

Abstract
Elucidating the response of breast cancer cells to chemotherapeutic and hormonal based drugs is clearly important as these are frequently used therapeutic approaches. A signaling pathway often involved in chemo- and hormonal-resistance is the Ras/PI3K/PTEN/Akt/mTOR cascade. In the studies presented in this report, we have examined the effects of constitutive activation of Akt on the sensitivity of MCF-7 breast cancer cells to chemotherapeutic- and hormonal-based drugs as well as mTOR inhibitors. MCF-7 cells which expressed a constitutively-activated Akt-1 gene [ΔAkt-1(CA)] were more resistant to doxorubicin, etoposide and 4-OH-tamoxifen (4HT) than cells lacking ΔAkt-1(CA). Cells which expressed ΔAkt-1(CA) were hypersensitive to the mTOR inhibitor rapamycin. Furthermore, rapamycin lowered the IC50s for doxorubicin, etoposide and 4HT in the cells which expressed ΔAkt-1(CA), demonstrating a potential improved method for treating certain breast cancers which have deregulated PI3K/PTEN/Akt/mTOR signaling. Understanding how breast cancers respond to chemo- and hormonal-based therapies and the mechanisms by which they can become drug resistant may enhance our ability to treat breast cancer. These results also document the potential importance of knowledge of the mutations present in certain cancers which may permit more effective therapies.
Keywords: Akt, mTOR, Targeted Therapy, Drug Resistance
INTRODUCTION
Growth factor receptors are critical for controlling the proliferation of cells. Once activated, these receptors induce multiple signaling pathways involved in growth, prevention of apoptosis, differentiation and other fundamental processes. The epidermal growth factor (EGF) receptor (EGFR) and isoforms such as HER2 are also associated with breast cancer development and resistance to anticancer agents [1]. The HER2 growth factor receptor is frequently amplified in human breast cancer and is a target of diverse therapeutic approaches as well as an important biomarker [2-4]. Among the signaling pathways downstream of these and other receptors, the PI3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK pathways have been shown to regulate growth and prevent apoptosis and their deregulation is often implicated in malignant transformation [5-12]. These pathways also play key roles in the genesis of cancer initiating cells (CICs, a.k.a. cancer stem cells) [13-22].
The PI3K/PTENAkt/mTOR pathway is frequently activated by mutations which may alter sensitivity to targeted therapy. The PI3K p110 catalytic subunit gene (PIK3CA) is one of the most frequently mutated genes in breast cancer [23]. Mutations in the kinase domain of PIK3CA will result in activation of Akt and contribute to malignant transformation, however the roles of these mutations in sensitivity to traditional chemo- and hormonal- based therapies and targeted therapies are not so well elucidated [24-26].
The regulation of Akt activation is complex. Akt is a key domino in determining whether a cell will survive and proliferate, undergo apoptosis or senescence [27-40]. Phosphatidylinositol (PI) (3,4)P2 and PI(3,4,5)P3 produced by class 1A PI3Ks recruit phosphoinositide dependent kinase-1 (PDK1) as well as Akt isoforms to the plasma membrane by interacting with their pleckstrin homology (PH) domains [41-44]. Co-localization of PDK1 with Akts at the plasma membrane causes PDK1 to phosphorylate Akts at a threonine residue (T308) and a serine residue (S473). The kinase which phosphorylates S473 is believed to be mTORC2, which actually is the rapamycin-insensitive complex which lies downstream of Akt [44]. Activation of PDK1 and Akt by class 1A PI3Ks is negatively regulated by phosphatase and tensin homologue deleted on chromosome ten (PTEN) [7, 9, 44-48]. PTEN removes phosphate groups from PI(3,4)P2 and PI(3,4,5)P3 added by PI3K as well as from tyrosine phosphorylated proteins including focal adhesion kinase (FAK) and Shc [7,11,44]. PTEN is a critical tumor suppressor gene and another key lynch-pin in regulation of this pathway. PTEN is frequently inactivated by various genetic mechanisms which results in Akt activation.
Diverse mechanisms regulate PTEN expression [10,47-50]. These range from gene deletion, alterations in mRNA splicing, subcellular localization or epigenetic mechanisms which prevent PTEN transcription. There are many microRNAs (miRNAs) which target the PTEN gene to inhibit its expression [10,47,51]. Furthermore there is a pseudo PTEN gene which serves as a decoy to bind and neutralize some of these miRNAs [10,47]. Mutations have been reported to occur at PTEN in breast cancer at varying frequencies (5-21%). While PTEN is deleted in certain cancers, loss of heterozygosity (LOH) is probably a more common genetic event (30%) leading to changes in PTEN expression [47,52]. PTEN promoter methylation leads to low PTEN expression [47]. In one study, 26% of primary breast cancers had low PTEN levels which correlated with lymph node metastases and poor prognoses [53]. PTEN has both plasma membrane and nuclear localized activities. Disruption of PTEN activity by various mechanisms frequently results in Akt activation. Activation of Akt could have vast effects on different processes affecting breast cancer drug resistance and sensitivity to targeted therapy.
A consequence of impaired PTEN expression is elevated activation of Akt. Elevated Akt activity can have many effects on cell growth, such as; the phosphorylation and activation/inactivation of transcription factors which control pivotal gene expression, the inactivation of anti-apoptotic molecules by their phosphorylation and subsequent protesomal degradation, or by the regulation of translation of key “weak” mRNAs involved in growth. One downstream molecule of mTOR is ribosomal S6 kinase (p70S6K). This kinase regulates the efficiency of translation of certain mRNAs and also functions in a negative feedback loop to control Akt activity [10,44,54-55]. The roles of Akt and p70S6K and other molecules are essential for the formation of the eIF4F translation complex. The eIF4F complex is necessary for the translation of mRNAs containing long 5’UTRs which are highly-structured and have an elevated G+C content. These mRNAs are considered “weak” mRNAs. These “weak” mRNAs often encode genes involved in oncogenesis and survival such as c-Myc, Mcl-1, cyclin-D, VEGF and survivin. Furthermore p70S6K has important roles in autophagy and cellular senescence [56].
Akt, mTOR and p70S6K activation have been associated with a more severe prognosis in breast and other cancers [53,57-64]. Targeting the PI3K/PTEN/Akt/mTOR pathway may prove effective in various cancer therapies [12,54,65-66]. High levels of activated Akt expression have been associated with both chemo- and hormonal resistance in breast cancer [58-59,67]. Indeed some studies have evaluated the effectiveness of targeting mTOR in PTEN-negative cells [60]. Cells which express high levels of activated Akt may be more sensitive to mTOR inhibitors and inhibition of mTOR activity by rapamycin may restore their sensitivity to chemo- and hormonal based therapies [9,60]. A distinct advantage to targeting mTOR with rapamycin is that rapamycin has been used many years to treat organ transplant patients (especially kidney transplant). Rapamycin is now being examined in treatment of certain cancers and in the prevention of aging and other diseases including AIDS [11-12,68].
