stimulate IL-10, a predominant toleregenic cytokine of Treg origin [29]. However, there is an increase in the Th1 (IFN-γ) and Th17 (IL-17) cytokine synthesis and T cell responses are restricted to epitopes (pep1, 4 and 5) from α1(V) at the diagnosis of BOS. Stimulation with hydroxyl-proline-modified peptides gave similar results, suggesting that post-translational modification of Col-V played a minimal role, if any, in the Th-specific immune response (data not shown). The Th phenotype precursor frequency switch was also correlated with serum cytokines at the respective time-points (Fig. 4). This agrees with our findings presented in Figs 1 and 2 that at 2 years prior to BOS the LTxR develop antibodies to both α1(V) and α2(V) chains of Col-V. However, at 6 months after diagnosis with BOS the immunodominant epitopes (pep1, 4 and 5) are entirely from α1(V). Furthermore, it is of interest to note that pep1–4 are predominantly part of procollagen α1(V), which is normally cleaved off, before its incorporation into tissues. Based on this, we postulate that during high turnover of the extracellular matrix in the lungs the ‘cryptic’ antigenic determinants of procollagen is most probably exposed, which is antigenic and stimulates both humoral and cellular immune responses to this self-antigen.
Studies have shown that an increase in Tregs levels in the circulation correlate with stable graft function of liver, lung and kidney allografts [28], and a significant reduction in Treg is often associated with rejection [45]. In this study we examined the FoxP3+ Treg cell frequency using a FoxP3 DNA methylation assay. A significant decrease in the frequency of peripheral Tregs with a concomitant increase in the frequency of Th17 response was noted in LTxR diagnosed with BOS. These results suggest strongly that Treg may also play a role in shifting the balance between T helper cells and the local inflammatory cytokine milieu.
One of the questions we explored is the mechanism for the change in the Col-V epitopes following LTx. As there are published reports indicating that the activity of MMP2 and MMP9 is up-regulated after LTx [46,47], and MMP9 has been reported to play a pathogenic role in the development of autoimmune encephalomyelitis, experimental bullous pemphigoid, experimental myasthenia gravis and experimental arthritis [48–50], we determined whether there is any correlation between MMP activity and change in the Col-V epitope. Although the enzymatic role of MMPs in the regulation of immune response are not fully understood, MMP2 and 9 are shown to cleave collagen molecules [31]. It has been shown that the MMP9 cleavage site for Col-V is in the of α1(V) at the N-terminus non-helical telopeptide [51] and Gly439-Val [52]. Hibbs et al. have demonstrated that human alveolar macrophages in culture elaborate matrix metalloproteinases that degrade both native Col-V and denatured collagens [53]. Therefore, we first determined whether the shift in Col-V epitope which we have noted correlated with the enzymatic action of MMPs. To this end, we determined the MMP activation in the BAL fluid collected from the BOS+ patients using gelatin zymography. Our data demonstrate that there are significant increases in MMP2 (16-fold) and MMP9 (eightfold) along the decrease in tissue inhibitor of MMP, TIMP1 and 2 (Fig. 5) activity in the BAL fluid of BOS+ patients. It is noteworthy that the activation of MMPs is seen prior to the shift in epitopes. Previous studies in human LTx with BOS and animal models of OAD have shown an increase in MMP levels prior to development of chronic rejection pathology [26,30], and MMP9-deficient animals do not develop OAD [54]. Therefore, based on this strong correlative evidence, we propose that the activation of MMPs causes differential cleavage and exposure of cryptic Col-V antigenic determinants which leads to the Th phenotype switch. It will be of interest to determine whether cleavage and liberation of specific pep1, 4 and 5 can take place by addition of MMP2 and 9 to Col-V and the BAL sample from LTxR diagnosed with BOS. In spite of this limitation, our current study agrees well with the previous reports of elevated MMPs in BOS and autoimmune diseases which ‘uncover’ different cryptic epitopes.
