

Abstract
Androgen plays an important role in blood pressure regulation. Epidemiological studies have shown that men have a higher prevalence for developing hypertension than aged-matched, premenopausal women. Interestingly, postmenopausal women and women with polycystic ovary syndrome, both of which have increased endogenous androgen production, have elevated risks for hypertension suggesting that androgen may contribute to its development. Studies from our laboratory and others have provided substantial evidence that 20-hydroxyeicosatetraenoic acid (20-HETE) mediates the hypertension seen in rodents treated with androgen. 20-HETE is the cytochrome P450 (CYP)-derived ω-hydroxylated metabolite of arachidonic acid. 20-HETE plays a complex role in blood pressure regulation. In the kidney tubules, 20-HETE decreases blood pressure by promoting natriuresis, while in the microvasculature it has a pressor effect. In the microcirculation, 20-HETE participates in the regulation of vascular tone by sensitizing the smooth muscle cells to constrictor stimuli and contributes to myogenic, mitogenic and angiogenic responses. In addition, 20-HETE acts on the endothelium to promote endothelial dysfunction and endothelial activation. Recently, we have demonstrated that 20-HETE induces endothelial ACE thus setting forth a potential feed forward mechanism through activation of the Renin-Angiotensin-Aldosterone-System. In this review, we will discuss the pro-hypertensive effects of 20-HETE and its role in androgen-induced vascular dysfunction and hypertension.
Keywords: Androgen, DHT, Hypertension, 20-HETE, CYP4A/F, ACE, IKK, Endothelial dysfunction, Vascular reactivity
1. Introduction
Gender differences are observed in many aspects of mammalian cardiovascular physiology and pathology. In particular, hypertension is more common in men than in premenopausal women of the same age [1, 2]. Possible contributing factors for this dimorphism include environmental factors, genetic polymorphisms, and gonadal hormones [3, 4]. Many epidemiological, clinical, and experimental studies have shown that androgens may be an important determinant of sex-specific differences in arterial blood pressure [3, 5]. Reports have shown that men younger than 60 years of age have a higher systolic BP than women [2]. Furthermore, the ratio of androgen/estrogen has also been suggested to play a role in hypertensive effects in postmenopausal women [6]. Androgens have been hypothesized to modulate blood pressure both at the cardiac level as well as the vascular level [5, 7]. Sex-specific differences in blood pressure are also observed in experimental animals including, spontaneously hypertensive rats (SHR)[8-10], Dahl salt-sensitive rats[11], deoxycorticosterone (DOCA) acetate-salt hypertensive rats [12, 13], New Zealand genetically hypertensive rats [14], and cyp4a14 (-/-) knockout mice [15]; in all, males demonstrated higher blood pressure than females. Treatment of rats or mice with androgen increased blood pressure in both male and female rats [16]. The cellular mechanism underlying the androgen-induced increase in blood pressure and susceptibility to cardiovascular morbidity is not completely known. Recent studies from our laboratory and others provided substantial evidence that androgen-induced hypertension is 20-HETE-dependent and suggest that CYP4-derived 20-HETE is a key mediator of androgen-induced vascular resistance and hypertension. In this review, we will discuss the pro-hypertensive properties of 20-HETE, its role in androgen-mediated vascular dysfunction and hypertension, and its contribution to hypertension in human.
2. Androgen and hypertension
Clinical studies have shown that elevated levels of androgen, both endogenous and exogenous, are correlated with an increase in blood pressure.
2.1 Endogenous androgen and hypertension
Epidemiological, clinical, and experimental studies have shown implicated androgens as an important determinant of gender-specific differences in arterial blood pressure. Men younger than 60 years of age have a higher systolic blood pressure than premenopausal women of the same age [2]. In a Danish study of 352 healthy, normotensive subjects, 24-hour ambulatory blood pressure was monitored as blood pressure was measured every 15 minutes from 7:00 AM to 22:59 PM and every 30 minutes from 23:00 PM to 6:59 AM [17]. These investigators found that blood pressure increased with age in both men and women, but men had a significantly higher systolic blood pressure than women. This sexually dimorphic pattern was also seen in a larger study, the Third National Health and Nutrition Evaluation Survey (NHANES III). Data from a total of 6635 men and 7189 women over the age of 18 years old who were not under treatment for high blood pressure was collected from 1988 through 1994. Results indicated that the mean systolic blood pressure in men is higher than women in early adulthood across ethnicities (non-Hispanic Blacks, non-Hispanic White, and Mexican Americans) [18]. Interestingly, sex-specific differences in blood pressure are also observed in various animal models including SHR [8-10], Dahl salt-sensitive rats [11], DOCA-salt hypertensive rats [12, 13], New Zealand genetically hypertensive rats [14], and Cyp4a14 (-/-) knockout mice [15]. Within all models, males demonstrated higher BP than females. There are various contributing factors for this dimorphism including environmental factors, genetic polymorphisms, sex chromosome, and gonadal hormones [3, 4, 19]. Importantly, epidemiological, clinical, and experimental studies have shown that androgens may be an important determinant of sex-specific differences in arterial blood pressure [3, 5].
Along with gender differences, the correlation between androgen and blood pressure is also seen in postmenopausal women [6]. It is generally accepted that postmenopausal women have a higher risk in developing high blood pressure equal to that of or greater than men. Many factors may contribute to this elevated risk including the loss of protective effects of estrogen. In addition to loss of estrogen, androgen levels are often elevated in postmenopausal women suggesting a possible correlation with blood pressure elevation [20]. In a study by Phillips and colleagues [21], 24 hypertensive and 19 healthy postmenopausal women were screened for levels of total testosterone and free testosterone. Mean serum free testosterone levels and free-to-total testosterone ratios were increased in women with hypertension. This suggests that androgen may play a role in promoting a hypertensive risk factor in these patients. Recently there is increasing evidence suggesting that women with polycystic ovary syndrome (PCOS) have a greater risk at developing cardiovascular disease. According to the NHLBI sponsored Women's Ischemia Syndrome Evaluation which studied 390 postmenopausal women, women with PCOS have a significantly higher risk for developing hypertension as compared to women without PCOS (60.1% vs 44.2% respectively) [22]. Given that women with PCOS often present with hyperandrogenemia, it is possible that the increased androgen levels contribute in part to the hypertension. However, the link between hypertension and 20-HETE in PCOS may relate to obesity. Obesity is seen in a high percentage (30-70%) of PCOS patients [23]. Interestingly, several studies reported a direct relationship between 20-HETE and BMI [24, 25], suggesting that obesity may be a confounder in the potential link between 20-HETE and hypertension in PCOS patients.
2.2 Exogenous androgen and hypertension
In addition to elevated endogenous androgens, exogenous androgens may also promote blood pressure elevation. Achar et al., [26] carried out a meta-analysis investigating the role of anabolic-androgenic steroid (AAS) abuse on lipids, blood pressure, left ventricular dimensions and rhythms. This study showed a significant association between AAS use and increases in systolic as well as diastolic blood pressures. Furthermore, in a retrospective study by Urhausen et al.,[27] body builders using anabolic steroids had a significantly higher mean systolic blood pressure (140±10mmHg) in comparison to ex-users and non-users (130±5 mmHg and 125 ±10mmHg respectively). Other studies suggested that there is no significant correlation between AAS abuse and blood pressure elevation [26]. Variations in results may be due to the variety of AAS used, the reliability of ex-users reporting discontinuation of AAS use, as well as other supplements taken in addition to AAS. Clearly additional studies are necessary to define the correlation between AAS and blood pressure; however, this does suggest a potential role between exogenous androgen and blood pressure.
Testosterone supplementation and its relationship to blood pressure elevation is also observed in young men with congenital hypogonadism. Sonmez et al. [28] reported that patients with congenital hypogonadism have elevated baseline blood pressure as compared to control patients. Interestingly, testosterone supplementation further elevated blood pressure and this increase was also correlated with arterial stiffness. Bernini et al. [29], showed that hypogonadal patients have impaired vascular reactivity, including decrease in endothelial-dependent vasorelaxation due to reduced nitric oxide availability. Similar to the results in the blood pressure study by Sonmez, testosterone administration further impaired nitric oxide availability in these patients. Exogenous androgen also increases vascular stiffness in female-to-male transsexuals receiving androgen supplementation [30]. These studies support a pressor role for androgen in blood pressure regulation.