Previously it was determined that mutated forms of Akt and PTEN can induce chemotherapeutic- and hormonal-based drug resistance in breast cancer [9,58,67]. PTEN mutation which eliminate lipid phosphatase activity will result in activated Akt expression which will lead to drug resistance and sensitivity to the mTOR inhibitor rapamycin [9].
In the following studies, the effects enforced expression of activated Akt-1 on drug resistance and sensitivity to targeted therapy. Inhibitors targeting mTOR suppressed drug resistance demonstrating the importance of mTOR in chemosensitivity. These experiments were performed to understand the effect of common chemotherapeutic drugs on anti-apoptotic signaling pathways as well as how these signaling pathways can change in drug resistant cells.
RESULTS
Effects of Constitutively-Activated Akt-1 Expression on the Sensitivity of MCF-7 Cells to Chemotherapeutic Drugs
A means to examine the effects of deregulated Akt-1 expression on the sensitivity of MCF-7 cells to chemotherapeutic- and hormonal-based drugs is to infect MCF-7 cells with a retrovirus encoding a constitutively-active Akt gene such as ΔAkt-1(CA) [69]. Pools of ΔAkt-1(CA)-infected MCF-7 cells were recovered after selection in medium containing G418 as that is the drug that the neoR gene present in the retroviral vector encodes resistance to. The ΔAkt-1(CA)-infected cells are named MCF7/ΔAkt-1(CA). The sensitivities of these cells to chemotherapeutic drugs were determined by MTT analysis after incubation for 4 days in the indicated culture conditions. Figure 1 illustrates the sensitivities of the MCF-7 and MCF7/ΔAkt-1(CA) cells to doxorubicin, etopside and paclitaxel. As an additional control, similar experiments were performed with MCF-7 cells transfected with the empty neor plasmid, pEGFP (MCF7/EGFP). These cells represent a control for cells transfected with just a neoR gene. MCF7/EGFP cells were similar in drug sensitivity as MCF-7 cells.
Figure 1. Effects of Akt-1 Expression on Chemotherapeutic Drug Sensitivity.
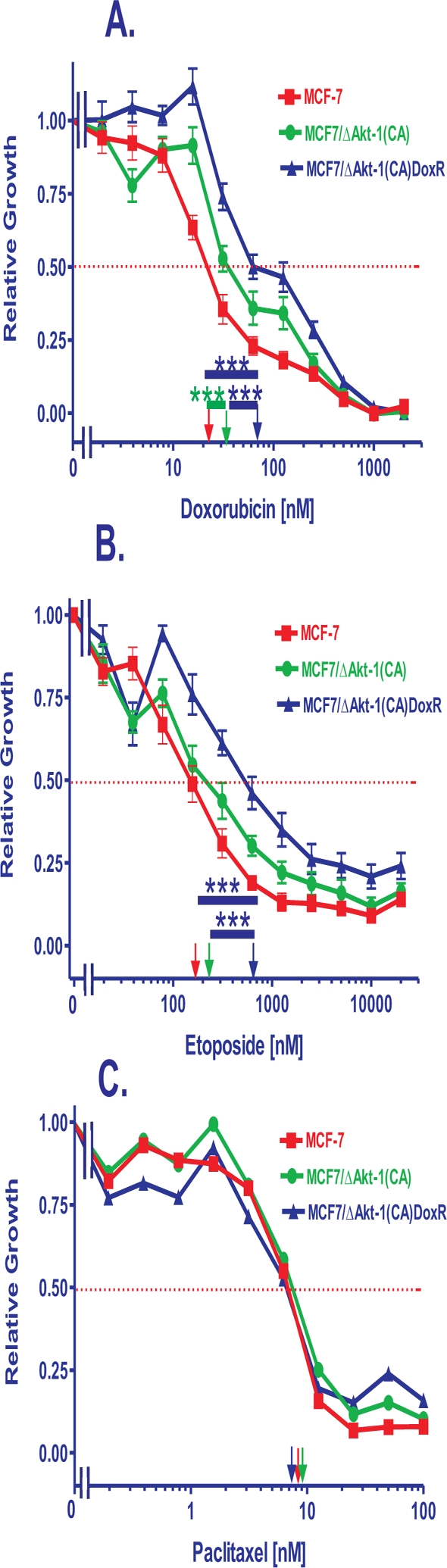
The effects of Akt-1(CA) expression on sensitivity of MCF-7 cells to: Panel A. doxorubicin, Panel B. etoposide and Panel C. paclitaxel were examined by MTT analysis after incubation of the cells in the indicated concentrations of the drugs. MCF7/ΔAkt-1(CA) were isolated after infection of MCF-7 cells with a retrovirus encoding ΔAkt-1(CA) and selection in G418 (geneticin) for 4 weeks. MCF7/ΔAkt-1(CA)DoxR cells were isolated after selection of MCF7/ΔAkt-1(CA) cells for 4 weeks in medium containing 25 nM doxorubicin. The dotted arrow represents where 50% inhibition of growth intercepts with the X axis and is used to estimate the IC50. The statistical significance was determined by the unpaired t test. Comparisons determined to be significant are indicated with (***). Panel A. The P value between doxorubicin treatment of MCF-7 and MCF7/ΔAkt-1(CA) cells was 0.0025. The P value between doxorubicin treatment of MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells was <0.0001. The P value between doxorubicin treatment of MCF7/ΔAkt-1(CA) and MCF7/ΔAkt-1(CA)DoxR cells was 0.0003. Panel B. The P value between etoposide treatment of MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells was 0.0003. The P value between etoposide treatment of MCF7/ΔAkt-1(CA) and MCF7/ΔAkt-1(CA)DoxR cells was 0.001.
Initially, the ΔAkt-1(CA)-infected cells were about 2- and 1.5-fold more resistant to doxorubicin (Figure 1, Panel A) and etoposide (Figure 1, Panel B) respectively than the parental MCF-7 (Table 1). In contrast the MCF7/ΔAkt-1(CA) cells did not show any difference in sensitivity to paclitaxel than the parental MCF-7 cell line from which they were derived (Figure 1, Panel C).
Table 1.