In conclusion, we demonstrate for the first time that a restrictive shift in the epitopes of self-antigen Col-V is critical for the Th phenotype switch leading to autoimmune responses which predisposes to chronic rejection following human LTx. We propose that following transplantation there is a loss of peripheral tolerance mechanism mediated primarily by a decrease in Treg and loss of IL-10 responses to self-antigen. This is probably mediated by a shift in the epitopes of Col-V, which is due most probably to differential cleavage of Col-V by increased activity of MMPs. This shift in the immunodominant peptides of Col-V results in induction of IFN-γ and Th17 responses in conjunction with the loss of Tregs and peripheral tolerance, leading to autoimmunity which initiates a cascade of cytokines and growth factors that are involved in the immunopathogenesis of chronic rejection following human LTx. Based upon the reports that immune responses to tissue-specific self-antigens may play a role in chronic rejection following cardiac and renal transplantation, it is likely that a mechanism similar to what we have noticed in this report for exposing the self-antigenic determinants of Col-V may also play a role in exposure of cryptic determinants of other self-antigens proposed to be involved in chronic rejection following all solid organ transplantations.
Acknowledgments
This work was supported by NIH/NHLBI RO1 HL056643, NIH/NHLBI RO1 HL092514 and BJC foundation (T.M.), NIH/NHLBI T32 HL007312 (J.W.) and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (D.B.). We thank Ms Billie Glasscock for her help in preparing this manuscript.
Disclosure
All the authors declare no competing financial interests.
References
- 1.Arcasoy SM, Kotloff RM. Lung transplantation. N Engl J Med. 1999;340:1081–91. doi: 10.1056/NEJM199904083401406. [DOI] [PubMed] [Google Scholar]
- 2.Trulock EP, Edwards LB, Taylor DO, et al. The Registry of the International Society for Heart and Lung Transplantation: twentieth official adult lung and heart-lung transplant report – 2003. J Heart Lung Transplant. 2003;22:625–35. doi: 10.1016/s1053-2498(03)00182-7. [DOI] [PubMed] [Google Scholar]
- 3.Sarahrudi K, Estenne M, Corris P, et al. International experience with conversion from cyclosporine to tacrolimus for acute and chronic lung allograft rejection. J Thorac Cardiovasc Surg. 2004;127:1126–32. doi: 10.1016/j.jtcvs.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 4.de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia–reperfusion-induced lung injury. Am J Respir Crit Care Med. 2003;167:490–511. doi: 10.1164/rccm.200207-670SO. [DOI] [PubMed] [Google Scholar]
- 5.Seetharam A, Tiriveedhi V, Mohanakumar T. Alloimmunity and autoimmunity in chronic rejection. Curr Opin Organ Transplant. 2010;15:531–6. doi: 10.1097/MOT.0b013e32833b31f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan SR, Paradis IL, Yousem SA, et al. Sequelae of cytomegalovirus pulmonary infections in lung allograft recipients. Am Rev Respir Dis. 1992;146:1419–25. doi: 10.1164/ajrccm/146.6.1419. [DOI] [PubMed] [Google Scholar]
- 7.Reid KR, McKenzie FN, Menkis AH, et al. Importance of chronic aspiration in recipients of heart–lung transplants. Lancet. 1990;336:206–8. doi: 10.1016/0140-6736(90)91734-r. [DOI] [PubMed] [Google Scholar]
- 8.Al-Githmi I, Batawil N, Shigemura N, et al. Bronchiolitis obliterans following lung transplantation. Eur J Cardiothorac Surg. 2006;30:846–51. doi: 10.1016/j.ejcts.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 9.Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha1 tubulin-specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180:4487–94. doi: 10.4049/jimmunol.180.7.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiriveedhi V, Weber J, Seetharam A, Mohanakumar T. Cross-talk of alloimmune response and autoimmunity: role in pathogenesis of chronic rejection. Discov Med. 2010;9:229–35. [PubMed] [Google Scholar]