3. Androgen, 20-HETE, and hypertension
20-hydroxyeicosatetraenoic acid (20-HETE) is an arachidonic acid metabolite of the cytochrome P450 (CYP) system that plays a complex role in blood pressure modification. In renal tubules, 20-HETE is anti-hypertensive; it promotes natriuresis and diuresis by inhibiting tubular epithelial ion transport mechanisms. In contrast, within the vasculature, 20-HETE is pro-hypertensive; it increases vascular resistance by increasing vascular smooth muscle cell contraction and impairing endothelial-dependent vasorelaxation [31]. Recent studies have shown that vascular 20-HETE production is stimulated by androgen suggesting that androgen-dependent hypertension is driven, at least in part, by 20-HETE [15, 32-34].
3.1. 20-HETE synthesis in the vasculature
20-HETE is the ω-hydroxylated metabolite of arachidonic acid that is produced by CYP enzymes, mainly by members of the CYP4 family. The CYP4 gene family is comprised of thirty proteins in three subfamilies, CYP4A, CYP4B and CYP4F, which catalyze the hydroxylation of the terminal co-carbon and the (ω-1) carbon of saturated and unsaturated fatty acids [35]. CYP4 proteins share high sequence similarity and common catalytic properties, but differ in their tissue localization and hormonal regulation [35]. Arachidonic acid metabolism by CYP4A and CYP4F occurs in the liver, kidney, heart, lung, brain, and the vasculature [36, 37]. Both the CYP4A and CYP4F subfamilies produce 20-HETE in animals and humans, although to varying degrees [38, 39]. The major 20-HETE producing CYP4 isoforms are CYP4F2 and CYP4A11 in human [40, 41], CYP4A1, CYP4A2/3, CYP4A8, CYP4F1 and CYP4F4 in rats [42, 43] and Cyp4a12 in mice [15, 44]. The catalytic activity of the five murine Cyp4f isoforms (4f13, 4f14, 4f15, 4f16, and 4f18) with regard to 20-HETE synthesis is unknown [45]. Changes in the production of 20-HETE have been observed in numerous pathological conditions including ischemic cerebrovascular diseases, polycystic kidney diseases, hypertension, diabetes, toxemia of pregnancy and cancer [46]. Of importance are the numerous studies linking CYP4A to the development of hypertension and to the severity of ischemic cerebral and cardiac disease in experimental models [46, 47], and reports of correlation between urinary 20-HETE, oxidative stress and endothelial dysfunction in human subjects [48-50] along with association between CYP4F2 polymorphism and hypertension [51, 52] and ischemic stroke [53].
The synthesis of 20-HETE has been localized to occur at the vascular smooth muscle [54]. It can also be formed in human and canine neutrophils (upon the addition of arachidonic acid) and human myeloid cells in the peripheral blood and bone marrow [55, 56]. However, except for the pulmonary circulation [57], the ability of 20-HETE to be produced in the endothelium is still unknown. In the vasculature, CYP4A proteins show a distinct distribution along the vascular tree, where their expression increases with decreased vascular diameter [58]. Likewise, synthesis of 20-HETE increases as vascular diameter decreases. Levels of 20-HETE are not detected in large conduit vessels [58]. It is largely believed that 20-HETE is an eicosanoid of the microcirculation.
3.2. 20-HETE prohypertensive mechanisms
20-HETE participates in the regulation of vascular tone by sensitizing the smooth muscle cells to constrictor stimuli such as angiotensin II (Ang II), phenylephrine, and endothelin, and contributes to myogenic, mitogenic and angiogenic responses. Its release is stimulated by Ang II, endothelin, and serotonin and is also increased following treatment with NOS inhibitors [31]. The vasoconstrictor action of 20-HETE was first documented in 1988 by Escalante et al. [59], and was determined to be cyclooxygenase-dependent in rat aortic rings. Further studies demonstrated that in the microcirculation including the renal, cerebral, mesenteric and skeletal muscle arterioles, the constrictor activity of 20-HETE is largely cyclooxygenase-independent [46]. It can activate protein kinase C (PKC), mitogen activated protein kinase (MAPK), and src-type typrosine kinase, all of which phosphorylate and inhibit the conductance Ca2+-activated K+ channels, leading to depolarization and elevation in cytosolic [Ca2+], as well as opening and increasing Ca2+ entry through the L-type Ca2+ channels [31]. 20-HETE alone also increases the conductance of L-type Ca2+ channels through activation of PKC. Recent work by Inoue et al., [60] demonstrated that synergistic activation of vascular transient receptor potential canonical 6 (TRPC6) channel in the presence of 20-HETE enhances the conductance of inward nonselective cation and amplifies myogenic tone. In some blood vessels, 20-HETE acts through Rho-kinase to preserve phosphorylated myosin light chain 20 (MLC20) and to sensitize the contractile apparatus to Ca2+ [61]. Previous studies have shown that suppression and overexpression of CYP4A proteins in small arteries and arterioles decreases and increases vascular reactivity and myogenic tone, respectively [62-64]. These effects may contribute to the increase in blood pressure and the development of hypertension seen in experimental models where vascular 20-HETE synthesis is increased [32, 34, 65-67].
In addition to acting on the smooth muscle cells, 20-HETE can also act on the endothelium. The endothelium is important in the regulation of vascular tone, vessel diameter, and blood flow. It acts as the first layer of defense against injurious stimuli. The function of the endothelium is dependent upon many factors, one of which is nitric oxide (NO), which is generated from L-Arginine by endothelial nitric oxide synthase (eNOS) yielding L-citrulline as a byproduct. Loss of NO bioavailability leads to “endothelial dysfunction”, a term that refers to the diminished production or bioavailability of NO and/or an imbalance in relative contribution of endothelium-derived relaxation and contraction. Endothelial dysfunction is a feature of hypertension and an early risk factor for cardiovascular disease [68]. The 20-HETE affects on NO homeostasis was first suggested in a report by Frisbee et al. [69] showing that 20-HETE attenuated acetylcholine-induced relaxation of cremasteric arterioles. Other studies suggested a link between 20-HETE levels and endothelial dysfunction in hypertensive individuals [50], whereas in the pulmonary circulation 20-HETE increases relaxation by activating eNOS [70, 71]. Our recent studies provided substantial evidence for a causative link between the CYP4A-20-HETE pathway and endothelial dysfunction in vitro and in vivo. We showed that intravenous injection of an adenovirus to overexpress CYP4A2 in rats caused hypertension and that renal arteries from these rats featured increased vascular CYP4A expression and 20-HETE production, and displayed endothelial dysfunction exemplified by reduced vasodilator response to acetylcholine, reduced levels of NO, and increased levels of superoxide anion [72]. Similar results were obtained in a rat model of androgen-induced hypertension in which the vascular expression of CYP4A8 and production of 20-HETE were upregulated [32, 33]. In both models, inhibition of 20-HETE synthesis abrogated androgen-induced vascular dysfunction and hypertension. Moreover, 20-HETE inhibition of acetylcholine-induced relaxation was limited to the NO component of the relaxation to acetylcholine suggesting that 20-HETE interferes with NO synthesis and/or bioavailability [72]. These initial studies prompted a thorough examination of the relationship between 20-HETE and the eNOS-NO pathway in the vascular endothelium. In cultured endothelial cells, 20-HETE causes eNOS uncoupling by inhibiting the association of HSP90 with eNOS, leading to reduced NO production and bioavailability [73]. This was further validated in isolated renal arteries using the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at serine 239 which has been shown to be an indicator of bioactive NO and the activity of the NO/cGMP/PKG signaling pathway [73]. Additional studies indicated that 20-HETE-mediated eNOS uncoupling and endothelial dysfunction is EGFR-, MAPK- and IκB Kinase (IKK)-dependent [74]. Interaction between 20-HETE and eNOS were also studied in endothelial cells of other vascular beds. Ward et al. [75] further showed that in human umbilical vein endothelial cells chronic activation of AMPK inhibited 20-HETE-mediated dissociation of eNOS from HSP90. In contrast, within pulmonary endothelial cells, 20-HETE activates eNOS by increasing the phosphorylation of eNOS at serine 1179 and AKT at serine 473 leading to increase NO production [71]. It should be noted that unlike the renal, cerebral and mesenteric arteries, pulmonary arteries relax in response to 20-HETE [46]. In all, our studies and studies in several animal models [32, 67, 72, 76] implicate 20-HETE as an important determinant of endothelial dysfunction in the microcirculation and this adds to the mechanisms underlying the pro-hypertensive effect of 20-HETE.