Effects of Activated Akt-1 Expression on Chemotherapeutic and Hormonal Drugs and Signal Transduction Inhibitors IC50s.1
| Cell Line | Drug Treatment | IC502 or Change in IC503 |
| MCF-7 | Doxorubicin | 20 nM2 |
| MCF-7/ΔAkt-1(CA) | Doxorubicin | 2 X↑3 |
| MCF-7/ΔAkt-1(CA)DoxR | Doxorubicin | 6.7 X↑3 |
| MCF-7 | Etoposide | 140 nM2 |
| MCF-7/ΔAkt-1(CA) | Etoposide | 1.5 X↑3 |
| MCF-7/ΔAkt-1(CA)DoxR | Etoposide | 3.1 X↑3 |
| MCF-7 | Paclitaxel | 10 nM2 |
| MCF-7/ΔAkt-1(CA) | Paclitaxel | -3 |
| MCF-7/ΔAkt-1(CA)DoxR | Paclitaxel | -3 |
| MCF-7 | 4HT | 7 nM2 |
| MCF-7/ΔAkt-1(CA) | 4HT | 4.3 X↑3 |
| MCF-7/ΔAkt-1(CA)DoxR | 4HT | 10 X↑3 |
| MCF-7 | Rapamycin | 7 nM2 |
| MCF-7/ΔAkt-1(CA) | Rapamycin | 2.5 X↓3 |
| MCF-7/ΔAkt-1(CA)DoxR | Rapamycin | 7 X↓3 |
We also isolated ΔAkt-1(CA) cells with increased resistance to chemotherapeutic drugs by culturing the cells for 4 weeks in medium containing 25 nM doxorubicin. These cells were named MCF7/ΔAkt-1(CA)DoxR and they were 5-fold more frequently recovered from cells infected with the ΔAkt-1(CA)-containing virus than from control cell lines. The MCF7/ΔAkt-1(CA)DoxR cells were 6.7- and 3.1-fold more resistant to doxorubicin and etoposide respectively than control MCF-7 cells (Figure 1, Panels A & B). In contrast, no difference in sensitivity to paclitaxel was observed (Figure 1, Panel C). The IC50s for doxorubicin ranged from approximately 10 to 120 nM in the doxorubicin sensitive MCF-7 and resistant MCF7/ΔAkt-1(CA)DoxR cells respectively. The IC50s for etoposide ranged from 150 to 600 nM in the doxorubicin sensitive MCF-7 and resistant MCF7/ΔAkt-1(CA)DoxR cells the respectively. In contrast, the IC50s for paclitaxel were approximately 10 nM in the doxorubicin sensitive MCF-7 and resistant MCF7/ΔAkt-1(CA)DoxR cells.
Effects of Constitutively-Activated Akt-1 Expression of the Sensitivity of MCF-7 Cells to the Hormone-Based Drug Tamoxifen
The effects of the hormonal drug 4-hydroxytamoxifen (4HT) were examined on the estrogen receptor positive (ER+) MCF-7 and MCF7/ΔAkt-1(CA) cells by MTT analysis (Figure 2, Panel A). The IC50s for 4HT in the ER+ MCF-7 and Akt-transduced cells ranged from 7 to 200 nM (Table 1). The MCF7/ΔAkt-1(CA) cells were approximately 4.3-fold more resistant to the effects of 4HT than MCF-7 cells. The MCF7/ΔAkt-1(CA)DoxR cells were approximately 10-fold more resistant the effects of 4HT than MCF-7 cells (Table 1). Thus constitutive Akt-1 expression elevated the concentration of 4HT required to achieve the IC50.
Figure 2. Effects of Akt Expression on Sensitivity to Tamoxifen and Rapamycin.
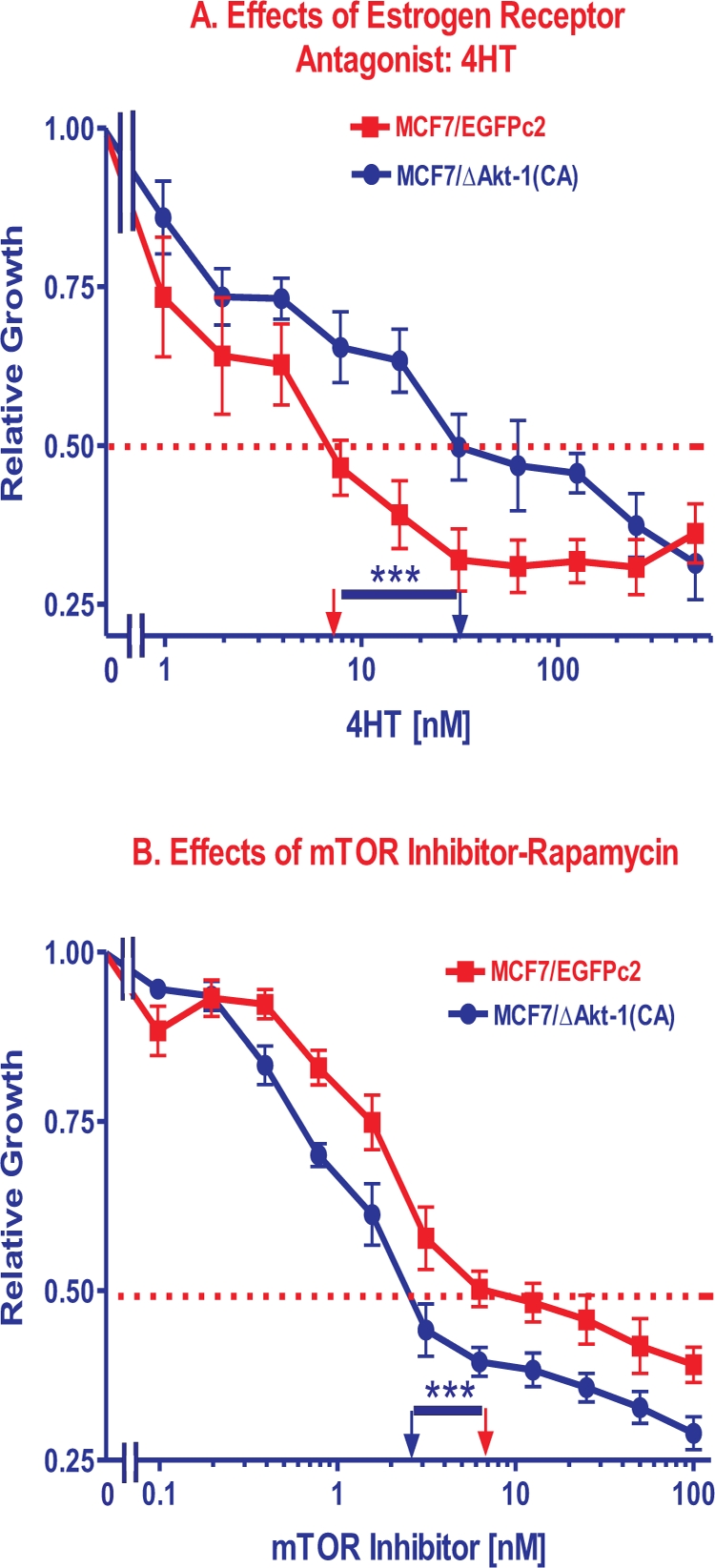
The effects of ΔAkt-1(CA) expression on sensitivity of MCF-7 cells to: Panel A. tamoxifen (4HT) and Panel B. rapamycin was examined by MTT analysis after incubation of the cells in the indicated concentrations of the drugs. The dotted arrow represents where 50% inhibition of growth intercepts with the X axis and is used to estimate the IC50. The statistical significance was determined by the unpaired t test. Comparisons determined to be significant are indicated with (***). Panel A. The P value between 4HT treatment of MCF7/EGFPc2 and MCF7/ΔAkt-1(CA) cells was 0.005. The P value between rapamycin treatment of MCF7/EGFPc2 and MCF7/ΔAkt-1(CA) cells was 0.019.