- 12.Vanderlugt CJ, Miller SD. Epitope spreading. Curr Opin Immunol. 1996;8:831–6. doi: 10.1016/S0952-7915(96)80012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–9. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 14.Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182:309–18. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–7. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 16.Ciubotariu R, Liu Z, Colovai AI, et al. Persistent allopeptide reactivity and epitope spreading in chronic rejection of organ allografts. J Clin Invest. 1998;101:398–405. doi: 10.1172/JCI1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bharat A, Fields RC, Trulock EP, Patterson GA, Mohanakumar T. Induction of IL-10 suppressors in lung transplant patients by CD4+25+ regulatory T cells through CTLA-4 signaling. J Immunol. 2006;177:5631–8. doi: 10.4049/jimmunol.177.8.5631. [DOI] [PubMed] [Google Scholar]
- 18.Bharat A, Mohanakumar T. Allopeptides and the alloimmune response. Cell Immunol. 2007;248:31–43. doi: 10.1016/j.cellimm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun RK, Molitor-Dart M, Wigfield C, et al. Transfer of tolerance to collagen type V suppresses T-helper-cell-17 lymphocyte-mediated acute lung transplant rejection. Transplantation. 2009;88:1341–8. doi: 10.1097/TP.0b013e3181bcde7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bharat A, Fields RC, Steward N, Trulock EP, Patterson GA, Mohanakumar T. CD4+25+ regulatory T cells limit Th1-autoimmunity by inducing IL-10 producing T cells following human lung transplantation. Am J Transplant. 2006;6:1799–808. doi: 10.1111/j.1600-6143.2006.01383.x. [DOI] [PubMed] [Google Scholar]
- 21.Ali MK, Bergson C. Elevated intracellular calcium triggers recruitment of the receptor cross-talk accessory protein calcyon to the plasma membrane. J Biol Chem. 2003;278:51654–63. doi: 10.1074/jbc.M305803200. [DOI] [PubMed] [Google Scholar]
- 22.Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37:2378–89. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 23.Wieczorek G, Asemissen A, Model F, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69:599–608. doi: 10.1158/0008-5472.CAN-08-2361. [DOI] [PubMed] [Google Scholar]
- 24.Lal G, Zhang N, van der Touw W, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–73. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woessner JF., Jr Quantification of matrix metalloproteinases in tissue samples. Methods Enzymol. 1995;248:510–28. doi: 10.1016/0076-6879(95)48033-1. [DOI] [PubMed] [Google Scholar]
- 26.Campbell LG, Ramachandran S, Liu W, et al. Different roles for matrix metalloproteinase-2 and matrix metalloproteinase-9 in the pathogenesis of cardiac allograft rejection. Am J Transplant. 2005;5:517–28. doi: 10.1111/j.1600-6143.2005.00744.x. [DOI] [PubMed] [Google Scholar]
- 27.Chanut-Delalande H, Fichard A, Bernocco S, Garrone R, Hulmes DJ, Ruggiero F. Control of heterotypic fibril formation by collagen V is determined by chain stoichiometry. J Biol Chem. 2001;276:24352–9. doi: 10.1074/jbc.m101182200. [DOI] [PubMed] [Google Scholar]
- 28.Bharat A, Fields RC, Mohanakumar T. Regulatory T cell-mediated transplantation tolerance. Immunol Res. 2005;33:195–212. doi: 10.1385/IR:33:3:195. [DOI] [PubMed] [Google Scholar]
- 29.O'Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest. 2004;114:1372–8. doi: 10.1172/JCI23215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith GN, Jr, Mickler EA, Payne KK, et al. Lung transplant metalloproteinase levels are elevated prior to bronchiolitis obliterans syndrome. Am J Transplant. 2007;7:1856–61. doi: 10.1111/j.1600-6143.2007.01850.x. [DOI] [PubMed] [Google Scholar]
- 31.Lauer-Fields JL, Sritharan T, Stack MS, Nagase H, Fields GB. Selective hydrolysis of triple-helical substrates by matrix metalloproteinase-2 and -9. J Biol Chem. 2003;278:18140–5. doi: 10.1074/jbc.M211330200. [DOI] [PubMed] [Google Scholar]
- 32.Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 33.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 34.Yamada Y, Sekine Y, Yoshida S, et al. Type V collagen-induced oral tolerance plus low-dose cyclosporine prevents rejection of MHC class I and II incompatible lung allografts. J Immunol. 2009;183:237–45. doi: 10.4049/jimmunol.0804028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 36.Kaczmarek I, Deutsch MA, Kauke T, et al. Donor-specific HLA alloantibodies: long-term impact on cardiac allograft vasculopathy and mortality after heart transplant. Exp Clin Transplant. 2008;6:229–35. [PubMed] [Google Scholar]
