

Abstract
The type I interferon (IFN) response plays a critical role in autoimmunity and is induced by innate receptor ligation and activation of IFN-regulatory factors (IRF). The present study investigated the roles and functional hierarchy of IRF3, IRF5, and IRF7 in expression of cytokines, chemokines, and matrix metalloproteinases in human THP1 monocytic cells. Targeted IRF knockdown was followed by evaluation of gene expression, promoter activation, and mRNA stability to determine the role of IRF as potential targets for modulating IFN responses in patients with autoimmune diseases. IRF played a distinct role in regulation of type I IFN gene expression in human monocytic cells and specifically regulated gene expression through the IFN-stimulated response element, with no contribution to transcription of traditionally AP-1 or NF-κB regulated genes. IRF7 regulated IL-6 gene expression by increasing IL-6 mRNA stability. IRF regulation of inflammation and induction of the IFN signature might contribute to the pathogenesis of autoimmune diseases and therefore represent novel therapeutic targets.
Keywords: autoimmune disease, interferon regulatory factors, transcription factors, monocytes, type I interferon
1. Introduction
Activation of the type I interferon (IFN) system contributes to the pathogenesis of many rheumatic diseases, including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). In SLE patients, autoantibody production and immune complex formation result in tissue injury and secretion of type I IFN and activation of innate immunity (1–4). Peripheral blood mononuclear cells (PBMC) from SLE patients demonstrate increased expression of type I IFN and other IFN-regulated genes known as the IFN signature (5, 6). Innate immune responses also play a critical role in synoviocyte activation and recruitment into the synovial tissue of RA patients (7–12). In fact, the gene expression profile in RA synovial tissue reflects exposure to toll-like receptor (TLR) ligands and displays characteristic features of the type I IFN signature (13–16). In addition to the synovium, an interferon profile has also been reported in peripheral blood cells of a subset of RA patients (17).
Innate signaling is triggered by TLR-dependent and independent recognition of bacterial and viral products as well as endogenous self-nucleic acids containing RNA, DNA, and nuclear protein in immune complexes (18–20). These immune complexes can activate membrane TLR and cytoplasmic innate pattern recognition receptors that induce the type I IFN response (21, 22). Binding to innate receptors results in activation of several transcription factors, including interferon regulatory factors (IRF), c-Jun/ATF2, and NF-κB, which are involved in production of the type I IFN response through formation of the IFN enhanceosome (23). The IRF family is composed of nine transcription factors that regulate the type I IFN system and host defense (24). Distinct functions and differential regulation of IRF have been elucidated in a variety of cell types and in response to viral infection and other triggers of innate receptors (9, 10, 25–27).
Because immune complexes contribute to the pathogenesis of RA and SLE and are often cleared from the circulation by the mononuclear phagocytic system, we evaluated the relative roles and functional hierarchy of IRF in the human monocytic cell line THP1. The present study investigated the role of IRF3, IRF5, and IRF7 in promoter activation and gene expression of cytokines, chemokines, and MMP in THP1 monocytic cells. We evaluated the human cell line THP1 because monocytes from patients with autoimmune diseases are functionally and phenotypically distinct from healthy controls and participate in clearance of immune complexes from the circulation, possibly contributing to disease pathogenesis. Initial studies of the pathogenesis of SLE were somewhat more focused on the adaptive immune system and lymphocyte abnormalities; however, this has shifted more recently toward innate immunity. Monocytes and macrophages are an essential part of the innate immune response and contribute to phagocytosis and cytokine and IFN production. Aberrations of monocyte or macrophage phenotype and function are increasingly recognized in SLE and other autoimmune diseases. Targeted IRF knockdown was followed by evaluation of protein and gene expression, promoter activation, and mRNA stability to determine the specific roles of IRF and potential as therapeutic targets in SLE or RA patients.
2. Materials and methods
2.1. Reagents
Monoclonal anti-IRF7 (sc-74472) and GAPDH (sc-32233) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti-IRF5 (3255) was purchased from Cell Signaling Technology (Danvers, MA). Polyinosinic and polycytidylic acid [poly (I-C)] and LPS were purchased from Sigma Aldrich (St. Louis, MO). CpG ODN2006 (tlrl-hodnb-1) was obtained from Invivogen (San Diego, CA). TNF and IL-1 were purchased from R&D systems (Minneapolis, MN).
2.2. Cell culture
Human monocytoid THP-1 cells were purchased from ATCC (Manassas, VA) and grown in serum-free RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 2mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL gentamicin and 10% heat inactivated fetal bovine serum (Gemini Bio-Products, Calabasas, CA).