Effects of Constitutively-Activated Akt-1 Expression of the Sensitivity of MCF-7 Cells to the mTOR Inhibitor Rapamycin
Constitutive expression of activated Akt-1 conferred increased sensitivity to the mTOR inhibitor rapamycin as the IC50s for rapamycin were 2.8- and 7-fold lower in MCF-7/ΔAkt-1(CA) and MCF-7/ΔAkt-1(CA)DoxR, respectively, than in MCF-7 cells (Figure 2, Panel B and Table 1). The IC50s for rapamycin ranged from approximately 1 to 10 nM. MCF-7 cells are inherently sensitive to rapamycin, perhaps due to a mutation at PIK3CA in this cell line [23]. The doxorubicin-selected MCF-7/ΔAkt-1(CA)DoxR cells were 2.5-fold more sensitive to rapamycin than the MCF-7/ΔAkt-1(CA) cells. Thus introduction of activated Akt-1 into MCF-7 cells conferred increased sensitivity to rapamycin and drug resistant MCF-7/ΔAkt-1(CA)DoxR cells were even more sensitive to rapamycin.
Enhancement of Doxorubicin Induced-Inhibition of Proliferation by Addition of Tamoxifen
The abilities to improve chemotherapy by potentially lowering dose-associated deleterious side effects were examined. Namely, we determined the ability of hormonal-based therapy to lower the doxorubicin IC50. In this particular experiment presented in Figure 3, Panel A, we added a constant dose of 50 nM 4HT and different doses of doxorubicin to examine the effects on MCF7/ΔAkt-1(CA) cells. It should be pointed out that 4HT by itself, like rapamycin, did not totally eliminate viability of the cells after 4 days of incubation, as approximately 25% relative growth was still observed (Figure 2, Panels A & B). In contrast, high concentrations of doxorubicin (>500 nM) almost totally eliminated the relative growth of the cells at sufficient concentrations (Figure 1). As shown in Figure 3, Panel A, addition of 50 nM 4HT reduced the doxorubicin IC50 approximately 10-fold. Moreover, when the doses of doxorubicin required for essentially total cell death (>95% inhibition) were examined, a 2-fold lower dose of doxorubicin was required (250 vs 500 nM doxorubicin) when 4HT was added than in its absence.
Figure 3. Effects of Co-Treatment with Doxorubicin, 4HT and a mTOR Inhibitor. Panel A.
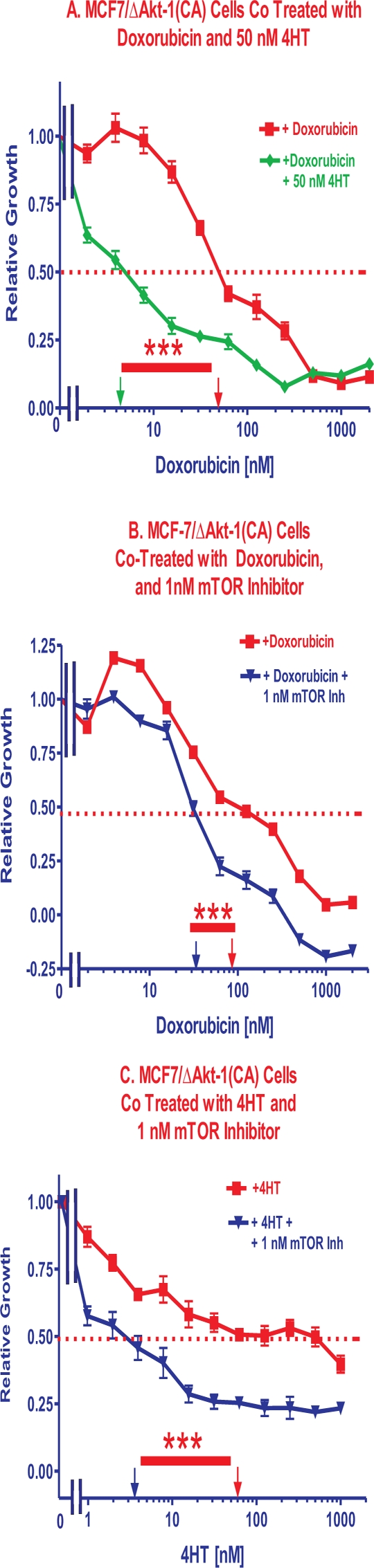
The effects of adding a constant dose of 50 nM 4HT on the doxorubicin IC50 were examined in MCF7/ΔAkt-1(CA) cells. Panel B. The effects of adding a constant dose of 1 nM rapamycin on the doxorubicin IC50 were examined in MCF7/ΔAkt-1(CA) cells. Panel C. The effects of adding a constant dose of 1 nM rapamycin on the 4HT IC50 were examined in MCF7/ΔAkt-1(CA) cells. The statistical significance was determined by the unpaired t test. Comparisons determined to be significant are indicated with (***). Panel A. The P value between doxorubicin vs. doxorubicin and 50 nM 4HT treatment of MCF7/ΔAkt-1(CA) cells was 0.0001. Panel B. The P value between doxorubicin vs. doxorubicin and 1 nM rapamycin treatment of MCF7/ΔAkt-1(CA) cells was 0.0008. Panel C. The P value between 4HT vs. 4HT and 1 nM rapamycin treatment of MCF7/ΔAkt-1(CA) cells was 0.0001.
Next we examined the effects of a constant, sub-optimal dose of 5 nM 4HT on the doxorubicin IC50s in MCF-7, MCF-7/ΔAkt-1(CA), and MCF-7/ΔAkt-1(CA)DoxR cells. 7.5-, 10.7- and 8.9-fold decreases in the IC50 for doxorubicin were observed (Table 2). These results indicated that suppressing the ER could promote the cytotoxic effects of doxorubicin at lower concentrations. Moreover, even though the MCF-7/ΔAkt-1(CA) cells required more 4HT to reach the IC50 than MCF-7 cells, they remained sensitive to 4HT.
Table 2.
