

Abstract
The identification of the factors that enable normally folded proteins to remain in their soluble and functional states is crucial for a comprehensive understanding of any biological system. We have determined a series of energy landscapes of the acylphosphatase from Drosophila melanogaster under a variety of conditions by combining NMR measurements with restrained molecular dynamics simulations. We thus analyzed the differences in the structures, dynamics, and energy surfaces of the protein in its soluble state or in situations where it aggregates through conformational states that have native-like structure, folding stability, and enzymatic activity. The study identifies the nature of the energy barriers that under normal physiological conditions prevent the protein ensemble from populating dangerous aggregation-prone states. We found that such states, although similar to the native conformation, have altered surface charge distribution, alternative topologies of the β-sheet region, and modified solvent exposure of hydrophobic surfaces and aggregation-prone regions of the sequence. The identified barriers allow the protein to undergo functional dynamics while remaining soluble and without a significant risk of misfolding and aggregation into nonfunctional and potentially toxic species.
Keywords: protein misfolding, free energy barriers, avoidance of protein aggregation, molecular simulations
The majority of proteins have evolved to adopt distinctive and well-defined functional states under physiological conditions, either as monomers or as complexes. The structures corresponding to these states are encoded in the sequence as is the crucial ability of the molecules to remain soluble within the crowded cellular environment (1–5). It is increasingly evident, however, that even under physiological conditions the aggregated states of proteins, such as the highly ordered amyloid form, can be thermodynamically more stable than native states (6–8), indicating that kinetic factors are of key importance in enabling protein homeostasis to be maintained (1, 9–11).
Proteins in vivo only rarely convert into aberrant aggregated states, such as those associated with pathological conditions such as Alzheimer’s disease and type II diabetes, despite their inherent tendency to do so in vitro (5). It is thus clear that all living systems rely on a large variety of regulatory mechanisms, such as molecular chaperones (12, 13) and quality control processes, which act to inhibit aggregation (14, 15) and for triggering the degradation of partially unfolded proteins (16). But the primary mechanism of maintaining functional and soluble states of proteins is encoded in the sequence and involves the existence of intrinsic energy barriers that prevent the conversion into an aggregation-prone state (1, 5, 17). It is for these barriers that the majority of proteins are able to avoid aggregation under normal physiological conditions despite considerable evidence that show that amyloid formation in vitro is a common characteristic of normally folded proteins (10, 18).
In the present study, we used a combination of NMR experiments and molecular dynamics simulations to identify the characteristic features of the free energy landscapes that enable the majority of the proteins to avoid aggregation under physiological conditions. We chose for this scope the acylphosphatase from Drosophila melanogaster (AcPDro2) because this is a particularly well-suited system for investigating the molecular strategies used by living systems for the maintenance of protein solubility. AcPDro2 in its native state is a globular and monomeric protein with a structure consisting of five β-strands (S1–S5), which form a single β-sheet, and two α-helices (H1 and H2) that lie adjacent to this α-sheet. The importance that subtle intrinsic factors play in enabling this protein to remain soluble is clearly shown by the fact that a very low concentration (5% vol/vol) of trifluoroethanol (TFE) is sufficient to induce rapid formation of amyloid fibrils, although the protein still populates a highly native-like conformational ensemble before aggregation occurs (19). Indeed, under these conditions, the hydrodynamic radius, intrinsic fluorescence, secondary structure content, and enzymatic activity of AcPDro2 in its monomeric state are indistinguishable with those of the protein in the absence of TFE, where the propensity of AcPDro2 to aggregate is extremely low (19). Moreover, within experimental error, AcPDro2 has the same thermodynamic stability (i.e., the same free energy of unfolding, ΔGU-F) in the presence and in the absence of 5% (vol/vol) TFE as determined using urea-induced denaturation at equilibrium (19). The similar thermodynamic stabilities in 0% and 5% (vol/vol) TFE originate from the fact that the folding and unfolding rates are accelerated to similar extents following the addition of 5% (vol/vol) TFE (19). By contrast to AcPDro2, most folded proteins aggregate in the presence of much higher concentrations of TFE (15–30%) where a significant portion of the molecules are unfolded or strongly destabilized.
In the present study, we have explored the factors that enable AcPDro2 in its native state to remain soluble for long periods of time under some conditions but to aggregate rapidly under others. We have therefore determined the structural ensembles and free energy surfaces under a series of different conditions by combining NMR data from relaxation and H/D exchange experiments, with statistical mechanics techniques based on restrained molecular dynamics simulations. Analysis of these energy surfaces has enabled us to define a series of specific structural and dynamical factors along with the energy barriers that enable the protein to maintain its functional native state.
Results
Assignment and Analysis of NMR Spectra.