2.3. Western blot analysis
Cells were cultured in 6-well plates at 5 × 105 cells/mL. Cells were incubated with poly (I-C) (10 μg/mL), LPS (1 μg/mL), CpG (10 μg/mL), IL-1 (1 ng/mL), and TNFα (10ng/mL) for 6h. Dose response studies were performed with poly (I-C) for various times points up to 24h. Cells were washed with cold PBS, and protein was extracted using kinase lysis buffer (50 mM HEPES, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM MgCl2, 1.5 mM EDTA pH 8.0, 20 mM β-glycerophosphate, 50 mM NaF, 1 mM Na3VO4, 10 μg/mL aprotinin, 1 μM pepstatin A, and 1 mM PMSF). The protein concentrations were determined using Micro BCA protein assay kit (Thermo Scientific, Rockford, IL). Samples containing 50 μg protein were resolved via 4–12% SDS-PAGE and transferred to a polyvinylidene diflouride membrane. The membranes were blocked and incubated with primary antibody at 4°C overnight, followed by HRP-conjugated secondary antibody for 1h. Proteins were visualized with chemiluminescence using the VersaDoc 4000 MP imaging system (BioRad, Hercules, CA).
2.4. Small interfering RNA transfection
Dose-response and kinetic studies were performed using 1, 3, and 5 μg siRNA at days 3, 5, and 7 to standardize and confirm knockdown. A total of 1 × 106 cells were transfected with 5 μg IRF3, IRF5, IRF7, or scramble control Smartpool small interfering RNA (Dharmacon, Lafayette, CO) using the Cell Line Nucelofector Kit V according to the manufacturer’s instruction (Amaxa, Gaithersburg, MD). Approximately 75–85% decrease in protein expression is achieved using this method. Transfected cells were allowed to recover overnight and stimulated 6h with poly (IC) prior to lysis on day 3 post-transfection.
2.5. Quantitative real-time PCR
After siRNA transfection, cells were cultured in RPMI 1640 with 10% fetal bovine serum at 37°C overnight. Cells were treated with either medium or poly (I-C) (10 μg/mL) for 6h. Total RNA isolation with RNA STAT and cDNA synthesis by RT-PCR were performed as previously described (28). Gene expression was evaluated using TaqMan PCR analysis and the GeneAmp 7300 Sequence Detection System as previously described (29). Forward and reverse primers as well as fluorogenic TaqMan FAM/TAMRA-labeled hybridization probes were used (Assay on Demand; Applied Biosystems, Foster City, CA). To control for sample cellularity, human GAPDH forward and reverse primers and labeled probe were included in separate PCR. The threshold cycle C(t) was determined for each sample using GeneAmp software. Standard curves are generated by linear regression using log (C[t]) versus log (cell number). The cell equivalent (CE) number for samples was calculated using the standard curve. Data are expressed as the ratio between gene of interest CE and GAPDH CE, yielding the relative gene expression (RE).
2.6. Reporter gene assay
After siRNA transfection, THP-1 monocytic cells were cultured for 3 days, and subsequently, 1 × 106 cells were transfected with 1 μg reporter plasmid DNA and 0.1 μg Renilla reniformis luciferase construct as internal control for transfection efficiency (a generous gift from Dr. M. David UCSD). Reporter constructs containing IFN-stimulated response element (ISRE)-luciferase, which has five repeats of the ISRE sequence from IFN-stimulated gene 15 kD gene promoter, NF-κB-luciferase, or activator 1 (AP-1)-luciferase, and the full length IL-6 promoter-luciferase were individually transfected with control. After overnight incubation, transfected cells were stimulated with 10 μg/mL poly (I-C) for 6h. Luciferase activity was measured using a dual luciferase assay kit (Promega, Madison, WI).
2.7. Analysis of mRNA stability
After siRNA transfection and poly (I-C) treatment, THP-1 cells were treated with 5μg/mL actinomycin D to inhibit transcription (A1410) purchased from Sigma (St. Louis, MO), for 0.5h, 1h, 2h, 6h, or 18h. Cells were harvested using RNA STAT-60 and cDNA was isolated for Q-PCR according to the methods described.
2.8. Statistical analysis
Statistics were performed using the paired students t test. A comparison was considered significant if p < 0.05.
3. Results
3.1. Activation of IRF5 and IRF7 in human THP1 monocytes
To characterize the protein expression of IRF5 and IRF7 in human THP1 responses, cells were stimulated with cytokines or TLR ligands followed by Western blot analysis to detect IRF5 and IRF7 induction (Figure 1, top panel). Because IRF5 and IRF7 were inducible, we measured the increase in IRF5 and IRF7 protein expression. Quantification by densitometry of IRF5 and IRF7 protein expression in THP1 stimulated with each ligand is also shown in Figure 1 (bottom panel). Poly (I-C), LPS, CpG, IL-1, and TNF induced IRF5 and IRF7 expression in human THP1 monocyte cell lines. Based on these results, the synthetic dsRNA innate receptor ligand poly (I-C) was used for subsequent studies of IRF activation of the type I IFN response. To determine the time course of IRF5 and IRF7 induction, we stimulated human THP1 monocytes with poly (I-C) for up to 18h (Figure 2). Inducible IRF7 and IRF5 protein expression was detected within 6h and persisted to 18h. Quantification of protein expression by densitometry is also shown (Figure 2, lower panel).