Vascular wall inflammation plays a key role in the pathogenesis of various diseases including atherosclerosis, cardiovascular disease, and hypertension. Blood pressure is determined by cardiac output and total peripheral resistance and an increase in either of these two factors can lead to hypertension. Hypertension is known to be associated with an increase in the wall/lumen ratio (W/L) of resistance arteries, and the greatest vascular resistance occurs in the small arteries and arterioles [77, 78]; hence, decrease in the lumen of the small arteries significantly increases resistance. Decrease in lumen size can be attributed to mechanical and functional changes as well as structural changes in the blood vessels [78]. Recent studies provided evidence to suggest that inflammation-mediated vascular remodeling leads to an increase in vascular resistance, thus implicating inflammation in the pathophysiology of hypertension [79]. Inflammation contributes to the dysfunction of the vascular endothelium that is characteristically observed in hypertension. Pro-inflammatory changes in endothelial phenotype (i.e., endothelial activation) lead to an increase in cellular adhesion molecules, endothelial-leukocyte interaction and permeability, as well as alterations in the secretion of autocrine and paracrine factors that are important for an inflammatory response. Up-regulation of selectins, vascular cell adhesion molecule-1 (VCAM-1), and intracellular adhesion molecule-1 (ICAM-1) promotes the adherence of monocytes [80, 81]. Furthermore, continued release of cytokines and chemokines such as Monocyte Chemoattractant Protein-1 (MCP-1) by activated endothelial cells further increases migration/adhesion of monocytes via interactions with the CCR2 receptor [82, 83]. Suppression or deletion of these inflammatory molecules ameliorates inflammation and the associated vascular dysfunction and activation in the model of AngII-induced hypertension [84, 85]. These studies and recent reports demonstrating a role for immune cells in blood pressure regulation advanced the notion that inflammation may be a key factor in the pathogenesis of hypertension [86].
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a key transcriptional activator of the inflammatory gene program [87, 88]. Recently, there is increasing evidence that NF-κB activation and reactive oxygen species (ROS) play a key role in endothelial activation and the associated inflammation in cardiovascular diseases including atherosclerosis, hypertension, stroke and with aging [81, 87, 89-94]. NF-κB activation induces the transcription of a large number of genes related to vascular inflammation. Inhibition of NF-κB attenuates endothelial activation in aged rats, ameliorates atherosclerosis, reduces Ang-II-induced hypertension and prevents Ang II-induced end-organ damage [79, 95, 96]. There are many activators of NF-κB, and many of them act via ROS-dependent mechanisms [81]. Increase in ROS leads to activation of the IKK complex that further leads to the phosphorylation and degradation of IκB, thus unmasking NF-κB and contributing to endothelial activation [81, 87]. Previous work from our laboratory demonstrated that an increase in the levels of vascular 20-HETE is associated with increased expression of the NADPH oxidase system and increased vascular superoxide anion production in adenovirus CYP4A2- and DHT-induced hypertensive rats [32, 72]. Furthermore, in vitro studies showed that treatment of endothelial cells with 20-HETE lead to increased ROS and NF-κB activity resulting in endothelial activation with increased expression of ICAM and IL-8 levels [97]. These findings suggest that 20-HETE plays a role in endothelial activation, thus contributing to hypertension.
The inhibitor of kappa-B kinase (IKK) complex plays an important role in 20-HETE mediated signaling. As mentioned above 20-HETE causes endothelial dysfunction in an IKK-dependent manner [74]. Interestingly, 20-HETE also causes NF-κB activation in both IKK dependent and independent manner [74]. Hence, IKK seems to be a key downstream second messenger for both 20-HETE-induced endothelial dysfunction and activation that characterize the vascular phenotype in hypertension models. In a recent study, we showed that in the androgen-induced hypertensive model, in which blood pressure elevation, increased smooth muscle reactivity, endothelial dysfunction and endothelial activation is 20-HETE dependent, and administration of parthenolide (an IKK inhibitor) attenuated DHT-mediated endothelial dysfunction and hypertension [33]. Interestingly, inhibition of IKK did not alter smooth muscle reactivity suggesting that DHT-mediated (20-HETE-dependent) endothelial dysfunction is IKK-dependent whereas the increase in smooth muscle reactivity is IKK-independent. To further confirm the effect of IKK inhibition on 20-HETE-mediated vascular effects, acetylcholine-induced relaxation and vascular reactivity to phenylephrine were measured in renal interlobar arteries from normotensive SD rats treated with or without parthenolide (5 μg/ml) in the presence and absence of 20-HETE (1 μM). As seen in Figure 1A, parthenolide did not affect 20-HETE-mediated increase in smooth muscle reactivity; however, it did negate 20-HETE-mediated inhibition of acetylcholine-induced relaxation (IKK-dependent mechanism) (Figure 1B). These results suggest that the mechanisms by which 20-HETE affect endothelial function differ from those activated by 20-HETE in the smooth muscle. In the endothelium 20-HETE causes endothelial dysfunction and activation via IKK-dependent pathway, whereas 20-HETE actions in the smooth muscle are not mediated through IKK activation.
Figure 1. IKK inhibition with parthenolide abrogates 20-HETE-mediated inhibition of acetylcholine-induced relaxations but does not affect 20-HETE-mediated increase in vascular reactivity to phenylephrine.

Rat renal interlobar arteries (∼150 μm) were microdissected and placed in a wire myograph. (A) Vascular reactivity to phenylephrine and (B) relaxation to acetylcholine were measured in the presence and absence of parthenolide (5 μg/ml) and 20-HETE (1 μM) as described [33]. Results were analyzed by ANOVA and are mean±SE (n=4); *p<0.05 vs control; #p<0.05 vs 20-HETE alone.
In addition to increasing vascular reactivity and myogenic tone, 20-HETE has been implicated in the generation of superoxide and other ROS [98], thus promoting oxidative stress. Western blot analysis of renal interlobar arteries from androgen-treated rats revealed a 3- and 2-fold increase in levels of the NADPH oxidase subunits, p47phox and gp91phox, respectively. This was accompanied with an increase in vascular superoxide levels [32]. An increase in gp91phox expression and superoxide levels was also observed in arteries from rats transduced with adenovirus or lentivirus carrying the CYP4A2 cDNA [67, 72]. This finding is of clinical significance as hypertensive patients have been shown to exhibit oxidative stress (measured as increase in F2-isoprostane levels) in association with an increase in urinary 20-HETE excretion [49]. In vitro, 20-HETE has been shown to stimulate superoxide production in pulmonary artery endothelial cells and in human dermal microvascular cells [73, 99, 100]. These results suggests that 20-HETE promotes oxidative stress.
The Renin-Angiotensin-Aldosterone system is a key regulator of blood pressure and fluid homeostasis. Components of the renin-angiotensin system (RAS) includes renin, angiotensin-converting enzyme (ACE), and angiotensin type 1 receptor (AT1R), which are generally expressed throughout the body in tissues that impact blood pressure modulation. Ang II, the product of sequential degradation of angiotensinogen by renin and ACE and the final effector of the RAS, increases blood pressure by mechanisms that include (i) vasoconstriction via AT1R activation in the vasculature as well as increasing sympathetic tone and the release of arginine vasopressin, (ii) modulation of renal sodium and water reabsorption by stimulating renal AT1R, the production and release of aldosterone from the adrenal glands, or the sensation of thirst in the central nervous system. Blocking the synthesis or actions of Ang II lowers BP in patients with hypertension. Mice null for angiotensinogen, renin, ACE and AT1A (the closest murine homologue to the single human AT1R gene) exhibit marked reduction in BP, indicating the role of RAS in normal BP homeostasis [101, 102]. Given the characteristics of the RAS and the CYP4A-20-HETE pathways, interactions between these two systems are highly conceivable. Ang II has been shown to stimulate the release of 20-HETE in isolated preglomerular vessels [103] and the renal synthesis of 20-HETE [104]. Increased production of 20-HETE in the peripheral vasculature contributes to the acute vasoconstrictor response to Ang II [105], acute inhibition of 20-HETE synthesis attenuates the renal pressor response to Ang II [104], and chronic inhibition of 20-HETE biosynthesis attenuates the development of Ang II-dependent hypertension [106]. In cultured aortic vascular smooth muscle cells, 20-HETE mediates Ang II- induced mitogenic effects and contributes to the vascular injury, hypertrophy and hypertension caused by Ang II in rats [107-109]. These studies suggest that 20-HETE may contribute to the hypertensive actions of Ang II. To this end, experimental models of hypertension that show increased vascular 20-HETE production, such as the SHR [65, 76] and the androgen-induced hypertension in rats and mice [15, 32, 34, 44] are also RAS mediated. Studies in our lab suggested that the interaction between 20-HETE and the RAS also includes induction of vascular ACE. In endothelial cells, 20-HETE is a potent inducer of ACE expression. Increased expression and synthesis of 20-HETE as in the CYP4A2-transduced rats [110] or the androgen-treated rats (Figure 2), is associated with increased expression of renal and vascular ACE which is abolished by treatment with 20-HETE synthesis inhibitors. In the model of 20-HETE-dependent hypertension (rats overexpressing the CYP4A2 in vascular endothelium), blood pressure is normalized by ACE inhibition or AT1R blockade [110]. This suggests the presence of a feed forward amplification of 20-HETE-induced vascular dysfunction by the RAAS. Thus, ACE induction by 20-HETE brings about increases in Ang II levels and its actions through the AT1R in vascular smooth muscle and endothelial cells. Such interactions may constitute at least in part the mechanism by which 20-HETE causes hypertension.