All NMR measurements were carried out at 298 K and pH 4.0. This pH is close to that used in the determination of the AcPDro2 crystal structure (pH 4.5, ref. 20), and under these conditions all protein states of interest in this study, including those in 5% TFE, are stable in solution for a sufficient time to enable the relevant NMR experiments to be performed prior to aggregation. We checked by electrospray ionization–mass spectrometry that under these conditions the H/D exchange of AcPDro2 occurs in an EX2 regime, which ensures that the exchange data provide a measure of the local equilibrium constant between “open” (solvent-accessible) and “closed” (solvent-inaccessible) states of the protein amide groups (21–25). Experiments were carried out in either 30 mM acetate buffer or 30 mM phosphate buffer and in the presence or absence of 5% (vol/vol) TFE. These conditions are denoted A (acetate buffer and 0% TFE), B (acetate buffer and 5% TFE), C (phosphate buffer and 0% TFE), and D (phosphate buffer and 5% TFE). In the case of phosphate buffer solutions, a phosphate ion is bound to the enzyme active site (26).
Assignments of the NMR spectra were made in 30 mM phosphate buffer (condition C) by means of triple resonance experiments [HNCA, CBCA(CO)NH, HNCACB, HNCO, HN(CA)CO, and 15N-total correlation spectroscopy–heteronuclear single quantum coherence (TOCSY-HSQC) spectra]. The analysis of the spectra, following the procedure described in ref. 27, resulted in the identification of main-chain resonances (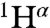 ,
, 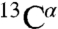 ,
, 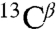 ,
, 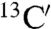 , 15N, 1HN) for 88 out of 101 residues (Fig. S1A). Most of the unassigned residues are in loop regions of the protein, although some are in helices H1 and H2 and strand S1 (Fig. S1A). Assignments in the 1H-15N-HSQC spectra in acetate buffer (condition A) were then derived by recording spectra at different concentrations of phosphate ions, such that the chemical shifts of the peaks could be correlated with those under condition C. Similarly, assignments of 1H-15N-HSQC spectra in the presence of 5% TFE were derived from the respective spectra in the absence of TFE. The 1H-15N-HSQC spectra of the ligand-free state (condition A) contain a smaller number (72) of resolved peaks than the spectra of the phosphate-bound state (condition C). The peaks that are not detected in the ligand-free state are mainly located in the ligand-binding pocket and in regions spatially close to it (Fig. S1B), and they are likely to be affected by exchange broadening resulting from the greater flexibility and disorder of these regions of the ligand-free state of the protein. It is worth noting that conditions A and B resulted in the same number of assigned main-chain resonances.
, 15N, 1HN) for 88 out of 101 residues (Fig. S1A). Most of the unassigned residues are in loop regions of the protein, although some are in helices H1 and H2 and strand S1 (Fig. S1A). Assignments in the 1H-15N-HSQC spectra in acetate buffer (condition A) were then derived by recording spectra at different concentrations of phosphate ions, such that the chemical shifts of the peaks could be correlated with those under condition C. Similarly, assignments of 1H-15N-HSQC spectra in the presence of 5% TFE were derived from the respective spectra in the absence of TFE. The 1H-15N-HSQC spectra of the ligand-free state (condition A) contain a smaller number (72) of resolved peaks than the spectra of the phosphate-bound state (condition C). The peaks that are not detected in the ligand-free state are mainly located in the ligand-binding pocket and in regions spatially close to it (Fig. S1B), and they are likely to be affected by exchange broadening resulting from the greater flexibility and disorder of these regions of the ligand-free state of the protein. It is worth noting that conditions A and B resulted in the same number of assigned main-chain resonances.
NMR Measurements of AcPDro2 Under Conditions A to D.
In order to begin to probe the differences between the solution conformations of AcPDro2 in the soluble or aggregation-prone states, we examined the chemical shift values in the spectra under conditions A and B (Fig. 1A). The analysis indicates small but significant changes for residues W42, N45, S85, I87, Q88, and I100 (from strands S2, S4, and S5), residues H32, R36, and L62 (from helices H1 and H2), and residues R47 and T96 (from loop regions).
Fig. 1.

NMR measurements of AcPDro2 under conditions where it is highly stable in the monomeric state (A, condition A) or readily self-assembles into amyloid fibrils (B, condition B) or bound to phosphate ions (C, conditions C and D). (A) Spectral changes upon addition of TFE (0% to 5%, condition A to B, respectively). The X-ray structure of AcPDro2 is shown on the left of the panel. Regions shown in red on the structure indicate residues with the largest (at least 1 ppm) changes in the 1H-15N-HSQC spectra. (B) Backbone amide protection factors reported as ΔG values. Red and green bars refer to 0% (condition A) and 5% (condition B) TFE, respectively. The structure of AcPDro2 is drawn on the left of the panel. Regions in red indicate residues showing the largest reductions of the protection factors (Fig. S2). (C) Phosphate-bound AcPDro2 solution states (condition C and D). Backbone amide protection factors are reported as ΔG values. Red and green bars refer to 0% (condition C) and 5% (condition D) TFE, respectively. (D) S2 order parameters measured for the ligand-free (black) and phosphate-bound states (red).