Effects of Combination of Chemotherapeutic, Hormone Based and Signal Transduction Inhibitors IC50s.1
| Cell Line | Drug Treatment | Change in IC502 |
| MCF-7 | Doxorubicin + 5 nM 4HT | 7.5 X↓2 |
| MCF-7/ΔAkt-1(CA) | Doxorubicin + 5 nM 4HT | 10.7 X↓2 |
| MCF-7/ΔAkt-1(CA)DoxR | Doxorubicin + 5 nM 4HT | 8.9 X↓2 |
| MCF-7 | Doxorubicin + 1 nM Rapamycin | -2 |
| MCF-7/ΔAkt-1(CA) | Doxorubicin + 1 nM Rapamycin | 2.5 X↓2 |
| MCF-7/ΔAkt-1(CA)DoxR | Doxorubicin + 1 nM Rapamycin | 60 X↓2 |
| MCF-7 | Etoposide + 1 nM Rapamycin | -2 |
| MCF-7/ΔAkt-1(CA) | Etoposide + 1 nM Rapamycin | 1.3 X↓2 |
| MCF-7/ΔAkt-1(CA)DoxR | Etoposide + 1 nM Rapamycin | 2.5 X↓2 |
| MCF-7 | 4HT + 1 nM Rapamycin | 5.8 X↓2 |
| MCF-7/ΔAkt-1(CA) | 4HT + 1 nM Rapamycin | 18.7 X↓2 |
| MCF-7/ΔAkt-1(CA)DoxR | 4HT + 1 nM Rapamycin | 60 X↓2 |
Enhancement of Doxorubicin and 4HT Induced Inhibition of Proliferation by the mTOR Inhibitor Rapamycin
The abilities to improve chemo- and hormonal-based therapy by potentially lowering dose-associated deleterious side effects were examined. Namely, we examined the ability of the mTOR inhibitor rapamycin to lower the doxorubicin and 4HT IC50s. In these experiments, we used a constant doses of 1 nM mTOR inhibitor and different doses of either 4HT or doxorubicin to examine the effects on MCF7/Akt-1(CA) cells (Figure 3, Panels B and C). Co-addition of constant doses of the mTOR inhibitor lowered the doses of doxorubicin and 4HT required to obtain the doxorubicin and 4HT IC50s by 2.6- and 14-fold respectively. However, even with the mTOR inhibitor, 4HT did not totally eliminate the relative growth of MCF7/ΔAkt-1(CA) cells (Panel C) while doxorubicin did (Panel B).
Combinatorial effects on 4HT IC50s were observed with rapamycin in MCF-7, MCF7/ΔAkt-1(CA) and MCF7/ΔAkt-1(CA)DoxR cells (Table 2). The highest level of rapamycin-mediated suppression of the 4HT IC50s was observed with the MCF7/ΔAkt-1(CA)DoxR cells although rapamycin-mediated suppression of the 4HT IC50s was also observed in doxorubicin sensitive MCF-7 cells. Thus rapamycin is able to synergize with 4HT in these ER+ cells (Table 2).
Effects of mTOR Inhibitors on Sensitivity of MCF-7 Cells to Doxorubicin
MCF-7 cells were more sensitive to doxorubicin than MCF7/ΔAkt-1(CA) cells (Figure 4, Panel A). However, low doses of rapamycin had greater effects on the doxorubicin IC50s in MCF7/ΔAkt-1(CA) than in MCF-7 cells. Essentially a constant dose of 1 nM rapamycin did not reduce the IC50 for doxorubicin in MCF-7 cells (Figure 4, Panel B), while it did reduce the IC50 for doxorubicin in MCF7/ΔAkt-1(CA) cells to approximately the same IC50 level observed in MCF-7 cells (Panel C).
Figure 4. Effects of Co-Treatment with Doxorubicin and a mTOR Inhibitor. Panel A.
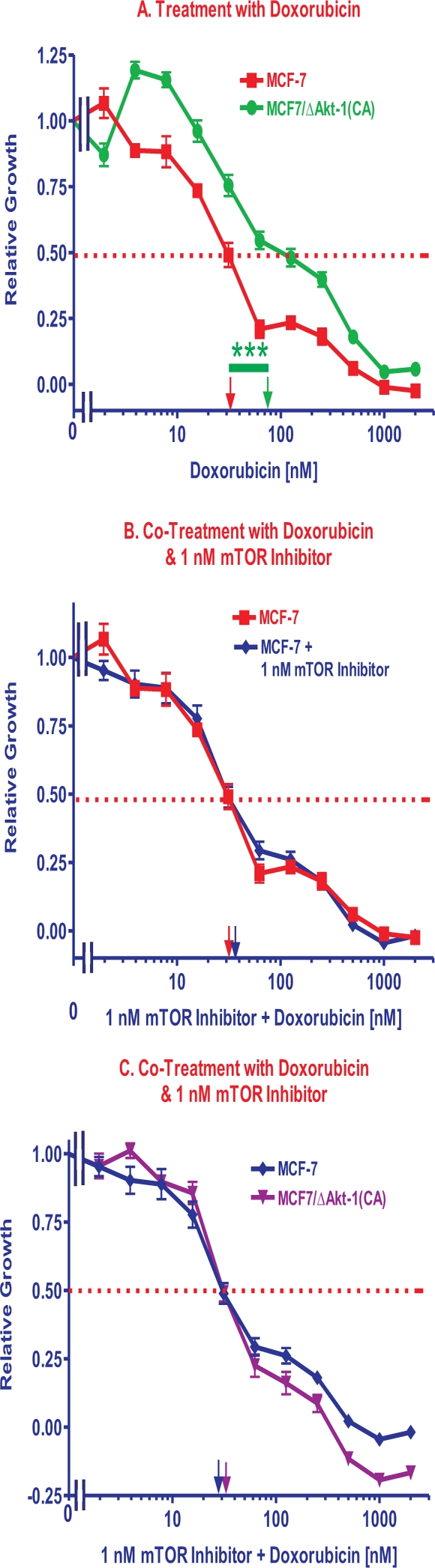
The sensitivities of MCF-7 and MCF7/ΔAkt-1(CA) cells to different doses of doxorubicin in presented. Panel B. The sensitivity of MCF-7 cells to different doses of doxorubicin is compared with the effects of doxorubicin and a suboptimal dose of 1 nM rapamycin. Panel C. The sensitivities of MCF-7 and MCF7/ΔAkt-1(CA) cells to different doses of doxorubicin and a suboptimal dose of 1 nM rapamycin in presented. The statistical significance was determined by the unpaired t test. Comparisons determined to be significant are indicated with (***). Panel A. The P value between doxorubicin treatment of MCF-7 and MCF7/ΔAkt-1(CA) cells was 0.0005.
Effects of mTOR Inhibitors on Sensitivity of MCF-7 and MCF7/ΔAkt-1(CA) Cells to Etoposide and 4HT
MCF-7 cells were more sensitive to etoposide than MCF-7/ΔAkt-1(CA)DoxR cells (Figure 5, Panel A). Rapamycin was able to lower the concentration of etoposide required to reach the IC50 in the MCF-7/ΔAkt-1(CA) and MCF-7/ΔAkt-1(CA)DoxR cells (Figure 5 and Table 2). Usually a higher degree of interaction was observed in the MCF-7/ΔAkt-1(CA)DoxR cells. The results of addition of 1 nM rapamycin on the etoposide IC50s in MCF-7 and MCF-7/ΔAkt-1(CA)DoxR cells are presented in Panel B. Rapamycin suppressed the etoposide IC50s the most in the MCF-7/ΔAkt-1(CA)DoxR cells, with approximately a 2-fold reduction as compared to MCF-7 cells (Table 2). 1 nM rapamycin did not suppress the IC50s of etoposide in MCF-7 cells (Panel C), but did suppress the IC50 for etoposide in MCF-7/ΔAkt-1(CA)DoxR cells approximately 2.5 fold (Panel D).
Figure 5. Effects of Co-Treatment with Etoposide and Rapamycin on Etoposide IC50s.