Figure 2. Androgen increases ACE protein levels in a 20-HETE-dependent manner.
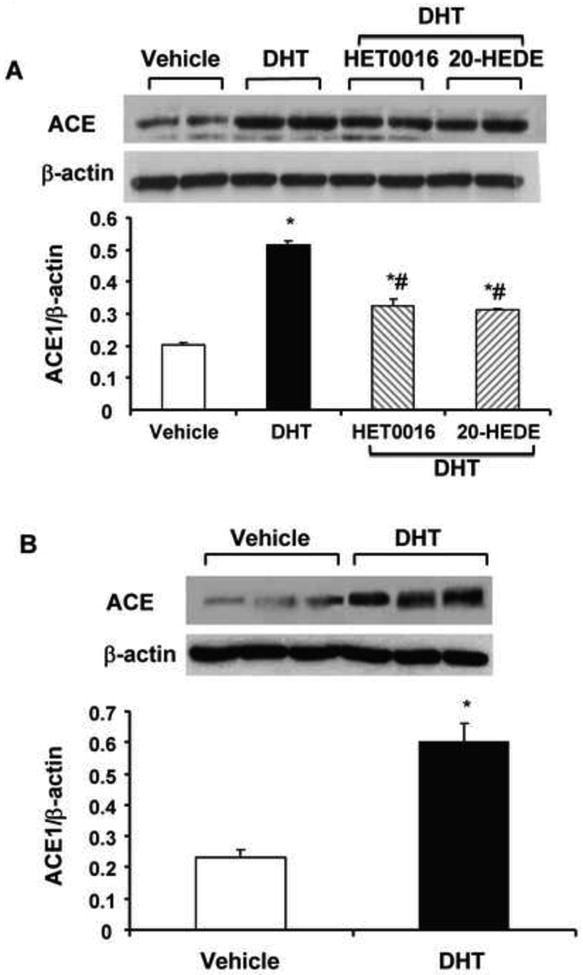
Rats were treated with DHT (56 mg/kg/day, IP in benzyl alcohol with corn oil) or its vehicle with and without HET0016 (10mg/kg/day) or 20-HEDE (10 mg/kg/day) for 14 days [33]. (A) Renal cortical tissue and (B) microdissected renal interlobar arteries were processed for Western blot analysis of ACE as described [110]. Results were analyzed by ANOVA are mean±SE (n=4); *p<0.05 vs Vehicle; #p<0.05 vs DHT alone.
3.3. Androgen-induced hypertension and 20-HETE
The report by Sacerdoti et al. [111] showing that depletion of CYP normalizes blood pressure in the SHR was the first to implicate 20-HETE in the pathogenesis of hypertension. Consequently, numerous studies in animal models and human provided ample evidence of a role for 20-HETE in the regulation of blood pressure [reviewed in [31]]. The possible role of 20-HETE in androgen-induced hypertension was first introduced by Capdevila and colleagues who undertook a genetic approach to substantiate a causal relationship between CYP4, 20-HETE and blood pressure [15, 34]. These investigators found that targeted disruption of the Cyp4a14 gene (the mouse homologue of the rat CYP4A2) resulted in spontaneous hypertension that was androgen-dependent [15]. The Cyp4a14 null mice also displayed increased plasma androgens, elevated expression of Cyp4a12, and increased urinary excretion of 20-HETE. A subsequent vascular function study showed that in Krebs-perfused isolated kidneys, perfusion pressure to phenylephrine was elevated in Cyp4a14 null mice and this effect was blunted using 12,12-dibromododec-11-enoic acid (DBDD) (a 20-HETE synthesis inhibitor) [112]. These results suggest that blood pressure elevation in Cyp4a14 (-/-) mice is mediated by increased vascular 20-HETE production through elevated expression of Cyp4a12 driven by androgen [15].
The correlation between androgen and Cyp4a12 expression is also seen in wild type mice. Muller et al [44] compared the level of Cyp4a12a expression in male and female wild type mice from NMRI, FVB/N, 129 Sv/J, Balb/C, and C57BL/6 background. In general, levels of Cyp4a12 were higher in male compared to female (45-fold for NMRI, 88-fold for FVB/N, 40-fold for 129 Sv/J, 7-fold for Balb/c and 48-fold for C57BL/6). These results correlated with 20-HETE production in mouse renal microsomes in which male had higher levels than female mice. Along with gender differences, exogenous implantation of a pellet containing 5α-dihydrotestosterone (DHT, 5 mg/day pellet) led to further increase in Cyp4a12 expression as well as microsomal 20-HETE production. In addition to the mice model, exogenous androgen also increases 20-HETE production and blood pressure in rats. Sprague Dawley (SD) rats, both male and female, treated with exogenous DHT resulted in elevated blood pressure as well as 20-HETE production in renal arteries [16, 32, 34]. RNA analysis from whole kidney as well as renal interlobar arteries showed that DHT elevated expression of CYP4A8 (the rat homologue to the mice Cyp4a12) [32]. Recent studies from our laboratory [33] have confirmed these findings and showed that upon androgen treatment, vascular 20-HETE production significantly increases at day 2 of treatment. This elevation seemed to precede that of androgen-induced blood pressure elevation, which significantly increases at day 3. Furthermore, inhibition of 20-HETE synthesis with HET0016 or its action with a synthetic 20-HETE antagonist (20-HEDE) prevented and reversed androgen-mediated blood pressure elevation, led to increase in smooth muscle reactivity to phenylephrine, and endothelial dysfunction suggesting that androgen-dependent hypertension is 20-HETE mediated. Given that androgen can mediate its signaling through androgen receptor dependent and independent mechanisms, we treated SD rats with vehicle, DHT (56 mg/kg/day, IP in benzyl alcohol with corn oil), DHT with Flutamide (an androgen receptor blocker), or Flutamide (50 mg/kg/day, IP in benzyl alcohol with corn oil). Co-treatment with Flutamide prevented DHT-mediated blood pressure elevation as well as vascular 20-HETE suggesting that androgen-mediated elevation in blood pressure and vascular 20-HETE is androgen receptor-dependent (Figure 3A and B).
Figure 3. Androgen receptor blockade with flutamide inhibits androgen-induced hypertension and 20-HETE synthesis.

Rats were treated with DHT (56 mg/kg/day, IP in benzyl alcohol with corn oil) or its vehicle with and without Flutamide (50 mg/kg/day, IP in benzyl alcohol with corn oil) for 3 days. (A) Systolic blood pressure and (B) 20-HETE levels in the renal interlobar arteries were measured as described [33]. Results were analyzed by ANOVA are mean±SE (n=4); *p<0.05 vs vehicle control; #p<0.05 vs DHT alone.
4. Summary and perspective
4.1. Clinical relevance
To date, several polymorphisms have been identified in the CYP4 family [113]. Some of the polymorphisms lead to gain of function while others lead to loss of function. Polymorphisms in CYP4A11 [41, 114, 115] and CYP4F2 [51 -53, 116-118] have been associated with cardiovascular diseases including hypertension, myocardial infarction, ischemic stroke and cerebral infarction However, at which extent do these polymorphisms contribute to hypertension is still unclear. The CYP4A11 gene is a highly polymorphic gene out of which the most important genetic variant is the T8590C variant that results in a significant reduction in the catalytic activity of CYP4A11 [41]; however, its role in blood pressure modulation is yet to be elucidated due to conflicting results from different cohort studies. In the Tennessee cohort, the 8590C allele was associated with an increased risk of hypertension in Caucasians; however, it had no effect in African Americans [41, 115]. In a following study screening 732 African Americans with hypertensive renal disease, men with the 8590CC genotype was associated with higher systolic blood pressures and pulse pressures [114]. Other studies have also shown that carriers of the C allele had higher blood pressures and prevalence of hypertension [119, 120]. On the other hand, Ward et al [52] studied a cohort consisted of 235 (129 men and 106 women) predominately white individuals (97%) and suggested that while the CYP4A11 T8590C variant is associated with reduction in urinary 20-HETE, it does not associated with hypertension. Given that 20-HETE has opposing effects in the tubules versus vasculature, the distribution of CYP4A11 expression within different ethnic groups may play a role in this contradicting data.