In addition to measurements of chemical shifts, the exchange rate constants kobs of the backbone amide hydrogens were determined for 59 residues under condition A (Fig. 1B) by direct observation of the decay in the intensities of peaks in the 1H-15N-HSQC spectra as a function of time after dilution into deuterated buffer. In the case of the remaining amides, the hydrogens of 13 residues exchanged effectively completely prior to acquisition of the first 1H-15N HSQC spectrum, preventing the determination of exchange rate constants kobs from the fitting of the decay curves of the 1H-15N HSQC signals. For some of these residues, however, the rate constants kobs could be estimated from phase-modulated clean chemical exchange (CLEANEX-PM, ref. 28) experiments (see Materials and Methods and Fig. S2). These values of kobs allowed the amide hydrogen protection factors to be determined for eight residues located at the disordered N terminus of the protein (residues A2, G3, S4, G5, V6) and in the loop between strands S2 and S3 (residues T46, R47, and D48). Under condition B, 58 kobs values could be measured by following the decays of 1H-15N-HSQC spectra, whereas 14 amide protons exchanged prior to acquisition of the first spectrum (Fig. 1B). Similar to condition A, kobs rate constants of eight backbone amides (residues A2, G3, S4, G5, V6, T46, R47, and D48) could be obtained by the CLEANEX-PM method.
Many of the protection factors are essentially unchanged in the aggregation-prone and resistant states but some clusters of residues show a higher degree of protection in the aggregation-resistant state (condition A) than in the aggregation-prone state (condition B, Fig. 1B and Figs. S3 and S4). These clusters include amide protons of residues G41, C43, N45, D99, and I101, with major reductions in the protections for residues C43 and D99 whose interactions represent the core of the network of H bonding at the interface between strands S2 and S5; this result indicates that the interface between these two strands is stabilized under conditions where the protein is aggregation resistant (Figs. S3B, and Fig. S4 for the decay profiles). Higher protection factors in the absence of 5% TFE are also found for residues clustered in helix H1 (residues 34–7) and in helix H2 (residues N61 and L62), albeit to a lesser extent (Figs. S3A, and Fig. S4 for the decay profiles). The lack of changes for the majority of the amide protection factors is consistent with previous measurements that revealed no significant differences in the folding stability in the presence or absence of 5% (vol/vol) TFE (19).
By following the decays of-HSQC spectra in the phosphate-bound state (conditions C and D), kobs values for 72 residues could be determined; in addition, amides of 16 residues were found to exchange prior to acquisition of the first spectrum, none of which was detectable in CLEANEX-PM experiments at mixing times up to 25 ms (Fig. S2B). The protection factors calculated from the 72 kobs values measured for the phosphate-bound condition C (Fig. 1C) are on average approximately 4 kJ/mol higher than those measured under the ligand-free condition A (Fig. 1B). Moreover, for the phosphate-bound state, protection factors measured in 0% and 5% TFE are virtually identical, a result that correlates with the finding that phosphate binding induces resistance to aggregation such that the protein is no longer prone to amyloid formation even in 5% (vol/vol) TFE.
Energy Landscapes of AcPDro2.
The exchange protection factors discussed above were then used as the basis for generating ensembles of structures of AcPDro2 under the four sets of conditions studied in this work. This approach combines experimental NMR data and molecular dynamics simulations in order to obtain detailed conformational descriptions that incorporate both structural and dynamical information (29–32); this is achieved by performing restrained molecular dynamics simulations that enforce a pseudoenergy term in such a way as to maximize the agreement between experimental data and the NMR observables back-calculated from the ensembles of structures (33). Such a procedure assists conformational sampling and ensures that an enhanced representation of the solution state free energies is generated (29). Because the present restrained samplings enforce NMR observables (i.e., H/D exchange protection factors) that reflect fluctuations occurring in the millisecond timescale (and beyond) (21–24), the resulting structural ensembles are particularly valuable for describing backbone dynamics relevant to the processes of protein folding and misfolding, which are those that affect the maintenance of protein solubility and are governed by events that typically occur in these timescales (34, 35).
The ensembles generated from the NMR data recorded under the different conditions were analyzed by defining structural parameters that provide independent measures of the key conformational features of the various states of the protein. The choice of these parameters is crucial for describing appropriately both the molecular conformations and the Boltzmann populations of the species composing the ensembles. Accordingly, we selected for this purpose three specific “coordinates” [namely, the rmsd of Cα atoms from the crystal structure (c1), the radius of gyration (c2), and the fraction of native contacts (c3)]. By calculating the distribution of states in these three dimensions, it has been possible to generate free energy surfaces of AcPDro2 from the Boltzmann populations of the ensembles as projected onto these three coordinates (Fig. 2). The free energy maps have been generated from 200,000 conformations in each sampling. A detailed error analysis has been performed in order to define the statistical significance of the energy barriers herein reported (see SI Materials and Methods).