The effects of adding a sub-optimal constant dose of 1 nM rapamycin on the etoposide IC50s were examined in MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells. Panel A. The sensitivity of MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells to different doses of etoposide in presented. Panel B. The sensitivities of MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells to different doses of etoposide and a suboptimal dose of 1 nM rapamycin in presented. Panel C. The sensitivity of MCF-7 cells to different doses of etoposide is compared with the effects of etoposide and a suboptimal dose of 1 nM rapamycin. Panel D. the sensitivity of MCF7/ΔAkt-1(CA)DoxR cells to different doses of etoposide is compared with the effects of etoposide and a suboptimal dose of 1 nM rapamycin. Comparisons determined to be significant are indicated with (***). Panel A. The P value between etoposide treatment of MCF-7 and MCF7/ΔAkt-1(CA)DoxR cells was 0.0003. Panel D. The P value between etoposide and etoposide and 1nM rapamycin treatment of MCF7/ΔAkt-1(CA)DoxR cells was 0.0005.
Rapamycin also suppressed the 4HT IC50s in the doxorubicin-sensitive and resistant- cells (Table 2). The highest level of rapamycin-mediated suppression of the 4HT IC50s was observed with the MCF-7/ΔAkt-1(CA)DoxR cells although rapamycin-mediated suppression of the 4HT IC50s was also observed in doxorubicin-sensitive MCF-7 cells.
DISCUSSION
In our studies, we examined the effects of three chemotherapeutic drugs (doxorubicin, etoposide and paclitaxel), one hormonal drug (tamoxifen, 4HT) and the signal transduction inhibitor rapamycin on MCF-7 and derivative cell lines which varied in their levels of activated Akt-1 expression. An advantage of our study is that all the cells had the same genetic background as they all were MCF-7 breast cancer cells, however they differed in the levels of activated Akt-1 expression due to introduction of an activated Akt-1 gene.
We have previously shown that introduction of dominant negative (DN) forms of PTEN into MCF-7 cells conferred resistance to doxorubicin and increased sensitivity to rapamycin [9]. Furthermore, rapamycin could synergize with doxorubicin to lower its IC50 [9]. In the MCF-7 cells transfected cells with the DN PTEN genes, increased levels of activated Akt were detected. However, these drug-resistant PTEN(DN) transfected cells were hypersensitive to rapamycin. These results have clinical significance as the PI3K/PTEN/Akt/mTOR pathway is often activated in breast cancer by mutations at PIK3CA or multiple genetic mechanisms leading to dysregulation of PTEN. Furthermore, drug resistance frequently develops in breast cancer after chemo- or hormonal-based therapies. Doxorubicin (a.k.a Adriamycin) is frequently used to treat breast cancer patients. In the studies present in this report, increased expression of activated Akt-1 resulted in the resistance of MCF-7 breast cancer cells to both chemotherapeutic drugs (doxorubicin and etoposide) as well as hormonal based drugs (4HT) and these drug resistant activated Akt-1 overexpressing cells were also hypersensitive to rapamycin, which reduced the concentrations of doxorubicin, etoposide and 4HT required to reach the IC50s in these cells.
The roles of Akt and downstream mTOR are critical. Hyperactivation of mTOR will induce cellular senescence which can be inhibited by mTOR inhibitors. mTOR appears to be an essential switch which stimulates p53 to promote cellular quiescence vs. senescence. [70-75].
Doxorubicin, etoposide and paclitaxel all have been used to treat breast cancer. However, doxorubicin and paclitaxel are more commonly employed in breast cancer therapy today than etoposide [76]. Tamoxifen is a common hormonal-based drug used to treat ER+ breast cancer. After chemotherapeutic drug and hormonal based drug treatment of breast cancer patients, resistance will develop [10,77]. Thus it is essential to develop new more effective and alternative methods to treat breast cancer.
Doxorubicin, etoposide and paclitaxel can all be transported by the same drug transporter (Mdr-1, a.k.a., Pgp, and ABCB1) [78]. Our results suggest that Mdr1 is not likely to be involved in the drug resistance of these cells but our studies do not exclude many other drug transporters. Both doxorubicin and etoposide target events involved in DNA replication, whereas paclitaxel targets microtubules. Therefore it is not surprising that cells selected for resistance to doxorubicin are also resistant to etoposide, but not paclitaxel. Our studies suggest that attention should be given during treatment of breast cancer patients who have become resistant to a particular chemotherapeutic drug, as these patients may be cross-resistant to other chemotherapeutic drugs which target similar events.
Interestingly, suboptimal doses of 4HT would interact with doxorubicin to lower the doxorubicin IC50 in MCF-7, MCF-7/ΔAkt-1(CA), and MCF-7/ΔAkt-1(CA)DoxR approximately 8- to 10-fold. This demonstrates the important effects of 4HT on ER cells. Even though cells that express activated Akt require more 4HT to reach the IC50 than cells which do not express activated Akt, they will still respond to 4HT when they are also treated with doxorubicin. Thus resistance to doxorubicin does not prevent a response to 4HT.
The mechanisms by which doxorubicin and rapamycin synergize to inhibit the growth of breast cancer cells are under further investigation. Doxorubicin can induce reactive oxygen species (ROS) which have many effects on cell growth and the prevention of apoptosis. A consequence of doxorubicin treatment may be the induction of the Raf/MEK/ERK pathway which may have some anti-apoptotic effects [79].
The ability of a 1 nM constant addition of the mTOR inhibitor rapamycin to interact with 4HT and lower the IC50 for 4HT was observed in MCF-7 and MCF-7/ΔAkt-1(CA) cells. In contrast, 1 nM rapamycin did not increase the sensitivity of MCF-7 cells to either doxorubicin or etoposide. Higher doses of the mTOR inhibitor were not examined in these studies as 1 nM rapamycin was close to the IC50s previously observed in the cell lines. 1 nM rapamycin was sufficient to increase the sensitivity of MCF-7/ΔAkt-1(CA) cells and not MCF-7 cells to doxorubicin and etoposide.
These results are relevant to potential cancer therapies as Akt is frequently activated by upstream PIK3CA or PTEN mutations or gene silencing. PTEN can be mutated or silenced by various mechanisms in human cancer. Mutations occur which either delete the PTEN gene or alter its activity. Sometimes these mutations actually make the cells sensitive to Akt and mTOR inhibitors as the growth of the cells becomes dependent upon elevated Akt levels and downstream mTOR and p70S6K activities [60].
MATERIALS AND METHODS
Cell Culture
MCF-7 cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). Cell culture medium for MCF-7 cells consisted of Roswell Park Memorial Institute-1640 (RPMI 1640) medium (Invitrogen. Carlsbad CA) supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS) as described [5].
Akt Plasmids
MCF-7 cells were infected with a retrovirus encoding the ΔAkt-1(CA) gene [69,80]. Stably ΔAkt-1(CA)- infected or EGFP-transfected MCF-7 cells were isolated after growth for 4 weeks in selective drug (2 mg/ml G418, Sigma-Aldrich, St. Louis, MO) as described [9,80].
Analysis of Sensitivity to Drugs and Signal Transduction Inhibitors.