Recent studies have shown that polymorphisms in CYP4F2, both in the regulatory region as well as in the coding region, are associated with hypertension. An initial study by Liu and collaborators [51] showed that G421C (c.-48G→C, rs3093100) polymorphism in the regulatory region of CYP4F2 gene is correlated with essential hypertension. Further investigation showed six additional variants in the regulatory region of CYP4F2: c.-91T→C (rs3093098), c.-77T→C (rs3093099), c.-43C→T (rs3093101), c.-23G→A (rs3093102), c.-13T→C (rs3093103), and c.+34T→G (Trp12Gly, rs3093105), within intron 1 and exon 2. These polymorphisms were commonly seen in two haplotypes, Hap I (c.-91T/c.-48G/c.-13T/c.+34T) and Hap II (c-91C/c.-48C/c.-13C/c.+34G), in which Hap I increased transcriptional activity of CYP4F2 via an NF-κB responsive element at c.-91T resulting in elevated urinary 20-HETE level. This increase in urinary 20-HETE was associated with hypertension in both the case-control and family based studies. Interestingly, levels of urinary 20-HETE were slightly higher in men than women. In addition to regulatory domain, Stec et al. [121] further identified two nonsynonymous single nucleotide polymorphism that lead to amino acid changes at position 12 (W12G) and 433 (V433M) in African and European Americans (n=24 and n=23 respectively). The M433 amino acid change was associated with a decrease in 20-HETE production (56-66% that of control). Since then numerous studies have shown association between V433M polymorphism and hypertension. The valine-to-methionine amino acid substation at 433 can occur due to SNP resulting in a guanine-to-adenine missense transition at nucleotide 1347. Ward and colleagues [52] performed a cohort study on 235 individuals and found that in the CYP4F2 coding region, 50% were homozygous for the G allele (GG), 40% were heterozygous with a single A transition (GA), and 9% were homozygous for the A allele (AA). Individuals with either GA or AA had significantly higher blood pressure than those with the GG alleles. Furthermore, patients with GA or AA alleles also had the highest levels of urinary 20-HETE. Similar findings were confirmed by Fava and colleagues in a cohort of the Malmö Diet and Cancer Study [53]. In their study, they found that the V433M polymorphism in CYP4F2 is associated with elevated BP level and hypertension prevalence in the male urban population-based sample of middle-aged Swedes. Recent studies by Hu et al. [122] showed that patients with either M433M or V433M polymorphism had elevated urinary 20-HETE concentration than V433V homozygotes. Furthermore, M433 allele carriers had a significantly greater peripheral augmentation index in men versus V433V homozygotes suggesting that 20-HETE may play a role in modulating arterial stiffness and contributing to hypertension.
4.2. Conclusions
20-HETE has been identified as a primary microcirculatory eicosanoid whose biological activities are conducive of a hypertensive factor. Numerous studies have documented a direct relationship between its vascular synthesis and blood pressure in animal models of hypertension and recent clinical studies suggested its involvement in human hypertension [49, 52]. Androgen has been implicated as a contributing factor to gender-specific differences in blood pressure and cardiovascular morbidity, postmenopausal hypertension and to the pathogenesis of polycystic ovary diseases [123]. In a series of studies, we and others have demonstrated that 20-HETE is a key mediator of the hypertension that follows administration of exogenous androgen such as DHT in rats and mice. The potential mechanisms by which 20-HETE mediates the actions of androgen in the vasculature and in the cardiovascular system are yet to be fully explored and have not been fully elucidated. Recent studies from our lab set forth a novel paradigm (Figure 4) where excessive production of 20-HETE within the vasculature triggered by increased androgen levels leads to hypertension via mechanisms that include smooth muscle contraction, endothelial dysfunction (eNOS uncoupling), endothelial activation (IKK-NF-kB stimulation, oxidative stress) and the induction of endothelial ACE, that further perpetuates an increase in vascular Ang II which, in turn and together with 20-HETE, promotes vascular dysfunction, increased vascular resistance and hypertension. A possible relationship between androgen, 20-HETE and menopausal hypertension was recently documented in rat model of menopause [124]. Such studies along with evidence for gender-specific and androgen-driven expression of CYP4-20-HETE synthases in animal models and in human as well as clinical association of polymorphism in these CYPs with hypertension suggest that 20-HETE and its downstream signaling molecules in the vasculature may be potential targets for treating androgen-related hypertension and cardiovascular disease.
Figure 4. Postulated vascular mechanisms underlying the pro-hypertensive actions of 20-HETE.

In most vascular beds, the synthesis of 20-HETE is primarily localized to the vascular smooth muscle where it is subjected to regulation by various autacoids [125] as well as androgen. In the vascular smooth muscle, 20-HETE elicits vasoconstriction largely via inhibition of the SMC large-conductance Ca2+-activated K+ channel, leading to depolarization and elevation in cytosolic [Ca2+] [54], and in some blood vessels via a Rho-kinase phosphorylation of MLC20 and the sensitization of the contractile apparatus to Ca2+ [61]. 20-HETE acts on the endothelium in a paracrine manner through a yet to be identified “receptor”; it stimulates IKK activation through EGFR and MAPK (ERK1/2). The activation of IKK is a key signaling mechanism of 20-HETE actions in the vascular endothelium as it leads to eNOS uncoupling, NF-kB activation and ACE induction [73, 74]. The induction of ACE by 20-HETE brings about increases in Ang II levels and its actions through the AT1R in vascular smooth muscle and endothelial cells, thus setting up a feed forward amplification of 20-HETE-induced vascular dysfunction by the RAS. Vasoconstriction, endothelial dysfunction and endothelial activation are all contributing to hypertension.
Highlights.
This review discusses the role of 20-HETE in androgen-induced hypertension.
20-HETE is the cytochrome P450-derived ω-hydroxylated metabolite of arachidonic acid.
20-HETE promotes vasoconstriction and endothelial dysfunction.
20-HETE activates the RAS via induction of endothelial ACE.
CYP4F2 polymorphism is associated with increased urinary 20-HETE and hypertension.
Acknowledgments
This study was supported by NIH grants HL034300 (MLS) and F30 HL097402 (CCW). We would like to thank Victor Garcia for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension. 2001;37:1199–208. doi: 10.1161/01.hyp.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 2.Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988-1991. Hypertension. 1995;25:305–13. doi: 10.1161/01.hyp.25.3.305. [DOI] [PubMed] [Google Scholar]
- 3.Coylewright M, Reckelhoff JF, Ouyang P. Menopause and hypertension: an age-old debate. Hypertension. 2008;51:952–9. doi: 10.1161/HYPERTENSIONAHA.107.105742. [DOI] [PubMed] [Google Scholar]
- 4.Ruixing Y, Jinzhen W, Shangling P, Weixiong L, Dezhai Y, Yuming C. Sex differences in environmental and genetic factors for hypertension. The American journal of medicine. 2008;121:811–9. doi: 10.1016/j.amjmed.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 5.Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocrine reviews. 2003;24:313–40. doi: 10.1210/er.2003-0005. [DOI] [PubMed] [Google Scholar]
- 6.Reckelhoff JF, Fortepiani LA. Novel mechanisms responsible for postmenopausal hypertension. Hypertension. 2004;43:918–23. doi: 10.1161/01.HYP.0000124670.03674.15. [DOI] [PubMed] [Google Scholar]
- 7.Kienitz T, Quinkler M. Testosterone and blood pressure regulation. Kidney & blood pressure research. 2008;31:71–9. doi: 10.1159/000119417. [DOI] [PubMed] [Google Scholar]
- 8.Chen YF, Meng QC. Sexual dimorphism of blood pressure in spontaneously hypertensive rats is androgen dependent. Life sciences. 1991;48:85–96. doi: 10.1016/0024-3205(91)90428-e. [DOI] [PubMed] [Google Scholar]
- 9.Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension. 2000;35:480–3. doi: 10.1161/01.hyp.35.1.480. [DOI] [PubMed] [Google Scholar]