Figure 1.

Western blot analysis of IRF5/7 induction. THP-1 cells were stimulated for 6 h with poly (I-C), LPS, CpG, IL-1, or TNF. Lysates were then analyzed by Western blot using anti-IRF5, anti-IRF7, and anti-GAPDH antibodies. Lysate from poly (I-C) stimulated fibroblast like synoviocytes (FLS) was used as a positive control. Stimulation with poly (I-C), LPS, IL-1, and TNF showed significant induction of IRF7 and IRF5. Poly (I-C) showed the most significant increase (10.26-fold ± 1.36 and 7.39-fold ± 0.85; n = 3 respectively). Top panel shows a representative Western blot, and the bottom panel shows combined quantification of protein expression by densitometry for three independent experiments.
Figure 2.

Time course of induction of IRF5 and IRF7 protein expression in poly (I-C) stimulated THP-1 cells. Cells were incubated with poly (I-C) for up to 18 h at 10 μg/mL and analyzed by Western blot analysis. Poly (I-C) stimulated fibroblast-like synviocyte (FLS) lysate was used as a positive control. Poly (I-C) was most effective at inducing IRF7 and IRF5 at 10 μg/mL for 18 h (22.65-fold ± 3.65; n = 3 and 29.65-fold ± 4.37; n = 3 respectively). Top panel shows a representative Western blot, and the bottom panel shows combined densitometry results for three independent experiments.
3.2. Targeted knockdown of IRF3, IRF5, and IRF7
The relative contribution of IRF3, IRF5, and IRF7 to the type I IFN response and production of other cytokines was evaluated by transfecting THP1 with IRF3, IRF5, or IRF7 siRNA or control smartpool siRNA (sc) followed by 6h poly (I-C) stimulation. Western blot analysis confirmed effective knockdown of IRF3, IRF5, and IRF7 protein expression (Figure 3). Constitutive expression of IRF3 was decreased to below baseline protein levels and the inducible IRF5 and IRF7 were decreased to unstimulated basal expression levels in THP1 cells. Of note, IRF5 silencing reduced the total IRF3 protein in this representative Western blot. However, this amount of decreased protein expression was not significantly different when all three experiments were analyzed with densitometry. Quantification by densitometry is shown as a bar graph (Figure 3, lower panel). To confirm siRNA knockdown of mRNA, Q-PCR was performed after IRF siRNA transfection and poly (I-C) stimulation of cultured THP1 cells. As shown in Figure 4, IRF3, IRF5, and IRF7 siRNA significantly and specifically decreased mRNA and the respective relative expression to below baseline levels.
Figure 3.

A, Effect of siRNA knockdown on IRF3, IRF5, and IRF7 protein expression. THP-1 cells were transfected with Smartpool control siRNA (sc), IRF3, IRF5, or IRF7. Cells were stimulated for 6 h with poly (I-C). Western blot analysis confirmed knockdown of IRF3, IRF5, and IRF7 protein. Top panel shows a representative Western blot, and the bottom panel shows combined densitometry results for three independent experiments.
Figure 4.
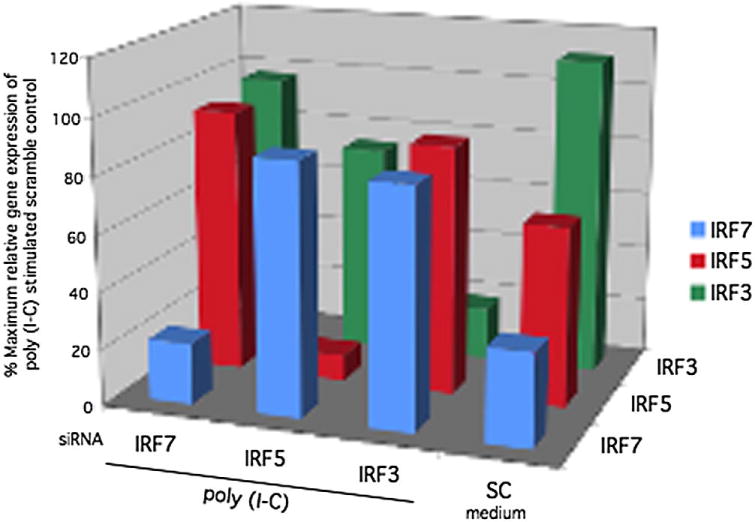
Effect of siRNA knockdown on IRF3, IRF5, and IRF7 gene expression. PCR was performed to determine the percent maximum relative gene expression of poly (I-C) stimulated scramble control of IRF3/5/7 genes after siRNA knockdown and 6 h poly (I-C) stimulation followed by quantitative analysis of mRNA. Transfection of siRNA significantly knocked down IRF3, IRF5, and IRF7 gene expression (86% ± 3%, n = 3; 74% ± 6%; n = 3; and 78% ± 4%; n = 3 respectively).