Fig. 2.

AcPDro2 solution state ensembles under conditions where it is highly stable in the monomeric state (A, condition A) or readily self-assembles into amyloid fibrils (B, condition B) or bound to phosphate ions (C, conditions C and D). The solution ensembles were generated by means of restrained molecular dynamics simulations employing the amide exchange NMR protection factors as restraints. The conformations are projected on to three reaction coordinates to define a three-dimensional free energy landscape. The coordinates employed are as follows: Cα rmsd from the crystal structure (c1), radius of gyration (c2), and the fraction of native contacts (c3). The free energy surfaces are drawn by means of contour levels embedding isosurfaces of free energy at levels −7.5, −10, −18, and -24 kJ/mol. The statistical errors for these isosurfaces are 2.17, 1.1, 0.21, and 0.06 kJ/mol, respectively. Representative ensembles in the minima of conformational wells are drawn by Cα traces. Color codes are yellow for β-strands, cyan for loops, red for α-helices.
Ligand-free soluble state of AcPDro2 (condition A).
The free energy surface of AcPDro2 under conditions in which the protein is stable in its monomeric state, condition A, is dominated by a single conformational well that covers a relatively broad region of free energy space and shows a substantial degree of structural variability indicative of the dynamics of the enzyme in the ligand-free state (Fig. 2A). The centroid of this energy well has a radius of gyration of 14.0 Å, an rmsd from the X-ray structure of 1.52 Å (calculated for all Cα atoms except those in loop regions), a hydrophobic accessible surface area of 5,470 Å2, and a fraction of native contacts equal to 0.82 (all values reported in this section refer to the centroids of the wells/lobes). The free energy surface determined under condition A also contains a protruding lobe (lobe 1) consisting of conformations with a low Cα rmsd (1.30 Å) from the X-ray structure and a large fraction (0.92) of native contacts (additional parameters are reported in Table 1).
Table 1.
Structural parameters of the free energy wells
| Cα rmsd,* Å | rgyr, Å | Fraction of native contacts | Hydrophobic SAS, nm2 | Main-chain SAS , nm2 | |
| Condition A main well | 1.52 | 14.0 | 0.82 | 54.7 | 4.71 |
| Condition A lobe 1 | 1.30 | 13.6 | 0.92 | 53.8 | 4.62 |
| Condition B main well | 1.57 | 14.2 | 0.83 | 55.0 | 4.73 |
| Condition B lobe 1 | 1.62 | 14.5 | 0.76 | 55.3 | 4.72 |
| Condition B lobe 2 | 2.75 | 15.0 | 0.72 | 58.6 | 5.12 |
| Conditions C and D | 1.08 | 13.7 | 0.96 | 53.2 | 4.56 |
The values refer to the centroids of the wells.
*Calculated by considering all Cα atoms except those located in loop regions of the protein.
Aggregation-prone state (condition B).
The energy surface under condition B, a state in which AcPDro2 readily aggregates, was again calculated by simulations restrained with NMR protection factors and reflects an ensemble of structures that is significantly more heterogeneous than that obtained from the equivalent restraints determined in the absence of TFE. The calculations did not explicitly include TFE; however, the effects of TFE are provided by the experimental data employed as restraints. The resulting surface is again characterized by a single dominant conformational well that is similar to the main well of the energy surface determined for condition A, although the well is generally broader and lacking structures with a very high degree of native-like content (i.e., as observed in the case of lobe 1 of condition A). In addition to the main well, however, the free energy surface under condition B shows two lobes, one of which (lobe 1) includes conformations with increased Cα-rmsd deviations from the crystal structure (1.62 vs. 1.57 Å) and a reduced fraction of native contacts (0.76 vs. 0.83) relative the main well (all values refer to the centroids of the wells).
The principal difference between the energy surfaces of AcPDro2 under the two conditions, however, is the existence of a second lobe under condition B that is not significantly accessible under condition A (Fig. 2). The conformations in this region have distinctive features compared to those in the other accessible regions of the surface, including a lower fraction of native contacts (0.72), a larger radius of gyration (15.0 Å), and a larger Cα rmsd from the crystal structure (2.75 Å). A particular distinctive trait is the more substantial exposure of regions of hydrophobic surface and main-chain atoms (512 Å2 compared to 471 Å2 of the main well of Ac0-TFE) as well as a lower content of secondary structure (defined by using the DSSP program; ref. 36 and Fig. S5).
Ligand-bound states (conditions C and D).