- 10.Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. 1998;31:435–9. doi: 10.1161/01.hyp.31.1.435. [DOI] [PubMed] [Google Scholar]
- 11.Crofton JT, Ota M, Share L. Role of vasopressin, the renin-angiotensin system and sex in Dahl salt-sensitive hypertension. Journal of hypertension. 1993;11:1031–8. doi: 10.1097/00004872-199310000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Crofton JT, Share L. Gonadal hormones modulate deoxycorticosterone-salt hypertension in male and female rats. Hypertension. 1997;29:494–9. doi: 10.1161/01.hyp.29.1.494. [DOI] [PubMed] [Google Scholar]
- 13.Ouchi Y, Share L, Crofton JT, Iitake K, Brooks DP. Sex difference in the development of deoxycorticosterone-salt hypertension in the rat. Hypertension. 1987;9:172–7. doi: 10.1161/01.hyp.9.2.172. [DOI] [PubMed] [Google Scholar]
- 14.Ashton N, Balment RJ. Sexual dimorphism in renal function and hormonal status of New Zealand genetically hypertensive rats. Acta endocrinologica. 1991;124:91–7. doi: 10.1530/acta.0.1240091. [DOI] [PubMed] [Google Scholar]
- 15.Holla VR, Adas F, Imig JD, et al. Alterations in the regulation of androgen-sensitive Cyp 4a monooxygenases cause hypertension. Proc Natl Acad Sci U S A. 2001;98:5211–6. doi: 10.1073/pnas.081627898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh H, Schwartzman ML. Renal vascular cytochrome P450-derived eicosanoids in androgen-induced hypertension. Pharmacol Rep. 2008;60:29–37. [PubMed] [Google Scholar]
- 17.Wiinberg N, Hoegholm A, Christensen HR, et al. 24-h ambulatory blood pressure in 352 normal Danish subjects, related to age and gender. Am J Hypertens. 1995;8:978–86. doi: 10.1016/0895-7061(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 18.Martins D, Nelson K, Pan D, Tareen N, Norris K. The effect of gender on age-related blood pressure changes and the prevalence of isolated systolic hypertension among older adults: data from NHANES III. J Gend Specif Med. 2001;4:10–3. 20. [PubMed] [Google Scholar]
- 19.Ji H, Zheng W, Wu X, et al. Sex Chromosome Effects Unmasked in Angiotensin II-Induced Hypertension. Hypertension. doi: 10.1161/HYPERTENSIONAHA.109.144949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanes LL, Reckelhoff JF. Postmenopausal Hypertension. Am J Hypertens. 2011 doi: 10.1038/ajh.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips GB, Jing TY, Laragh JH. Serum sex hormone levels in postmenopausal women with hypertension. Journal of human hypertension. 1997;11:523–6. doi: 10.1038/sj.jhh.1000481. [DOI] [PubMed] [Google Scholar]
- 22.Shaw LJ, Bairey Merz CN, Azziz R, et al. Postmenopausal women with a history of irregular menses and elevated androgen measurements at high risk for worsening cardiovascular event-free survival: results from the National Institutes of Health--National Heart, Lung, and Blood Institute sponsored Women's Ischemia Syndrome Evaluation. J Clin Endocrinol Metab. 2008;93:1276–84. doi: 10.1210/jc.2007-0425. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Vrbikova J, Hainer V. Obesity and polycystic ovary syndrome. Obes Facts. 2009;2:26–35. doi: 10.1159/000194971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laffer CL, Laniado-Schwartzman M, Nasjletti A, Elijovich F. 20-HETE and circulating insulin in essential hypertension with obesity. Hypertension. 2004;43:388–92. doi: 10.1161/01.HYP.0000112224.87290.3a. [DOI] [PubMed] [Google Scholar]
- 25.Ward NC, Hodgson JM, Puddey IB, Beilin LJ, Croft KD. 20-Hydroxyeicosatetraenoic acid is not associated with circulating insulin in lean to overweight humans. Diabetes Res Clin Pract. 2006;74:197–200. doi: 10.1016/j.diabres.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Achar S, Rostamian A, Narayan SM. Cardiac and metabolic effects of anabolic-androgenic steroid abuse on lipids, blood pressure, left ventricular dimensions, and rhythm. Am J Cardiol. 2010;106:893–901. doi: 10.1016/j.amjcard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urhausen A, Albers T, Kindermann W. Are the cardiac effects of anabolic steroid abuse in strength athletes reversible? Heart. 2004;90:496–501. doi: 10.1136/hrt.2003.015719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonmez A, Haymana C, Bolu E, et al. Metabolic Syndrome and The Effect of Testosterone Treatment In Young Men With Congenital Hypogonadotrophic Hypogonadism. Eur J Endocrinol. 2011 doi: 10.1530/EJE-10-0951. [DOI] [PubMed] [Google Scholar]
- 29.Bernini G, Versari D, Moretti A, et al. Vascular reactivity in congenital hypogonadal men before and after testosterone replacement therapy. J Clin Endocrinol Metab. 2006;91:1691–7. doi: 10.1210/jc.2005-1398. [DOI] [PubMed] [Google Scholar]
- 30.Emi Y, Adachi M, Sasaki A, Nakamura Y, Nakatsuka M. Increased arterial stiffness in female-to-male transsexuals treated with androgen. J Obstet Gynaecol Res. 2008;34:890–7. doi: 10.1111/j.1447-0756.2008.00857.x. [DOI] [PubMed] [Google Scholar]
- 31.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. Journal of cardiovascular pharmacology. 2010;56:336–44. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh H, Cheng J, Deng H, et al. Vascular Cytochrome P450 4A Expression and 20-Hydroxyeicosatetraenoic Acid Synthesis Contribute to Endothelial Dysfunction in Androgen-Induced Hypertension. Hypertension. 2007;50:123–9. doi: 10.1161/HYPERTENSIONAHA.107.089599. [DOI] [PubMed] [Google Scholar]
- 33.Wu CC, Cheng J, Zhang FF, et al. Androgen-Dependent Hypertension Is Mediated by 20-Hydroxy-5,8,11,14-Eicosatetraenoic Acid-Induced Vascular Dysfunction: Role of Inhibitor of {kappa}B Kinase. Hypertension. 2011;57:788–94. doi: 10.1161/HYPERTENSIONAHA.110.161570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa K, Marji JS, Schwartzman ML, Waterman MR, Capdevila JH. Androgen-mediated induction of the kidney arachidonate hydroxylases is associated with the development of hypertension. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1055–R62. doi: 10.1152/ajpregu.00459.2002. [DOI] [PubMed] [Google Scholar]
- 35.Hsu MH, Savas U, Griffin KJ, Johnson EF. Human cytochrome p450 family 4 enzymes: function, genetic variation and regulation. Drug Metab Rev. 2007;39:515–38. doi: 10.1080/03602530701468573. [DOI] [PubMed] [Google Scholar]
- 36.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. FASEB J. 1992;6:731–6. doi: 10.1096/fasebj.6.2.1537463. [DOI] [PubMed] [Google Scholar]
- 37.Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol. 2005;45:413–38. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- 38.Hardwick JP. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochemical pharmacology. 2008;75:2263–75. doi: 10.1016/j.bcp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Kalsotra A, Strobel HW. Cytochrome P450 4F subfamily: at the crossroads of eicosanoid and drug metabolism. Pharmacology & therapeutics. 2006;112:589–611. doi: 10.1016/j.pharmthera.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Lasker JM, Chen WB, Wolf I, Bloswick BP, Wilson PD, Powell PK. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J Biol Chem. 2000;275:4118–26. doi: 10.1074/jbc.275.6.4118. [DOI] [PubMed] [Google Scholar]
- 41.Gainer JV, Bellamine A, Dawson EP, et al. Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation. 2005;111:63–9. doi: 10.1161/01.CIR.0000151309.82473.59. [DOI] [PubMed] [Google Scholar]
- 42.Xu F, Falck JR, Ortiz de Montellano PR, Kroetz DL. Catalytic activity and isoform-specific inhibition of rat cytochrome p450 4F enzymes. J Pharmacol Exp Ther. 2004;308:887–95. doi: 10.1124/jpet.103.059626. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol. 1999;276:R1691–R700. doi: 10.1152/ajpregu.1999.276.6.R1691. [DOI] [PubMed] [Google Scholar]
- 44.Muller DN, Schmidt C, Barbosa-Sicard E, et al. Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. The Biochemical journal. 2007;403:109–18. doi: 10.1042/BJ20061328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stec DE, Flasch A, Roman RJ, White JA. Distribution of cytochrome P-450 4A and 4F isoforms along the nephron in mice. Am J Physiol Renal Physiol. 2003;284:F95–102. doi: 10.1152/ajprenal.00132.2002. [DOI] [PubMed] [Google Scholar]