3.3. Decreased IFN-regulated gene expression in IRF deficient THP1 monocytes
We then studied the role of individual IRF in poly (I-C)-induced gene expression. IRF3, IRF5, and IRF7 siRNA knockdown was performed and cells were stimulated with poly (I-C). Q-PCR was used to measure IFN-regulated IFNβ, IP-10, MCP-1, RANTES, and MIP1α mRNA levels. These genes are considered IFN-regulated or IFN-stimulated because they are partially controlled by the upstream ISRE, although other response elements are present in the promoter region. Figure 5A shows that IRF3, IRF5, and IRF7 deficiency all significantly decreased IFNβ (66.6%, 60.0%, 73.3% respectively, p < 0.02; n = 3), IP-10 (78.9%, 47.3%, 39.5% respectively, p < 0.01), and RANTES (59.9%, 65.9%, 35.1%, respectively, p < 0.03). IRF3 and IRF5 deficiency significantly decreased MCP1 gene expression (75.7%, 57.6% respectively, p < 0.02). IRF3 deficiency specifically decreased MIP1α gene expression (85.7%, p < 0.01) compared with poly (IC) stimulated control, but IRF5 and IRF7 deficiency did not significantly regulate MIP1α. Data is summarized in Table 1.
Figure 5.

A, Inhibition of IFN-response gene expression by IRF3/5/7 knockdown. Quantitative PCR was performed to determine the percent maximum relative gene expression of poly (I-C) stimulated scramble control of IFN-regulated gene (ISRE promoter) after IRF3, IRF5, or IRF7 siRNA knockdown. After transfection, cells were stimulated for 6 h with poly (I-C), followed by quantitative analysis of IFNβ, RANTES, IP-10, MCP-1, and MIP-1α mRNA. Percent inhibition for IRF siRNA compared with scrambled siRNA stimulated with poly (I-C) was calculated. Figure 5A shows that IRF3, IRF5, and IRF7 deficiency all significantly decreased IFNβ (66.6%, 60.0%, 73.3% respectively, p < 0.02; n = 3), IP-10 (78.9%, 47.3%, 39.5% respectively, p < 0.01), and RANTES (59.9%, 65.9%, 35.1%, respectively, p < 0.03). IRF3 and IRF5 deficiency significantly decreased MCP1 gene expression (75.7%, 57.6% respectively, p < 0.02). IRF3 deficiency specifically decreased MIP1α gene expression (85.7%, p < 0.01) compared with poly (I-C) stimulated control, but IRF5 and IRF7 deficiency did not significantly regulate MIP1α. B. Inhibition of cytokine and MMP gene expression by IRF3, IRF5, and IRF7 knockdown. Quantitative PCR was performed to determine the percent maximum relative gene expression of poly (I-C) stimulated scramble control of cytokines and MMP with IRF7, IRF5, or IRF3 siRNA knockdown. After transfection, cells were stimulated for 6h with poly (I-C), followed by quantitative analysis of IL-6, IL-8, IL-10, MMP3, and MMP9 gene expression by Q-PCR (n = 3). IRF3 and IRF5 had no significant effect on poly (I-C) induced gene expression of IL-6, IL-8, MMP3, and MMP9 in THP1 cells. Surprisingly, IRF7 inhibition significantly decreased IL-6 gene expression (76% p < 0.001; n = 3) but did not regulate expression of other genes. IRF3, IRF5, and IRF7 deficiency resulted in decreased IL-10 expression (p < 0.004; n = 3).
Table 1.
Distinct roles for IRF in poly (I-C) induced gene expression in human cell lines
| Fibroblast-like synoviocyte | THP1 Monocyte | |
|---|---|---|
| IRF3 |
|
|
| IRF5 | Not determined |
|
| IRF7 | No effect |
|
Figure 5B demonstrates inhibition of pro-inflammatory cytokine and MMP gene expression by IRF3, IRF5, and IRF7 knockdown. IRF3 and IRF5 had no significant effect on poly (I-C) induced gene expression of IL-6, IL-8, MMP3, and MMP9 in THP1 cells. Surprisingly, IRF7 inhibition significantly decreased IL-6 gene expression (76% p < 0.001; n = 3) but did not regulate expression of IL-8, MMP3, and MMP9. IRF3, IRF5, and IRF7 deficiency resulted in decreased IL-10 expression (45.8%, 43.8%, 37% p < 0.004; n = 3).