The protection factors measured in phosphate buffer in the absence or presence of 5% TFE (conditions C and D, Fig. 1C) were employed for generating energy surfaces representative of the ligand-bound state of AcPDro2. Phosphate ions were not explicitly considered in the calculations but the effects of the phosphate binding are incorporated in the information carried in the experimental data. The resulting free energy surfaces indicate that in both cases the dynamics of the phosphate-bound state are significantly more limited than those of the corresponding ligand-free ensembles (Fig. 2). The free energy landscapes under conditions C and D (Fig. 2C) show a single deep conformational well with similar features to lobe 1 of the energy surface determined under condition A, indicating that the accessible conformations in the phosphate-bound state are very close to the crystal structure of the enzyme (Cα rmsd of 1.08 Å for the centroid of the basin). Moreover, in neither of the phosphate-bound structural ensembles, conformations similar to those found for lobe 2 of the energy surface of condition B are populated at any detectable level.
The rms fluctuation profiles indicate that the most significant reductions of the backbone dynamics associated with binding are located in the loop connecting strand S1 and helix H1, the region involved in binding the phosphate ions (Fig. S6). Relaxation experiments reveal significantly higher order parameters (S2) for the phosphate-bound state compared to the ligand-free state (Fig. 1D), suggesting that the entire protein acquires a more rigid status upon binding and that such local effects in the binding site dynamics are eventually propagated throughout the protein. This result provides the detailed mechanism of the mode of actions of a small molecule that, upon binding, alters the energy landscape of the protein in a way to particularly disfavor dangerous aggregation-prone regions to be populated.
Energy Barriers for the Maintenance of Protein Solubility.
The free energy profiles calculated along a single coordinate (rmsd from the crystal structure of the Cα atoms) provides a direct estimation of the energy barriers that have been selected during evolution to maintain protein solubility (Fig. 3A). Although the energy landscape determined under condition A shows a symmetric well around a Cα-rmsd value of 1.5 Å, the profile determined under condition B shows an additional accessible region with Cα-rmsd values ranging from 2.5 to 3.6 Å (Fig. 3A). At an rmsd value of 2.5 Å, a free energy difference between conditions A and B is 18.2 ± 2.4 kJ/mol (see Fig. 3A, dashed line), thus providing an estimate for the energy barrier that prevents the native state (condition A) to explore regions having higher values of rmsd, which are populated exclusively in aggregation-prone conditions (condition B). As regions at rmsd values higher than 2.5 Å are accessible only under condition B, we designate this part of the conformational ensemble as an activated state “N∗” for aggregation, whereas the conformations ranging between 1.0 and 2.5 Å of Cα-rmsd will be referred as the native state “N” and include the conformations with high solubility (Fig. 3A).
Fig. 3.

Characteristics of the N and N∗ states of AcPDro2. (A) Energy barriers for the aggregation-prone conformations of AcPDro2. Free energy profiles are computed as a function of the Cα rmsd from the crystal structure (loop residues are not considered in the calculation). Red and blue lines represent the free energy profiles determined under conditions A and B, respectively. Statistical errors in the energies are reported. The free energies have been shifted in order to present the minimum at 0 kJ/mol. N∗ conformations range from rmsd of 2.5 to 3.6 Å. In the three-dimensional energy landscapes (Fig. 2), the N states populate the main wells and lobe 1 of the surfaces of both conditions A and B, whereas the N∗ species correspond to the conformations clustered in lobe 2 of the free energy surface determined under condition B. (B) Differences in SAS area for N and N∗ (upper histogram). The lower graph reports the Zaggregator profile for the AcPDro2 sequence. Aggregation-prone regions (Zaggregator values > 1) are marked in green. These regions are designated as Z1 (residues 11–17), Z2 (residues 39–43), and Z3 (residues 92–100). The location of Z1, Z2, and Z3 are marked in red on the ribbon representations of the structures of N and N∗ (right of the panel).
To assess the influence of the missing backbone N-H resonances in the spectra recorded under conditions A and B, we performed control calculations in which some missing protection factors were estimated by scaling the data obtained under condition C (see caption of Fig. S7). This analysis indicates that missing protection factors are not likely to affect the energy barrier separating the state N∗ from N when the protein is in the native state (condition A). Moreover, a control simulation performed by using randomly decreased protection factors maintains energy barriers that are similar to those showed in condition A (Fig. S8). This finding indicates that the three-dimensional clustering of residues that become less protected in aggregation-prone conditions is a key factor for influencing the energy landscapes.
