

Abstract
Chronic hypoxic pulmonary hypertension (HPH) is associated with large pulmonary artery (PA) stiffening, which is correlated with collagen accumulation. However, the mechanisms by which collagen contributes to PA stiffening remain largely unexplored. Moreover, HPH may alter mechanical properties other than stiffness, such as pulse damping capacity, which also affects ventricular workload but is rarely quantified. We hypothesized that collagen content and cross-linking differentially regulate the stiffness and damping capacity of large PAs during HPH progression. The hypothesis was tested with transgenic mice that synthesize collagen type I resistant to collagenase degradation (Col1a1R/R). These mice and littermate controls (Col1a1+/+) were exposed to hypoxia for 10 days; some were treated with β-animopropionitrile (BAPN), which prevents new cross-link formation. Isolated PA dynamic mechanical tests were performed and collagen content and cross-linking were measured. In Col1a1+/+ mice, HPH increased both collagen content and cross-linking and BAPN treatment prevented these increases. Similar trends were observed in Col1a1R/R mice except that collagen content further increased with BAPN treatment. Mechanical tests showed that in Col1a1+/+ mice, HPH increased PA stiffness and damping capacity and these increases were impeded by BAPN treatment. In Col1a1R/R mice, HPH led to a smaller but significant increase in PA stiffness and a decrease in damping capacity. These mechanical changes were not affected by BAPN treatment. Vessel-specific correlations for each strain showed that the stiffness and damping capacity were correlated with the total content rather than cross-linking of collagen. Our results suggest that collagen total content is critical to extralobar PA stiffening during HPH.
Introduction
Hypoxic pulmonary hypertension (HPH) occurs in patients with lung diseases such as chronic obstructive pulmonary disease (Barbera et al. 2003), cystic fibrosis (Fraser et al. 1999), and sleep apnea (Hiestand and Phillips 2008), and contributes significantly to morbidity and mortality. HPH has been traditionally viewed as a disease of the peripheral lung vasculature with excessive vasoconstriction and structural remodeling. The consequent increase in pulmonary vascular resistance leads to right ventricular (RV) overload and ultimately right heart failure. Recently, large conduit pulmonary artery (PA) stiffening has been recognized as a critical pathophysiological factor in pulmonary hypertension and accounts for over a third of the RV workload increase (Stenmark et al. 2006b). In clinical studies, an increase in PA stiffness – or a loss of distension during systole – was found to be an excellent predictor of mortality in patients with pulmonary hypertension (Mahapatra et al. 2006; Gan et al. 2007). Despite its increasingly recognized clinical significance, the biological mechanisms that lead to large PA stiffening are not well understood.
It has been reported that stiffening of large PAs is linked with accumulation of collagen (Tozzi et al. 1994; Kobs et al. 2005; Kobs and Chesler 2006; Drexler 2008) and, in some studies, elastin (Kobs et al. 2005; Kobs and Chesler 2006; Lammers et al. 2008). However, if collagen and elastin concomitantly increase during HPH, one cannot discern which is more responsible for arterial stiffening. A recent study suggests elastin remodeling contributes to HPH-induced PA stiffening in neonatal calves (Lammers et al. 2008), but discrepant observations are also reported in other species in adults. For example, there was no change in elastin content in rodent large PAs after chronic hypoxia (Merklinger et al. 2005; Drexler 2008). Our latest study using a novel transgenic mouse model (Col1a1) suggests changes in collagen rather than elastin track large PA stiffness during HPH progression and recovery (Ooi et al. 2010). Therefore, in the present study, we focus on the mechanisms by which collagen accumulation contributes to large PA stiffening.
To determine how collagen accumulation affects arterial stiffness, it is critical to examine factors such as mass production (i.e. total content), cross-linking, orientation, and cell-matrix interactions (Humphrey 2008a). Previous studies have suggested a correlation between collagen content and the overall stiffness of large PAs (Tozzi et al. 1994; Ooi et al. 2010). This does not rule out collagen cross-linking because as the overall production increases, the cross-linking also increases as the newly synthesized collagen fibrils need to form intermolecular cross-links to be stabilized and provide tensile strength (Fratzl 2008). Prior work has shown that the slope of elastic stress-strain curve for self-assembled collagen fibers increases with increased degree of cross-linking (Silver et al. 2001). Therefore, we hypothesized that large PA stiffening is affected by both collagen content and cross-linking. In addition, our previous studies have shown an increase in arterial pulse damping capacity during HPH (Kobs et al. 2005; Kobs and Chesler 2006), which also affects ventricular workload but is rarely studied. Thus we further hypothesized that the increase in large PA damping capacity is correlated to the increase in collagen content.