3.4. Regulation of ISRE promoter activity by IRF3
The upstream binding of IRF3, IRF5, and IRF7 to the ISRE in the promoter of the classic IFN-response gene ISG15, dependent on the ISRE, was evaluated using an ISRE luciferase reporter construct (Figure 6A). Poly (I-C) stimulated ISRE promoter luciferase activity was significantly lower in IRF3, IRF5, and IRF7 deficient THP1 (p < 0.001; n = 3 each). In contrast to human RA synoviocytes (9), IRF3, IRF5, and IRF7 deficiency in THP1 monocytes had no effect on AP-1 or NF-κB luciferase activity in THP1 cells (Figure 6A and B). To further evaluate the regulation of IL-6 gene expression by IRF7 in THP1, the IL-6 promoter luciferase construct was transfected after IRF7 siRNA followed by luciferase assay. Surprisingly, IRF7 inhibition did not decrease IL-6 promoter activity despite a significant and specific decrease in IL-6 gene expression (Figure 6D).
Figure 6.

Regulation of ISRE promoter activity by IRF. IRF3, IRF5, IRF7, or sc siRNA-treated THP-1 cells were transfected with an IRSE luciferase reporter construct. Cells were stimulated with poly (I-C) (10 μg/mL) for 6 h, and THP1 lysates were assayed for luciferase activity normalized to Renilla reniformis luciferase. Cells transfected with sc siRNA were used as control. A. ISRE promoter activity was decreased to baseline by IRF3 knockdown (p < 0.001; n = 3). There was also a significant reduction of approximately 50% in ISRE promoter activation with IRF7 and IRF5 deficiency (p < 0.001). B. IRF3/5/7 deficiency had no effect on AP-1 promoter activity (n = 3). C. IRF3/5/7 did not contribute to NF-κB promoter activation as determined by luciferase activity. D. IRF7 deficiency did not regulate IL-6 promoter activation as measured by relative luciferase activity.
3.5. IRF7 regulation of IL-6 mRNA stability
Because IRF7 inhibition did not affect IL-6 promoter activation, we investigated post-transcriptional regulation of IL-6 mRNA stability by IRF7 as a potential mechanism for the effects on IL-6 gene expression. IL-6 levels are regulated in part by stability of mRNA (30). We measured the half-life of IL-6 mRNA after inhibition of transcription with actinomycin D treatment of cultured cells. THP1 cells were transfected with IRF7 siRNA, stimulated with poly (I-C), treated with actinomycin D to block transcription and THP1 RNA was harvested at various time points up to 18h to determine the half-life of IL-6 mRNA. IRF7 deficiency decreased the half-life of IL-6 mRNA from 2.3h to 0.75h (Figure 7). These experiments suggest that IRF7 contributes to regulation of IL-6 gene expression through stabilization of IL-6 mRNA.
Figure 7.
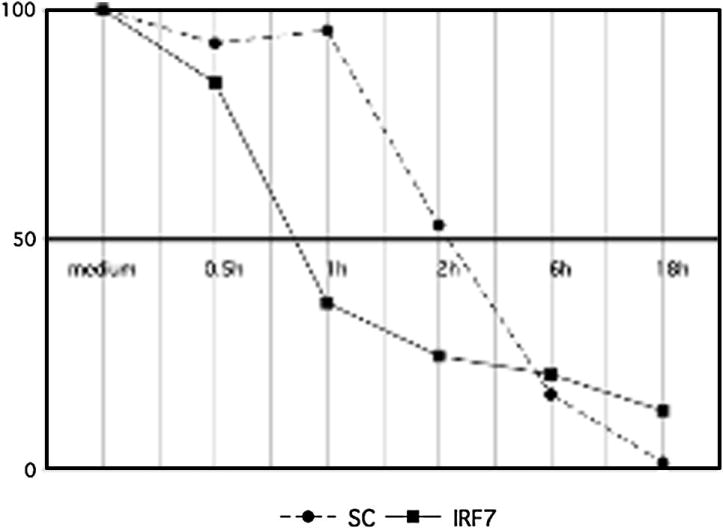
IRF7 decreases IL-6 mRNA stability. THP1 cells were transfected with IRF7 siRNA, stimulated with poly (I-C) and harvested at various time points after treatment with actinomycin D to block transcription and determine half-life of IL-6 mRNA. IRF7 deficiency decreased the half-life of IL-6 mRNA from 2.2 h to 0.69 h (p < 0.04; n=3).