Comparison of the N and N∗ ensembles reveals that the former possesses specific structural features that are likely to reduce the aggregation propensity. First, N fully preserves the structural features of a β-bulge extending from residues 85 to 87 that has been identified both generally (2) and specifically for AcP (37) as a negative designed element for reducing the aggregation propensity of edge strands, here S4. Because this element is partially disrupted in N∗ (three times lower than in the N ensemble), strand S4 is expected to be more prone to engage in intermolecular β-sheet interactions with the edge strands of other molecules (2). Second, in N∗ the interface between strands S2 and S5 is disrupted, giving rise to an increase in solvent accessible surface of 40 Å2 from N to N∗. Hence, where N is able to maintain the integrity of the native edge strand S5, N∗, by disrupting the native interface of strand S5, exposes strand S2 which becomes a new, nonnative edge strand. This altered topology of the β-sheet is particularly crucial in terms of the average aggregation propensity of the protein, as strand S2 does not possess the elements of negative design for avoiding protein aggregation that are normally carried by edge strands (2). Third, although local surface charges from strand S5 and the N-terminal segment of helix H1 are exposed to the solvent in the N state (Fig. 3A), in N∗, the partial disruption of this helix and the displacement of strand S5 enhance the hydrophobicity of the local surface by increasing the exposure of residues W42, C43, M44, V52, and I100.
In order to quantify the changes in the solubility behavior of the two ensembles, we calculated the intrinsic aggregation propensity profile from the AcPDro2 sequence by using Zyggregator (17) and combined this information with the solvent accessible surface areas (SAS) of the main chains of the N and N∗ states (Fig. 3B). This analysis highlights three regions of the AcPDro2 sequence with a high aggregation propensity (Zyggregator values larger than one; ref. 17); these are the fragment 11–17 (spanning strand S1), the fragment 39–43 (spanning strand S2), and the fragment 92–100 (spanning strand S5 and part of the preceding loop), and we designate these regions as Z1, Z2, and Z3, respectively. Although Z1 is completely buried in both N and N∗ (see Fig. S9 for absolute SAS values), both Z2 and Z3 are significantly more buried in N than in N∗, as shown by the difference in SAS of the two states (Fig. 3B). This finding indicates that N shields the aggregation-prone regions of its sequence more effectively within its three-dimensional fold than does N∗ and underlines how the energy landscapes of native states are designed to limit fluctuations leading to the exposure of such sequences by means of specific energy barriers.
Discussion
The internal motions associated with proteins are not only important for protein function (38–41) but also because they act as crucial determinants of the ability of a protein to avoid aggregation (18, 42). The present study has addressed this point in detail by showing the manner by which the native free energy landscape limits the accessibility of aggregation-prone regions of conformational space, thereby allowing the protein to maintain a soluble and functional state in solution. The analyses show how minor perturbations to the Boltzmann populations of specific conformational states in the native ensemble (here resulting from addition of 5% TFE or binding a phosphate ion) can change fundamentally the ability of the protein to remain soluble. In particular, under aggregation-prone conditions (condition B), the energy landscape lacks barriers for preventing an aggregation-prone state to be populated. This state is featured by the exposure of hot spots for aggregation as (nonnative) edge strands (2) or sequences predicted to have high aggregation profiles (17). We also determined at high resolution the effects of the binding of small molecules acting as inhibitors for protein aggregation, here the phosphate ion, on the local and global dynamics of the protein. We showed how such binding can significantly perturb the free energy surface and the conformational dynamics of the protein so as to make the population of aggregation-prone states energetically unfavorable, even under conditions where aggregation is otherwise favored.
The sensitivity of the energy surfaces of proteins to minor perturbations supports the view that there is a delicate balance between functionality, stability, and solubility, which is encapsulated by the concept of “life on the edge” (3, 42). Access to regions of conformational space that can readily be modulated by external factors (for example, the binding of small molecules or partner proteins) is crucial for enabling proteins to carry out their functions and also to allow complex processes such as molecular signalling or degradation to occur under cellular conditions. Such conformational access must, however, be moderated to avoid the population of any states of the protein that enable aberrant and nonfunctional interactions to occur. The manifestation of this balance is exemplified by the present finding that modifications of the correct positioning of a small fragment (strand S5) can enable the protein to maintain a fully soluble state and suppress the population of strongly aggregation-prone species (e.g., N∗).
In conclusion, the present investigation describes and illustrates at high resolution, by a method to define energy surfaces, the nature of the energy barriers that evolution has designed to allow proteins to maintain a functional dynamics without having access to dangerous aggregation-prone conformational states. The principles emerging here add to our previous understanding of how proteins avoid aggregation (2, 17, 43) and emphasize previously unidentified strategies adopted by proteins. The approach described in this study should be generally applicable to other systems, and illustrates how exploration of such surfaces can identify specific factors that serve to maintain proteins in their soluble and functional states or to avoid protein aggregation and its consequences.
Materials and Methods
NMR Assignments.