Cells were seeded in 96-well cell culture plates (BD Biosciences) at a density of 5,000 cells/well in 100 μl/well of phenol red free RPMI-1640 containing 5% charcoal stripped (CS) FBS as described [5]. Cells were incubated for 1 day to permit cells to adhere to the bottom of each well. Cells were subsequently treated with serial 2-fold dilutions of the chemotherapeutic drugs (doxorubicin, etoposide or paclitaxel), the hormonal drug (4HT), signal transduction inhibitor, rapamycin, (all purchased from Sigma-Aldrich) or a constant suboptimal concentration of the hormonal drug or signal transduction inhibitor. Cells were incubated for an additional 4 days at 37 °C until extent of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma-Aldrich) reduction in each well was quantified at 530 nm [5].
Acknowledgments
This work was supported in part by a grant from the US National Institutes of Health to JAM (R01CA098195). This work was also supported in part by grants from: Fondazione del Monte di Bologna e Ravenna, MinSan 2008 entitled “Molecular therapy in pediatric sarcomas and leukemias against IGF-1 receptor system”, PRIN 2008 and FIRB 2010 (RBAP10447J) to AMM. This work was also supported in part by a grant from the Italian Ministry of Health, Ricerca Finalizzata Stemness 2008 entitled “Molecular Determinants of Stemness and Mesenchymal Phenotype in Breast Cancer” to ML.
REFERENCES
- 1.Shelton JG, Steelman LS, Abrams SL, Bertrand FE, Franklin RA, McMahon M, McCubrey JA. The epidermal growth factor receptor gene family as a target for therapeutic intervention in numerous cancers: what's genetics got to do with it? Expert Opin Ther Targets. 2005;9:1009–1030. doi: 10.1517/14728222.9.5.1009. [DOI] [PubMed] [Google Scholar]
- 2.Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle. 2009;8:18–22. doi: 10.4161/cc.8.1.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 4.Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, Zabezhinski MA, Anikin IV, Karkach AS, Romanyukha AA. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188–197. doi: 10.4161/cc.9.1.10407. [DOI] [PubMed] [Google Scholar]
- 5.Weinstein-Oppenheimer CR, Henriquez-Roldan CF, Davis JM, Navolanic PM, Saleh OA, Steelman LS, Franklin RA, Robinson PJ, McMahon M, McCubrey JA. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin Cancer Res. 2001;7:2898–2907. [PubMed] [Google Scholar]
- 6.Davis JM, Navolanic PM, Weinstein-Oppenheimer CR, Steelman LS, Hu W, Konopleva M, Blagosklonny MV, McCubrey JA. Raf-1 and Bcl-2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin Cancer Res. 2003;9:1161–1170. [PubMed] [Google Scholar]
- 7.Steelman LS, Bertrand FE, McCubrey JA. The complexity of PTEN: mutation, marker and potential target for therapeutic intervention. Expert Opin Ther Targets. 2004;8:537–550. doi: 10.1517/14728222.8.6.537. [DOI] [PubMed] [Google Scholar]
- 8.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it's implication in cancer. Cell Cycle. 2005;4:1309–1312. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 9.Steelman LS, Navolanic PN, Sokolosky M, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman K, Terrian DM, Leslie N, Martelli AM, Stivala F, Libra M, Franklin RA, McCubrey JA. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity of mTOR inhibitors. Oncogene. 2008;27:4086–4095. doi: 10.1038/onc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCubrey JA, Steelman LS, Kempf CR, Chappell W, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra M, Bäsecke J, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Cocco L, Martelli AM. Therapeutic Resistance Resulting from Mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Signaling Pathways. J Cell Physiol. 2011 doi: 10.1002/jcp.22647. doi: 0.1002/jcp.22647. [DOI] [PubMed] [Google Scholar]
- 11.Steelman LS, Chappell WH, Abrams SL, Kempf CR, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chappell WH, Steelman LS, Long JM, Kempf CR, Abrams SL, Franklin RA, Basecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Miatovic S, Montalto G, Cervello M, Laidler P, Milella M, Tafuri A, Bonati A, Evangelisti C, Cocco L, Martelli AM, McCubrey JA. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rational and importance to inhibiting these pathways in human health. Oncotarget. 2011:135–64. doi: 10.18632/oncotarget.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011;2:435–447. doi: 10.18632/oncotarget.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pajic M, Kersbergen A, van Diepen F, Pfauth A, Jonkers J, Borst P, Rottenberg S. Tumor-initiating cells are not enriched in cisplatin-surviving BRCA1; p53-deficient mammary tumor cells in vivo. Cell Cycle. 2010;9:3780–91. doi: 10.4161/cc.9.18.13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A, Cervello M, Martelli AM, McCubrey JA. Targeting the leukemic stem cell – the holy grail of leukemia therapy. Leukemia. 2009;23:25–42. doi: 10.1038/leu.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buitenhuis M, Coffer PJ. The role of the PI3K-PKB signaling module in regulation of hematopoiesis. Cell Cycle. 2009;8:560–566. doi: 10.4161/cc.8.4.7654. [DOI] [PubMed] [Google Scholar]
- 17.Tamburini J, Green AS, Chapuis N, Bardet V, Lacombe C, Mayeux P, Bouscary D. Targeting translation in acute myeloid leukemia: a new paradigm for therapy? Cell Cycle. 2009;8:3893–3899. doi: 10.4161/cc.8.23.10091. [DOI] [PubMed] [Google Scholar]
- 18.Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 2009;8:403–413. doi: 10.4161/cc.8.3.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–3837. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 20.Sabisz M, Skladonowski A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: implications for drug resistance and in vitro drug screening models. Cell Cycle. 2009;8:3208–3217. doi: 10.4161/cc.8.19.9758. [DOI] [PubMed] [Google Scholar]
- 21.McCubrey JA, Abrams SL, Stadelman K, Chappell WH, Lahair M, Ferland RA, Steelman LS. Targeting signal transduction pathways to eliminate chemotherapeutic drug resistance and cancer stem cells. Advances in Enzyme Regulation. 2010;50:285–307. doi: 10.1016/j.advenzreg.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCubrey JA, Chappell WH, Abrams SL, Franklin RA, Long JM, Sattler JA, Kempf CR, Laidler P, Steelman LS. Targeting the cancer initiating cells: the Achilles’ heel of cancer. Advances in Enzyme Regulation. 2011;51:152–62. doi: 10.1016/j.advenzreg.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors. Cell Cycle. 2009;8:1352–1358. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L, Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010;9:596–600. doi: 10.4161/cc.9.3.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Cappellini A, Ognibene A, McCubrey JA. The emerging role of the phosphatiylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim Biophys Act. 2010;1803:991–1001. doi: 10.1016/j.bbamcr.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narasimhan SD, Mukhopadhyay A, Tissenbaum HA. InAKTivation of insulin/IGF-1 signaling by dephosphorylation. Cell Cycle. 2009;8:3878–3884. doi: 10.4161/cc.8.23.10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brauer PM, Tyner AL. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle. 2009;8:2728–2732. doi: 10.4161/cc.8.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010;9:3151–3156. doi: 10.4161/cc.9.16.13120. [DOI] [PubMed] [Google Scholar]
- 31.Harikumar KB, Aggarwal BB. Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle. 2008;7:1020–1035. doi: 10.4161/cc.7.8.5740. [DOI] [PubMed] [Google Scholar]