4. Discussion
Innate pathways activate production of chemokines, cytokines, and degradative enzymes that promote inflammation, cell recruitment, joint destruction, and organ damage in autoimmune diseases. These signaling cascades have been implicated in RA and SLE, and the interferon signature induced by innate receptor activation has been observed in diverse autoimmune disorders. IFN-regulated gene expression in certain subsets of patients correlates with disease activity and in others disease severity. In many patients the IFN signature remains unchanged and is not predictive of disease activity, but in subgroups of SLE patients it can be used as a biomarker of disease activity. Because the type I IFN response has been implicated in SLE and RA, we hypothesized that the IFN-regulating transcription factors IRF3, IRF5, and IRF7, contribute to innate signaling that leads to production of IFN-stimulated gene expression and autoimmunity in rheumatic diseases. Regulation of IRF might be a factor in the pathogenesis of SLE or RA and induction of the IFN signature. As a result, IRF might serve as biomarkers for disease activity or therapeutic targets.
Genetic studies identified IRF5 and IRF7 polymorphisms that are associated with increased risk of SLE. IRF5 polymorphisms result in altered mRNA and IRF5-mediated transcription of target genes (31). Therefore, IRF5 might contribute to the IFN response and the pathogenesis of SLE. IRF7 variants in conjunction with SLE autoantibodies were found to result in increased serum type I IFN, suggesting a pathogenic role for IRF7 in SLE (32). However, IRF3 gene polymorphisms were not associated with susceptibility or severity of SLE. Targeted inhibition of IRF in SLE lymphocyte or monocyte subpopulations of PBMC has not been described, however case study results suggest that IRF7 induction in SLE PBMC correlated with increased disease activity and production of the IFN signature (33). In RA synoviocytes, targeted knockdown of IRF3 and IRF7 was performed to evaluate the hierarchy of these IRF in synoviocyte gene expression (9). In contrast to mouse embryonic fibroblasts and bone marrow-derived dendritic cells where IRF7 is the key IRF, IRF3 is the master regulator of type I IFN responses in poly (I-C) stimulated human RA FLS (9). Surprisingly, IRF3 deficiency also suppressed expression of some genes in RA FLS that are predominantly regulated by AP-1 (MMP) or NF-κB (IL-8, IL-6) promoter elements rather than an ISRE site (see Table 1).
Targeted knockdown of IRF was followed by evaluation of protein and gene expression, promoter activation, and mRNA stability. IRF3, IRF5, and IRF7 all contributed to IFNβ, IP-10, and RANTES gene expression in response to poly (I-C) stimulation of THP1. IRF3 and IRF5 deficiency decreased MCP-1 and only IRF3 knockdown decreased MIP-1α expression. The THP1 data is summarized and compared with the fibroblast-like synoviocyte results in Table 1.
IRF5 played a critical role in TLR signaling and pro-inflammatory cytokine production including TNF, IL-12, and IL-6 in hematopoietic cells from genetically deficient mice (34). The functions of IRF5 are probably species and cell specific, however the role in human THP1 monocytes also included regulation of pro-inflammatory cytokine and type I IFN responses. Similar to RA FLS, IRF3 played a key role in regulation of type I IFN responses in THP1 cells. In contrast to RA FLS, IRF3 more selectively regulated IFN-stimulated genes in THP1 through ISRE activation and did not contribute to traditionally AP-1 or NF-κB regulated gene expression. With the exception of IFNβ and IP-10, IRF7 did not significantly reduce expression of the other IFN-stimulated genes evaluated. Surprisingly, IRF7 regulated IL-6 gene expression in THP1 cells (Table 1).
Studies of promoter activity confirmed that in THP1 cells, IRF deficiency significantly decreased ISRE luciferase, confirming transcriptional regulation of the type I IFN response through IRF activation of the ISRE. Promoter assays also confirmed that IRF do not regulate AP-1 and NF-κB promoter activity in THP1 monocytic cells, in contrast to RA FLS. Because IRF7 regulated IL-6 gene expression, we used a full length IL-6 promoter construct to evaluate transcriptional regulation of IL-6 by IRF7. However, IL-6 promoter activity was not changed by IRF7 deficiency. Cytokines are regulated at many levels including mRNA transcription but also post-transcriptional regulation including stability of mRNA and translation of mRNA into protein. IL-6 levels are regulated in part by stability of mRNA and therefore changes in mRNA half-life (30). Because IRF7 inhibition did not affect IL-6 transcription, we investigated post-transcriptional regulation of IL-6 mRNA stability by IRF7 as a potential mechanism for the effects on IL-6 gene expression. IRF7 deficiency decreased the half-life of IL-6 mRNA and these results suggested that IRF7 contributes to regulation of IL-6 through stabilization of IL-6 mRNA. This regulation of IL-6 represents a novel mechanism of regulation of production of the cytokine IL-6 by IRF7. Several RNA binding proteins have been identified that recognize adenine and uridine rich elements in the untranslated region of mRNA. Studies are currently underway to further evaluate the mechanism of IL-6 mRNA stabilization by IRF7 including assessment of the RNA binding proteins tristetraprolin, HuR, and AUF1 known to contribute to mRNA stability.