Standard triple resonance experiments (HNCA, CBCA(CO)NH, HNCACB, HNCO, HN(CA)CO spectra) as well as HNHA and 15N-TOCSY-HSQC were recorded at 25 °C on a Bruker Avance 700 MHz spectrometer equipped with a cryogenic triple resonance probe (Bruker BioSpin). The chemical shifts of individual spin systems (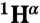 ,
, 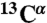 ,
, 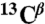 ,
, 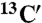 , 15N, 1HN) were determined by using a computer-aided procedure as described previously (27). The triple resonance spectra needed for the full backbone assignment were recorded in phosphate buffer, condition C, because the protein is in a particularly well-defined conformational state as a result of the binding of phosphate ions and is highly resistant to aggregation to permit the use of concentrated samples. Assignment of the 1H-15N-HSQC spectrum under condition A was achieved by following the peaks in spectra measured at different concentrations of phosphate ions, which link the 1H-15N-HSQC spectra under condition A to that under condition C. Assignments of spectra in 5% (vol/vol) TFE solutions were derived directly from the assignments of 1H-15N-HSQC spectra recorded in the absence of TFE.
, 15N, 1HN) were determined by using a computer-aided procedure as described previously (27). The triple resonance spectra needed for the full backbone assignment were recorded in phosphate buffer, condition C, because the protein is in a particularly well-defined conformational state as a result of the binding of phosphate ions and is highly resistant to aggregation to permit the use of concentrated samples. Assignment of the 1H-15N-HSQC spectrum under condition A was achieved by following the peaks in spectra measured at different concentrations of phosphate ions, which link the 1H-15N-HSQC spectra under condition A to that under condition C. Assignments of spectra in 5% (vol/vol) TFE solutions were derived directly from the assignments of 1H-15N-HSQC spectra recorded in the absence of TFE.
Measurements of Protein Dynamics.
The conformational dynamics of AcPDro2 were assessed by means of several NMR techniques covering the subnanoseconds to microseconds timescales and beyond. All the NMR experiments were conducted at 298 K and pH 4.0. Backbone dynamics were probed by means of H/D exchange and relaxation experiments.
H/D Exchange.
H/D exchange was monitored by following the intensities of 1H-15N-HSQC signals upon exposure of the protein to D2O. The settings employed enabled a full 2D acquisition to be recorded in approximately 2 min. The kobs values were extracted by fitting the signal decay curves to single exponential functions. Data obtained under conditions promoting AcPDro2 aggregation (i.e., 5% TFE) were processed with a baseline correction from the decay curves recorded in 90% H2O–10% D2O. Rapidly exchanging amide protons were characterized by CLEANEX-PM (28). This technique allows an estimation of the exchange rates from the slope of the linear interpolation of the intensities of amide peaks from spectra recorded at different mixing times τm (5, 10, 15, 20, and 25 ms). In particular, the volumes Vi of the peaks were normalized relative to those of the corresponding 1H-15N-HSQC peaks V0. By plotting Vi/V0 as a function of τm, kobs can be defined from the slopes of the linear interpolation.
Relaxation Experiments.
Longitudinal (T1) and transverse (T2) 15N-spin relaxation times were measured at 1H frequencies of 500 and 700 MHz (15N frequencies of 50.6 and 70.8 MHz, respectively) at 298 K. Further details are provided in SI Materials and Methods.
Structural Ensemble Refinement by H/D Exchange Data.
The protection factors determined from measurements of amide exchange rates were used to define ensembles of protein structures for the various conditions studied in this work by using the data as restraints in molecular dynamics samplings (29). Further details are reported in SI Materials and Methods.
Supplementary Material
Acknowledgments.
This work was supported by grants from the Medical Research Council (C.M.D.), Engineering and Physical Sciences Research Council (A.D.S. and C.M.D.), European Molecular Biology Organization (A.D.S.), Marie Curie (A.D.S.), Human Science Frontier Program (S.T.H.), Italian Ministero dell’Istruzione dell’Università e della Ricerca (F.C.), Boehringer Ingelheim Foundation and Murray Edwards College, Cambridge (A.D.), National Science Council of the Republic of China–Taiwan (S.T.H.), Biotechnology and Biological Sciences Research Council (C.M.D. and M.V.), and the Royal Society (M.V.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112197108/-/DCSupplemental.