- 32.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–1904. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 33.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–1900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 34.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 35.Blagosklonny MV. Aging-suppressants: Cellular senescence (hyperactivation) and its pharmacologic decleration. Cell Cycle. 2009;8:1883–1887. doi: 10.4161/cc.8.12.8815. [DOI] [PubMed] [Google Scholar]
- 36.Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010;9:487–497. doi: 10.4161/cc.9.3.10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lal MA, Bae D, Camilli TC, Patierno SR, Ceryak S. AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8:1589–1602. doi: 10.4161/cc.8.10.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin J, Wang GL, Timchenko L, Timchenko NA. GSK3beta and aging liver. Aging. 2009;1:582–585. doi: 10.18632/aging.100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009;8:1003–1006. doi: 10.4161/cc.8.7.8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010;2:344–352. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 42.Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474–881. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- 43.Brauer PM, Tyner AL. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle. 2009;8:2728–2732. doi: 10.4161/cc.8.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steelman LS, Martelli AM, Nicoletti F, McCubrey JA. Exploiting p53 Status to Enhance Effectiveness of Chemotherapy by Lowering Associated Toxicity. Oncotarget. 2011;2:109–12. doi: 10.18632/oncotarget.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965–970. doi: 10.4161/cc.7.8.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, Kumar R, Feng Z, Powell SN, Bhat A, Yaguchi T, Wadhwa R, Kaul SC, Parsons R, Khanna KK, Pandita TK. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8:2198–2210. doi: 10.4161/cc.8.14.8947. [DOI] [PubMed] [Google Scholar]
- 47.Leslie NR, Foti M. Not in my genes: non-genomic loss of PTEN function in cancer. Trends in Pharmacological Sciences. 2011;32:131–140. doi: 10.1016/j.tips.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 48.Diaz-Meco MT, Abu-Baker S. The Par-4/PTEN connection in tumor suppression. Cell Cycle. 2009;8:2518–2522. doi: 10.4161/cc.8.16.9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shan W, Liu J. Epithelial ovarian cancer: focus on genetics and animal models. Cell Cycle. 2009;8:731–735. doi: 10.4161/cc.8.5.7848. [DOI] [PubMed] [Google Scholar]
- 50.Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle. 2010;9:4450–4454. doi: 10.4161/cc.9.22.13936. [DOI] [PubMed] [Google Scholar]
- 51.Sayed D, Abdellatif M. AKT-ing via microRNA. Cell Cycle. 2010;9:3213–3217. doi: 10.4161/cc.9.16.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh B, Ittmann MM, Krolewski JJ. Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chrom Cancer. 1998;21:166–171. [PubMed] [Google Scholar]
- 53.Tsutsui S, Inoue H, Yasuda K, Suzuki K, Higashi H, Era S, Mori M. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 2005;68:398–404. doi: 10.1159/000086981. [DOI] [PubMed] [Google Scholar]
- 54.Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo V, Ruvolo P, Kempf CR, Steelman LS, McCubrey JA. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011;25:1064–1079. doi: 10.1038/leu.2011.46. [DOI] [PubMed] [Google Scholar]
- 55.Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo P, Ruvolo V, Evangelisti C, Martelli AM, McCubrey JA. Roles of the Ras/Raf/MEK/ERK Pathway in Leukemia Therapy. Leukemia. 2011;25:1080–1094. doi: 10.1038/leu2011.66. [DOI] [PubMed] [Google Scholar]
- 56.Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging. 2009;1:515–528. doi: 10.18632/aging.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bose S, Crane A, Hibshoosh H, Mansukhani M, Sandweis L, Parsons R. Reduced expression of PTEN correlates with breast cancer progression. Hu Pathol. 2002;33:405–409. doi: 10.1053/hupa.2002.124721. [DOI] [PubMed] [Google Scholar]
- 58.Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–717. [PubMed] [Google Scholar]
- 59.Tokunaga E, Kimura Y, Mashino K, Oki E, Oki E, Kataoka A, Ohno S, Morita M, Kakeji Y, Baba H, Maehara Y. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137–144. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 60.deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059–8067. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 61.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 62.Lin HJ, Hsieh FC, Song H, Lin J. Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br J Cancer. 2005;12:1372–1381. doi: 10.1038/sj.bjc.6602862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P, Yang W, Yin G, Hittelman WN, Yu D. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;66:2028–2037. doi: 10.1158/0008-5472.CAN-04-4559. [DOI] [PubMed] [Google Scholar]
- 64.Yim EK, Siwko S, Lin SY. Exploring Rak tyrosine kinase function in breast cancer. Cell Cycle. 2009;8:2360–2364. doi: 10.4161/cc.8.15.9264. [DOI] [PubMed] [Google Scholar]
- 65.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 66.Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, Henske EP, El-Deiry WS. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle. 2009;8:4168–4175. doi: 10.4161/cc.8.24.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faridi J, Wang L, Endemann G, Roth RA. Expression of Constitutively Active Akt-3 in MCF-7 Breast Cancer Cells Reverses the Estrogen and Tamoxifen Responsivity of these Cells in Vivo. Clin Cancer Res. 2003;9:2933–2939. [PubMed] [Google Scholar]
- 68.Donia M, McCubrey JA, Bendtzen K, Nicoletti F. Potential use of rapamycin in HIV infection. British Journal of Clinical Pharmacology. 2010;70:784–793. doi: 10.1111/j.1365-2125.2010.03735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 70.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010;2:344–352. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 72.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–1900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 73.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–1904. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 74.Wesierska-Gadek J. mTOR and its link to the picture of Dorian Gray - re-activation of mTOR promotes aging. Aging. 2010;2:892–893. doi: 10.18632/aging.100240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, Tesori V, Antonini A, Maulucci G, De Spirito M, Galeotti T, Pani G. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging. 2010;2:487–503. doi: 10.18632/aging.100183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Navolanic PM, McCubrey JA. Pharmacological breast cancer therapy. Int J Oncol. 2005;27:1341–1344. [PubMed] [Google Scholar]
- 77.Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy. Int J Oncol. 2003;22:237–252. [PubMed] [Google Scholar]
- 78.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 79.Steelman LS, Navolanic PM, Chappell WH, Abrams SL, Wong EWT, Franklin RA, McCubrey JA. Involvement of Akt and mTOR in Chemotherapeutic- and Hormonal-Based Drug Resistance and Response to Radiation in Breast Cancer Cells. Cell Cycle. 2011 doi: 10.4161/cc.10.17.17119. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shelton JG, Steelman LS, Lee JT, Knapp SL, Blalock WL, Moye PM, Franklin RA, Pohnert SC, Mizra AM, McMahon M, McCubrey JA. Effects of the Raf/MEK/ERK and PI3K signal transduction pathways on the abrogation of cytokine dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;24:2478–2492. doi: 10.1038/sj.onc.1206321. [DOI] [PubMed] [Google Scholar]