5. Conclusions
We have shown that the IRF transcription factors differentially regulate pro-inflammatory cytokine gene expression and induction of the type I IFN response in human monocytic cells. Cellular expression of IFN-regulated genes in subgroups of patients with SLE and RA is consistent with active IFN signaling. IRF controlled IFN-regulated gene expression through specific activation of the ISRE and represent a potential target for modulation of the IFN signature as treatment for autoimmune diseases. In both RA fibroblast-like synoviocyte and human monocytic cell lines, IRF3 was central to IFN-regulated gene expression. IRF5 and IRF7 both contributed to expression of a subset of IFN-regulated genes and IRF7 played novel role in stabilization of IL-6 mRNA in human monocytic cells. Targeting innate pathways and the IFN response requires a detailed understanding of the positive and negative regulatory roles of the IRF family. Vital and broad roles for each IRF in host defense, activation or suppression of immune responses, immune cell differentiation, and regulation of cell growth or death appear to overlap functionally (35). Considering the potential pathogenic role of IFN in autoimmune disease and the delicate balance between antiviral and cancer fighting effects, dissecting the role of IRF in the IFN response could have important therapeutic and safety implications.
Highlights.
Interferon regulatory factors activate the type I IFN response in THP1 monocytic cells
IRF3 was central to IFN-regulated gene expression, however IRF7 regulated IL-6
IRF represent therapeutic targets for RA and SLE
Acknowledgments
This publication was supported by grants from NIH/NIAMS K08 grant AR052800 and K08 ARRA AR052800-04S1 supplement, the Clinical Translational Research Institute Pilot Project grant number 1UL1RR031980 from the NIH National Center for Research Resources, and the Lupus Foundation of America
The author would like to thank Trevor B. Kimbler for technical assistance and statistical analysis.
Abbreviations
- AP-1
Activator protein 1
- FLS
fibroblast-like synoviocyte
- INF
interferon
- IKK
IκB kinase
- IP-10
IFN inducible protein 10
- ISRE
IFN-stimulated response element
- IRF
IFN regulatory factor
- MIP
macrophage inflammatory protein
- MMP
matrix metalloproteinase
- MCP1
monocyte chemotactic protein-1
- PBMC
peripheral blood mononuclear cell
- poly (I-C)
polyinosinic and polycytidylic acid
- RANTES
regulated upon activation, normal T-cell expressed, and secreted
- RA
rheumatoid arthritis
- siRNA
small interfering RNA
- SLE
systemic lupus erythematosus
- TNF
tumor necrosis factor
- TLR
toll-like receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vallin H, Perers A, Alm G, Ronnblom L. Anti-double stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol. 1999;163:6306–6313. [PubMed] [Google Scholar]
- 2.Bennett L, Palucka A, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crow M. Interferon pathway activation in systemic lupus erythematosus. Curr Rheumatol Rep. 2005;7:463–468. doi: 10.1007/s11926-005-0053-4. [DOI] [PubMed] [Google Scholar]
- 4.Baechler E, Gregersen P, Behrens T. The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol. 2004;16:801–807. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Baechler E, Batliwalla F, Karypis G, Gaffney P, Ortmann W, Espe K, Shark K, Grande W, Hughes K, Kapur V, Gregersen P, Behrens T. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5:279–287. doi: 10.1186/ar1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis. Cell Immunol. 2005;233:90–96. doi: 10.1016/j.cellimm.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 8.Sweeney S, Mo L, Firestein G. Antiviral gene expression in rheumatoid arthritis: role of IKK epsilon and interferon regulatory factor 3. Arthritis Rheum. 2007;56:743–752. doi: 10.1002/art.22421. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney S, Kimbler T, Firestein G. Synoviocyte Innate Immune Responses: II. Pivotal Role of IFN Regulatory Factor 3. J Immunol. 2010;15:7162–7168. doi: 10.4049/jimmunol.0903944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshizawa T, Hammaker D, Sweeney S, Boyle D, Firestein G. Synoviocyte innate immune responses: I. Differential regulation of interferon responses and the JNK pathway by MAPK kinases. J Immunol. 2008;181:3252–3258. doi: 10.4049/jimmunol.181.5.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney SE, Firestein GS. Signal transduction in rheumatoid arthritis. Curr Opin Rheumatol. 2004;16:231–237. doi: 10.1097/00002281-200405000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Firestein G. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol. 2005;11:S39–44. doi: 10.1097/01.rhu.0000166673.34461.33. [DOI] [PubMed] [Google Scholar]