References
- 1.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 2.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci. 2007;32:204–206. doi: 10.1016/j.tibs.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Lopez de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA. 2004;101:87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 6.Chatani E, Goto Y. Structural stability of amyloid fibrils of beta(2)-microglobulin in comparison with its native fold. Biochim Biophys Acta. 2005;1753:64–75. doi: 10.1016/j.bbapap.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Knowles TP, et al. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc Natl Acad Sci USA. 2007;104:10016–10021. doi: 10.1073/pnas.0610659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldwin AJ, et al. Metastability of native proteins and the phenomenon of amyloid formation. J Am Chem Soc. 2011;133:14160–14163. doi: 10.1021/ja2017703. [DOI] [PubMed] [Google Scholar]
- 9.Gianni S, et al. Structural characterization of a misfolded intermediate populated during the folding process of a PDZ domain. Nat Struct Mol Biol. 2010;17:1431–1437. doi: 10.1038/nsmb.1956. [DOI] [PubMed] [Google Scholar]
- 10.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 11.Douglas PM, Cyr DM. Interplay between protein homeostasis networks in protein aggregation and proteotoxicity. Biopolymers. 2010;93:229–236. doi: 10.1002/bip.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 13.Liberek K, Lewandowska A, Zietkiewicz S. Chaperones in control of protein disaggregation. EMBO J. 2008;27:328–335. doi: 10.1038/sj.emboj.7601970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behrends C, et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3:313–320. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia GG, et al. Prediction of aggregation-prone regions in structured proteins. J Mol Biol. 2008;380:425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 19.Soldi G, et al. Amyloid formation of a protein in the absence of initial unfolding and destabilization of the native state. Biophys J. 2005;89:4234–4244. doi: 10.1529/biophysj.105.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuccotti S, et al. Three-dimensional structural characterization of a novel Drosophila melanogaster acylphosphatase. Acta Crystallogr D Biol Crystallogr. 2004;60:1177–1179. doi: 10.1107/S0907444904006808. [DOI] [PubMed] [Google Scholar]
- 21.Best RB, Vendruscolo M. Structural interpretation of hydrogen exchange protection factors in proteins: characterization of the native state fluctuations of CI2. Structure. 2006;14:97–106. doi: 10.1016/j.str.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Clarke J, Itzhaki LS. Hydrogen exchange and protein folding. Curr Opin Struct Biol. 1998;8:112–118. doi: 10.1016/s0959-440x(98)80018-3. [DOI] [PubMed] [Google Scholar]
- 23.Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Annu Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 24.Veglia G, Zeri AC, MA C, Opella SJ. Deuterium/hydrogen exchange factors measured by solution nuclear magnetic resonance spectroscopy as indicators of the structure and topology of membrane proteins. Biophys J. 2002;82:2176–2183. doi: 10.1016/s0006-3495(02)75564-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez G, Anderson JS, LeMaster DM. Assessing the native state conformational distribution of ubiquitin by peptide acidity. Biophys Chem. 2010;153:70–82. doi: 10.1016/j.bpc.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soldi G, Plakoutsi G, Taddei N, Chiti F. Stabilization of a native protein mediated by ligand binding inhibits amyloid formation independently of the aggregation pathway. J Med Chem. 2006;49:6057–6064. doi: 10.1021/jm0606488. [DOI] [PubMed] [Google Scholar]
- 27.Fusco G, et al. 1H, 13C and 15N resonance assignments of human muscle acylphosphatase. Biomol NMR Assign. 2011 doi: 10.1007/s12104-011-9318-1. 10.1007/s12104-011-9318-1. [DOI] [PubMed] [Google Scholar]
- 28.Hwang TL, van Zijl PC, Mori S. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J Biomol NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 29.De Simone A, Richter B, Salvatella X, Vendruscolo M. Toward an accurate determination of free energy landscapes in solution states of proteins. J Am Chem Soc. 2009;131:3810–3811. doi: 10.1021/ja8087295. [DOI] [PubMed] [Google Scholar]
- 30.Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Gsponer J, et al. Determination of an ensemble of structures representing the intermediate state of the bacterial immunity protein Im7. Proc Natl Acad Sci USA. 2006;103:99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Best RB, Vendruscolo M. Determination of protein structures consistent with NMR order parameters. J Am Chem Soc. 2004;126:8090–8091. doi: 10.1021/ja0396955. [DOI] [PubMed] [Google Scholar]
- 33.Vendruscolo M, Paci E, Dobson CM, Karplus M. Rare fluctuations of native proteins sampled by equilibrium hydrogen exchange. J Am Chem Soc. 2003;125:15686–15687. doi: 10.1021/ja036523z. [DOI] [PubMed] [Google Scholar]
- 34.Schanda P, Forge V, Brutscher B. Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc Natl Acad Sci USA. 2007;104:11257–11262. doi: 10.1073/pnas.0702069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kern G, Handel T, Marqusee S. Characterization of a folding intermediate from HIV-1 ribonuclease H. Protein Sci. 1998;7:2164–2174. doi: 10.1002/pro.5560071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 37.Soldi G, Bemporad F, Chiti F. The degree of structural protection at the edge beta-strands determines the pathway of amyloid formation in globular proteins. J Am Chem Soc. 2008;130:4295–4302. doi: 10.1021/ja076628s. [DOI] [PubMed] [Google Scholar]
- 38.Mittermaier A, Kay L. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- 40.Gao YQ, Yang W, Karplus M. A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase. Cell. 2005;123:195–205. doi: 10.1016/j.cell.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Freiburger LA, et al. Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nat Struct Mol Biol. 2011;18:288–294. doi: 10.1038/nsmb.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 43.Monsellier E, Chiti F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 2007;8:737–742. doi: 10.1038/sj.embor.7401034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.