- 46.Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res. 2005;41:175–93. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- 47.Roman RJ. P-450 metabolites of arachidonic Acid in the control of cardiovascular function. Physiological reviews. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 48.Barden A, Zilkens RR, Croft K, et al. A reduction in alcohol consumption is associated with reduced plasma F(2)-isoprostanes and urinary 20-HETE excretion in men. Free Radic Biol Med. 2007;42:1730–5. doi: 10.1016/j.freeradbiomed.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Ward NC, Puddey IB, Hodgson JM, Beilin LJ, Croft KD. Urinary 20-hydroxyeicosatetraenoic acid excretion is associated with oxidative stress in hypertensive subjects. Free Radic Biol Med. 2005;38:1032–6. doi: 10.1016/j.freeradbiomed.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 50.Ward NC, Rivera J, Hodgson J, et al. Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation. 2004;110:438–43. doi: 10.1161/01.CIR.0000136808.72912.D9. [DOI] [PubMed] [Google Scholar]
- 51.Liu H, Zhao Y, Nie D, et al. Association of a functional cytochrome P450 4F2 haplotype with urinary 20-HETE and hypertension. J Am Soc Nephrol. 2008;19:714–21. doi: 10.1681/ASN.2007060713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward NC, Tsai IJ, Barden A, et al. A single nucleotide polymorphism in the CYP4F2 but not CYP4A11 gene is associated with increased 20-HETE excretion and blood pressure. Hypertension. 2008;51:1393–8. doi: 10.1161/HYPERTENSIONAHA.107.104463. [DOI] [PubMed] [Google Scholar]
- 53.Fava C, Montagnana M, Almgren P, et al. The V433M variant of the CYP4F2 is associated with ischemic stroke in male Swedes beyond its effect on blood pressure. Hypertension. 2008;52:373–80. doi: 10.1161/HYPERTENSIONAHA.108.114199. [DOI] [PubMed] [Google Scholar]
- 54.Zou AP, Fleming JT, Falck JR, et al. 20-HETE is an endogenous inhibitor of the large-conductance Ca2+-activated K+ channel in renal arterioles. AmJPhysiol. 1996;270:R228–R37. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 55.Abraham NG, Feldman E, Falck JR, Lutton JD, Schwartzman ML. Modulation of erythropoiesis by novel human bone marrow cytochrome P450-dependent metabolites of arachidonic acid. Blood. 1991;78:1461–6. [PubMed] [Google Scholar]
- 56.Hill E, Murphy RC. Quantitation of 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) produced by human polymorphonuclear leukocytes using electron capture ionization gas chromatography/mass spectrometry. Biol Mass Spectrom. 1992;21:249–53. doi: 10.1002/bms.1200210505. [DOI] [PubMed] [Google Scholar]
- 57.Zhu D, Zhang C, Medhora M, Jacobs ER. CYP4A mRNA, protein, and product in rat lungs: novel localization in vascular endothelium. J Appl Physiol. 2002;93:330–7. doi: 10.1152/japplphysiol.01159.2001. [DOI] [PubMed] [Google Scholar]
- 58.Marji JS, Wang MH, Laniado-Schwartzman M. Cytochrome P-450 4A isoform expression and 20-HETE synthesis in renal preglomerular arteries. Am J Physiol Renal Physiol. 2002;283:F60–F70. doi: 10.1152/ajprenal.00265.2001. [DOI] [PubMed] [Google Scholar]
- 59.Escalante B, Sessa WC, Falck JR, Yadagiri P, Schwartzman ML. Vasoactivity of 20-hydroxyeicosatetraenoic acid is dependent on metabolism by cyclooxygenase. JPharmacolExpTher. 1988;248:229–32. [PubMed] [Google Scholar]
- 60.Inoue R, Jensen LJ, Jian Z, et al. Synergistic Activation of Vascular TRPC6 Channel by Receptor and Mechanical Stimulation via Phospholipase C/Diacylglycerol and Phospholipase A2/{omega}-Hydroxylase/ 20-HETE Pathways. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 61.Randriamboavonjy V, Busse R, Fleming I. 20-HETE-induced contraction of small coronary arteries depends on the activation of Rho-kinase. Hypertension. 2003;41:801–6. doi: 10.1161/01.HYP.0000047240.33861.6B. [DOI] [PubMed] [Google Scholar]
- 62.Kaide J, Wang MH, Wang JS, et al. Transfection of CYP4A1 cDNA increases vascular reactivity in renal interlobar arteries. Am J Physiol Renal Physiol. 2003;284:F51–6. doi: 10.1152/ajprenal.00249.2002. [DOI] [PubMed] [Google Scholar]
- 63.Wang MH, Zhang F, Marji J, Zand BA, Nasjletti A, Laniado-Schwartzman M. CYP4A1 antisense oligonucleotide reduces mesenteric vascular reactivity and blood pressure in SHR. Am J Physiol Regul Integr Comp Physiol. 2001;280:R255–61. doi: 10.1152/ajpregu.2001.280.1.R255. [DOI] [PubMed] [Google Scholar]
- 64.Zhang F, Wang MH, Wang JS, et al. Transfection of CYP4A1 cDNA decreases diameter and increases responsiveness of gracilis muscle arterioles to constrictor stimuli. Am J Physiol Heart Circ Physiol. 2004;287:H1089–H95. doi: 10.1152/ajpheart.00627.2003. [DOI] [PubMed] [Google Scholar]
- 65.Zhang F, Wang MH, Krishna UM, Falck JR, Laniado-Schwartzman M, Nasjletti A. Modulation by 20-HETE of phenylephrine-induced mesenteric artery contraction in spontaneously hypertensive and Wistar-Kyoto rats. Hypertension. 2001;38:1311–5. doi: 10.1161/hy1201.096116. [DOI] [PubMed] [Google Scholar]
- 66.Imig JD, Falck JR, Gebremedhin D, Harder DR, Roman RJ. Elevated renovascular tone in young spontaneously hypertensive rats: Role of cytochrome P450. Hyperten. 1993;22:357–64. doi: 10.1161/01.hyp.22.3.357. [DOI] [PubMed] [Google Scholar]
- 67.Inoue K, Sodhi K, Puri N, et al. Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am J Physiol Renal Physiol. 2009;297:F875–84. doi: 10.1152/ajprenal.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 69.Frisbee JC, Falck JR, Lombard JH. Contribution of cytochrome P-450 omega-hydroxylase to altered arteriolar reactivity with high-salt diet and hypertension. Am J Physiol Heart Circ Physiol. 2000;278:H1517–H26. doi: 10.1152/ajpheart.2000.278.5.H1517. [DOI] [PubMed] [Google Scholar]
- 70.Yu M, McAndrew RP, Al-Saghir R, et al. Nitric oxide contributes to 20-HETE-induced relaxation of pulmonary arteries. J Appl Physiol. 2002;93:1391–9. doi: 10.1152/japplphysiol.00247.2002. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Medhora MM, Falck JR, Pritchard KA, Jacobs ER. Mechanisms of activation of eNOS by 20-hydroxyeicosatetraenoic acid and VEGF in bovine pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L369–L77. doi: 10.1152/ajplung.00424.2005. [DOI] [PubMed] [Google Scholar]
- 72.Wang JS, Singh H, Zhang F, et al. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ Res. 2006;98:962–9. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- 73.Cheng J, Ou JS, Singh H, et al. 20-Hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2008;294:H1018–26. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 74.Cheng J, Wu CC, Gotlinger KH, et al. 20-HETE Mediates Endothelial Dysfunction Via Ikk-Dependent Enos Uncoupling. J Pharmacol Exp Ther. 2009;332:57–65. doi: 10.1124/jpet.109.159863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward NC, Chen K, Li C, Croft KD, Keaney JF., Jr Chronic AMPK activation prevents 20-HETE induced endothelial dysfunction. Clin Exp Pharmacol Physiol. 2011 doi: 10.1111/j.1440-1681.2011.05509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:H2455–65. doi: 10.1152/ajpheart.00512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Izzard AS, Rizzoni D, Agabiti-Rosei E, Heagerty AM. Small artery structure and hypertension: adaptive changes and target organ damage. Journal of hypertension. 2005;23:247–50. doi: 10.1097/00004872-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 78.Schiffrin EL. Remodeling of resistance arteries in essential hypertension and effects of antihypertensive treatment. Am J Hypertens. 2004;17:1192–200. doi: 10.1016/j.amjhyper.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 79.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–74. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 80.Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S. New markers of inflammation and endothelial cell activation: Part I. Circulation. 2003;108:1917–23. doi: 10.1161/01.CIR.0000089190.95415.9F. [DOI] [PubMed] [Google Scholar]
- 81.Alom-Ruiz SP, Anilkumar N, Shah AM. Reactive oxygen species and endothelial activation. Antioxid Redox Signal. 2008;10:1089–100. doi: 10.1089/ars.2007.2007. [DOI] [PubMed] [Google Scholar]
- 82.Schober A, Zernecke A. Chemokines in vascular remodeling. Thromb Haemost. 2007;97:730–7. [PubMed] [Google Scholar]