- 13.van Holten J, Smeets TJ, Blankert P, Tak PP. Expression of interferon beta in synovial tissue from patients with rheumatoid arthritis: comparison with patients with osteoarthritis and reactive arthritis. Ann Rheum Dis. 2005;64:1780–1782. doi: 10.1136/ard.2005.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheum Dis. 2006;65:294–300. doi: 10.1136/ard.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Vicuna R, Gomez-Gaviro MV, Dominguez-Luis MJ, Pec MK, Gonzalez-Alvaro I, Alvaro-Gracia JM, Diaz-Gonzalez F. CC and CXC chemokine receptors mediate migration, proliferation, and matrix metalloproteinase production by fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Rheum. 2004;50:3866–3877. doi: 10.1002/art.20615. [DOI] [PubMed] [Google Scholar]
- 16.Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, Burdick MD, Pope RM, Strieter RM. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90:772–779. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsen N, Sokka T, Seehorn C, Kraft B, Maas K, Moore J, Aune T. A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann Rheum Dis. 2004;63:1387–1392. doi: 10.1136/ard.2003.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell Immunol. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Chi H, Flavell R. Innate recognition of non-self nucleic acids. Genome Biol. 2008;9:211. doi: 10.1186/gb-2008-9-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diebold S, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 21.Means T, Latz E, Hayashi F, Murali M, Golenbock D, Luster A. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ardoin S, Pisetsky D. Developments in the scientific understanding of lupusArthritis. Res Ther. 2008;10:218. doi: 10.1186/ar2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim TK, Kim TH, Maniatis T. Efficient recruitment of TFIIB and CBP-RNA polymerase II holoenzyme by an interferon- enhanceosome in vitro. Proc Natl Acad Sci U S A. 1998;95:12191–12196. doi: 10.1073/pnas.95.21.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 25.Romieu-Mourez R, Solis M, Nardin A, Goubau D, Baron-Bodo V, Lin R, Massie B, Salcedo M, Hiscott J. Distinct roles for IFN regulatory factor (IRF)-3 and IRF-7 in the activation of antitumor properties of human macrophages. Cancer Res. 2006;66:10576–10585. doi: 10.1158/0008-5472.CAN-06-1279. [DOI] [PubMed] [Google Scholar]
- 26.Génin P, Lin R, Hiscott J, Civas A. Differential regulation of human interferon a gene expression by interferon regulatory factors 3 and 7. Mol Cell Biol. 2009;29:3435–3450. doi: 10.1128/MCB.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goubau D, Romieu-Mourez R, Solis M, Hernandez E, Mesplède T, Lin R, Leaman D, Hiscott J. Transcriptional re-programming of primary macrophages reveals distinct apoptotic and anti-tumoral functions of IRF-3 and IRF-7. Eur J Immunol. 2009;39:527–540. doi: 10.1002/eji.200838832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sweeney SE, Hammaker D, Boyle DL, Firestein GS. Regulation of c-Jun phosphorylation by the I kappa B kinase-epsilon complex in fibroblast-like synoviocytes. J Immunol. 2005;174:6424–6430. doi: 10.4049/jimmunol.174.10.6424. [DOI] [PubMed] [Google Scholar]
- 29.Boyle DL, Rosengren S, Bugbee W, Kavanaugh A, Firestein GS. Quantitative biomarker analysis of synovial gene expression by real-time PCR. Arthritis Res Ther. 2003;5:R352–360. doi: 10.1186/ar1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paschoud S, Dogar A, Kuntz C, Grisoni-Neupert BLR, Kuhn L. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol. 2006;26:8228–8241. doi: 10.1128/MCB.01155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niewold T, Kelly J, Flesch M, Espinoza L, Harley J, Crow M. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salloum R, Franek B, Kariuki S, Rhee L, Mikolaitis R, Jolly M, Utset T, Niewold TB. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62:553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sweeney SE. Hematopoietic stem cell transplant for systemic lupus erythematosus: Interferon regulatory factor 7 activation correlates with the IFN signature and recurrent disease. Lupus. 2010 doi: 10.1177/0961203310394897. [DOI] [PubMed] [Google Scholar]
- 34.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak T, Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 35.Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother. 2010;59:489–510. doi: 10.1007/s00262-009-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]