- 37.Rose ML, Smith JD. Clinical relevance of complement-fixing antibodies in cardiac transplantation. Hum Immunol. 2009;70:605–9. doi: 10.1016/j.humimm.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 38.Nath DS, Ilias Basha H, Tiriveedhi V, et al. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant. 2010;29:1277–85. doi: 10.1016/j.healun.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leong HS, Mahesh BM, Day JR, et al. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. J Leukoc Biol. 2008;83:263–71. doi: 10.1189/jlb.0607339. [DOI] [PubMed] [Google Scholar]
- 40.Fotheringham J, Angel CA, McKane W. Transplant glomerulopathy: morphology, associations and mechanism. Nephron Clin Pract. 2009;113:c1–7. doi: 10.1159/000228069. discussion c. [DOI] [PubMed] [Google Scholar]
- 41.Li C, Yang CW. The pathogenesis and treatment of chronic allograft nephropathy. Nat Rev Nephrol. 2009;5:513–19. doi: 10.1038/nrneph.2009.113. [DOI] [PubMed] [Google Scholar]
- 42.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 43.Haque MA, Mizobuchi T, Yasufuku K, et al. Evidence for immune responses to a self-antigen in lung transplantation: role of type V collagen-specific T cells in the pathogenesis of lung allograft rejection. J Immunol. 2002;169:1542–9. doi: 10.4049/jimmunol.169.3.1542. [DOI] [PubMed] [Google Scholar]
- 44.Bradley JA. Indirect T cell recognition in allograft rejection. Int Rev Immunol. 1996;13:245–55. doi: 10.3109/08830189609061751. [DOI] [PubMed] [Google Scholar]
- 45.Dai Z, Li Q, Wang Y, et al. CD4+CD25+ regulatory T cells suppress allograft rejection mediated by memory CD8+ T cells via a CD30-dependent mechanism. J Clin Invest. 2004;113:310–17. doi: 10.1172/JCI19727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trello CA, Williams DA, Keller CA, Crim C, Webster RO, Ohar JA. Increased gelatinolytic activity in bronchoalveolar lavage fluid in stable lung transplant recipients. Am J Respir Crit Care Med. 1997;156:1978–86. doi: 10.1164/ajrccm.156.6.9704044. [DOI] [PubMed] [Google Scholar]
- 47.Hubner RH, Meffert S, Mundt U, et al. Matrix metalloproteinase-9 in bronchiolitis obliterans syndrome after lung transplantation. Eur Respir J. 2005;25:494–501. doi: 10.1183/09031936.05.00091804. [DOI] [PubMed] [Google Scholar]
- 48.Dubois B, Masure S, Hurtenbach U, et al. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest. 1999;104:1507–15. doi: 10.1172/JCI6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faber-Elmann A, Grabovsky V, Dayan M, Sela M, Alon R, Mozes E. An altered peptide ligand inhibits the activities of matrix metalloproteinase-9 and phospholipase C, and inhibits T cell interactions with VCAM-1 induced in vivo by a myasthenogenic T cell epitope. FASEB J. 2001;15:187–94. doi: 10.1096/fj.99-0976com. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Shipley JM, Vu TH, et al. Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med. 1998;188:475–82. doi: 10.1084/jem.188.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okada Y, Naka K, Kawamura K, et al. Localization of matrix metalloproteinase 9 (92-kilodalton gelatinase/type IV collagenase = gelatinase B) in osteoclasts: implications for bone resorption. Lab Invest. 1995;72:311–22. [PubMed] [Google Scholar]
- 52.Niyibizi C, Chan R, Wu JJ, Eyre D. A 92 kDa gelatinase (MMP-9) cleavage site in native type V collagen. Biochem Biophys Res Commun. 1994;202:328–33. doi: 10.1006/bbrc.1994.1931. [DOI] [PubMed] [Google Scholar]
- 53.Hibbs MS, Hoidal JR, Kang AH. Expression of a metalloproteinase that degrades native type V collagen and denatured collagens by cultured human alveolar macrophages. J Clin Invest. 1987;80:1644–50. doi: 10.1172/JCI113253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandez FG, Campbell LG, Liu W, et al. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am J Transplant. 2005;5:671–83. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]