- 83.Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103:398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. The Journal of experimental medicine. 2007;204:2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liao TD, Yang XP, Liu YH, et al. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008;52:256–63. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harrison DG, Guzik TJ, Lob HE, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–40. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol. 2008;105:1333–41. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baeuerle PA. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell. 1998;95:729–31. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 89.Ungvari Z, Orosz Z, Labinskyy N, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 90.Sung B, Park S, Yu BP, Chung HY. Amelioration of age-related inflammation and oxidative stress by PPARgamma activator: suppression of NF-kappaB by 2,4-thiazolidinedione. Exp Gerontol. 2006;41:590–9. doi: 10.1016/j.exger.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 91.Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100:1659–66. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- 92.Chung HY, Sung B, Jung KJ, Zou Y, Yu BP. The molecular inflammatory process in aging. Antioxid Redox Signal. 2006;8:572–81. doi: 10.1089/ars.2006.8.572. [DOI] [PubMed] [Google Scholar]
- 93.de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–14. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- 94.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) 2007;112:375–84. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 95.Theuer J, Dechend R, Muller DN, et al. Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord. 2002;2:3. doi: 10.1186/1471-2261-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Henke N, Schmidt-Ullrich R, Dechend R, et al. Vascular endothelial cell-specific NF-kappaB suppression attenuates hypertension-induced renal damage. Circ Res. 2007;101:268–76. doi: 10.1161/CIRCRESAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 97.Ishizuka T, Cheng J, Singh H, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–10. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- 98.Guo AM, Arbab AS, Falck JR, et al. Activation of Vascular Endothelial Growth Factor through Reactive Oxygen Species Mediates 20-Hydroxyeicosatetraenoic Acid-Induced Endothelial Cell Proliferation. J Pharmacol Exp Ther. 2007;321:18–27. doi: 10.1124/jpet.106.115360. [DOI] [PubMed] [Google Scholar]
- 99.Medhora M, Chen Y, Gruenloh S, et al. 20-HETE increases superoxide production and activates NAPDH oxidase in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L902–11. doi: 10.1152/ajplung.00278.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guo AM, Scicli G, Sheng J, Falck JC, Edwards PA, Scicli AG. 20-HETE can act as a nonhypoxic regulator of HIF-1alpha in human microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H602–13. doi: 10.1152/ajpheart.00874.2008. [DOI] [PubMed] [Google Scholar]
- 101.Le TH, Coffman TM. Targeting genes in the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2008;17:57–63. doi: 10.1097/MNH.0b013e3282f2fd39. [DOI] [PubMed] [Google Scholar]
- 102.Cvetkovic B, Sigmund CD. Understanding hypertension through genetic manipulation in mice. Kidney international. 2000;57:863–74. doi: 10.1046/j.1523-1755.2000.057003863.x. [DOI] [PubMed] [Google Scholar]
- 103.Croft KD, McGiff JC, Sanchez-Mendoza A, Carroll MA. Angiotensin II releases 20-HETE from rat renal microvessels. Am J Physiol Renal Physiol. 2000;279:F544–F51. doi: 10.1152/ajprenal.2000.279.3.F544. [DOI] [PubMed] [Google Scholar]
- 104.Alonso-Galicia M, Maier KG, Greene AS, Cowley AW, Jr, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2002;283:R60–R8. doi: 10.1152/ajpregu.00664.2001. [DOI] [PubMed] [Google Scholar]
- 105.Joly E, Seqqat R, Flamion B, et al. Increased renal vascular reactivity to ANG II after unilateral nephrectomy in the rat involves 20-HETE. Am J Physiol Regul Integr Comp Physiol. 2006;291:R977–86. doi: 10.1152/ajpregu.00401.2005. [DOI] [PubMed] [Google Scholar]
- 106.Chabova VC, Kramer HJ, Vaneckova I, et al. Effects of chronic cytochrome P-450 inhibition on the course of hypertension and end-organ damage in Ren-2 transgenic rats. Vascular pharmacology. 2007;47:145–59. doi: 10.1016/j.vph.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 107.Muthalif MM, Karzoun NA, Gaber L, et al. Angiotensin II-induced hypertension: contribution of Ras GTPase/Mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension. 2000;36:604–9. doi: 10.1161/01.hyp.36.4.604. [DOI] [PubMed] [Google Scholar]
- 108.Parmentier JH, Muthalif MM, Nishimoto AT, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates angiotensin ii-induced phospholipase d activation in vascular smooth muscle cells. Hypertension. 2001;37:623–9. doi: 10.1161/01.hyp.37.2.623. [DOI] [PubMed] [Google Scholar]
- 109.Yaghini FA, Zhang C, Parmentier JH, et al. Contribution of arachidonic acid metabolites derived via cytochrome P4504A to angiotensin II-induced neointimal growth. Hypertension. 2005;45:1182–7. doi: 10.1161/01.HYP.0000168051.04275.ea. [DOI] [PubMed] [Google Scholar]
- 110.Sodhi K, Wu CC, Cheng J, et al. CYP4A2-Induced Hypertension Is 20-Hydroxyeicosatetraenoic Acid- and Angiotensin II-Dependent. Hypertension. 2010;56:871–8. doi: 10.1161/HYPERTENSIONAHA.110.154559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sacerdoti D, Escalante B, Abraham NG, McGiff JC, Levere RD, Schwartzman ML. Science. Vol. 243. New York, NY: 1989. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats; pp. 388–90. [DOI] [PubMed] [Google Scholar]
- 112.Fidelis P, Wilson L, Thomas K, Villalobos M, Oyekan AO. Experimental biology and medicine. Vol. 235. Maywood, NJ: 2010. Renal function and vasomotor activity in mice lacking the Cyp4a14 gene; pp. 1365–74. [DOI] [PubMed] [Google Scholar]
- 113.Zordoky BN, El-Kadi AO. Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacology & therapeutics. 2010;125:446–63. doi: 10.1016/j.pharmthera.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 114.Gainer JV, Lipkowitz MS, Yu C, et al. Association of a CYP4A11 variant and blood pressure in black men. J Am Soc Nephrol. 2008;19:1606–12. doi: 10.1681/ASN.2008010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laffer CL, Gainer JV, Waterman MR, et al. The T8590C polymorphism of CYP4A11 and 20-hydroxyeicosatetraenoic acid in essential hypertension. Hypertension. 2008;51:767–72. doi: 10.1161/HYPERTENSIONAHA.107.102921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fu Z, Nakayama T, Sato N, et al. A haplotype of the CYP4F2 gene is associated with cerebral infarction in Japanese men. Am J Hypertens. 2008;21:1216–23. doi: 10.1038/ajh.2008.276. [DOI] [PubMed] [Google Scholar]
- 117.Fu Z, Nakayama T, Sato N, et al. Haplotype-based case-control study of the human CYP4F2 gene and essential hypertension in Japanese subjects. Hypertens Res. 2008;31:1719–26. doi: 10.1291/hypres.31.1719. [DOI] [PubMed] [Google Scholar]
- 118.Fu Z, Nakayama T, Sato N, et al. A haplotype of the CYP4F2 gene associated with myocardial infarction in Japanese men. Mol Genet Metab. 2009;96:145–7. doi: 10.1016/j.ymgme.2008.11.161. [DOI] [PubMed] [Google Scholar]
- 119.Mayer B, Lieb W, Gotz A, et al. Association of a functional polymorphism in the CYP4A11 gene with systolic blood pressure in survivors of myocardial infarction. Journal of hypertension. 2006;24:1965–70. doi: 10.1097/01.hjh.0000244944.34546.8e. [DOI] [PubMed] [Google Scholar]
- 120.Fu Z, Nakayama T, Sato N, et al. A haplotype of the CYP4A11 gene associated with essential hypertension in Japanese men. Journal of hypertension. 2008;26:453–61. doi: 10.1097/HJH.0b013e3282f2f10c. [DOI] [PubMed] [Google Scholar]
- 121.Stec DE, Roman RJ, Flasch A, Rieder MJ. Functional polymorphism in human CYP4F2 decreases 20-HETE production. Physiological genomics. 2007;30:74–81. doi: 10.1152/physiolgenomics.00003.2007. [DOI] [PubMed] [Google Scholar]
- 122.Hu BC, Li Y, Li FH, et al. Peripheral and central augmentation indexes in relation to the CYP4F2 polymorphisms in Chinese. Journal of hypertension. 2011;29:501–8. doi: 10.1097/HJH.0b013e328342673c. [DOI] [PubMed] [Google Scholar]
- 123.Reckelhoff JF. Sex steroids, cardiovascular disease, and hypertension: unanswered questions and some speculations. Hypertension. 2005;45:170–4. doi: 10.1161/01.HYP.0000151825.36598.36. [DOI] [PubMed] [Google Scholar]
- 124.Yanes LL, Lima R, Moulana M, et al. Postmenopausal hypertension: Role of 20-HETE. Am J Physiol Regul Integr Comp Physiol. 2011 doi: 10.1152/ajpregu.00387.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Muthalif MM, Benter IF, Karzoun N, et al. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. ProcNatlAcadSci USA. 1998;95:12701–6. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]