To test our hypotheses about the relative contributions of increases in collagen content and cross-linking in the large PA stiffening, we employed a transgenic mouse model (Col1a1) that is resistant to collagen type I degradation (Liu et al. 1995), but not cross-linking, and treated the mice with an anti-fibrotic drug (β-animopropionitrile, BAPN) that prevents new cross-link formation. With the BAPN treatment during chronic hypoxia, we expected that wild-type mice (Col1a1+/+) would have reduced collagen accumulation (both in content and cross-linking). We expected the homozygous collagen degradation-resistant mice (Col1a1R/R) would have persistently elevated collagen content but impaired cross-linking formation. Then, we performed isolated vessel dynamic mechanical tests on the large PAs from each group and examined the correlations between structural (collagen content and cross-linking) and mechanical (stiffness and damping capacity) properties. We performed these tests with arteries in a passive state (i.e., with smooth muscle cells relaxed) to focus on the role of extracellular matrix proteins, collagen specifically, in mechanical behavior. Our results suggest that the viscoelastic behavior of PAs in a passive state is largely related to collagen content rather than cross-linking and that collagen total content is critical to extralobar PA stiffening during HPH.
Methods
All procedures were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee. Breeding pairs of Col1a1tmJae mice on the B6/129 background were obtained from Jackson Laboratory (Bar Harbor, ME). Genotyping by polymerase chain reaction was performed as described previously (Zhao et al. 1999). 16–18 week old Col1a1+/+ and Col1a1R/R mice were randomized into three groups: normoxia, 10 days of hypoxia with or without BAPN treatment (Sigma-Aldrich Corp., intraperitoneal injection of 5 mg dissolved in 0.5 ml of PBS, twice per day). During hypoxia, animals were maintained in normobaric hypoxic chamber at controlled O2 concentration of 10% with 4 L min−1 air flow to maintain the carbon dioxide level at < 600 ppm (Ooi et al. 2010). The chambers were opened for less than 30 minutes at a time for regular animal care or BAPN injections.
Vessel harvest and biochemical assays
Extralobar PAs were excised from the pulmonary trunk to the first branches under microscopy. Right PAs were flash frozen for a cross-linking assay and left PAs were mounted in a vessel chamber (Living System Instrumentation (LSI), Burlington, VT) for dynamic mechanical testing. After the mechanical tests, left PAs were flash frozen and measured for hydroxyproline (OHP) to quantify collagen content following established methods (Ooi et al. 2010). PA collagen cross-linking was measured by pyridinoline (PYD) using a rapid enzyme immunoassay (Metra® PYD EIA kit, Quidel Corporation, San Deigo, CA) (Boutten et al. 2000).
Isolated vessel dynamic mechanical tests
Left PAs were stretched 140% to prevent buckling at higher pressures and tested at this fixed length in a passive state using calcium- and magnesium-free PBS as both perfusate and superfusate. The perfusate was supplied via a steady flow pump (LSI; Burlington, VT) and an oscillatory flow pump (EnduraTec TestBench; Bose Corporation; Eden Prairie, MN) to achieve sinusoidal pressurization at 10–25 mmHg and 25–40 mmHg at the frequency of 0.01Hz. Superfusate was continuously circulated and maintained at 37°C. The vessels were preconditioned at 0.014Hz for 10 cycles before data recording as done previously (Kobs et al. 2005). During the dynamic test, pressure-outer diameter data were recorded simultaneously by IonWizard software (Version 6.0, IonOptix, Milton, MA) using in-line pressure transducers at an acquisition frequency of 1Hz and a CCD camera (IonOptix, Milton, MA) connected to an inverted microscope (Olympus, Center Valley, PA) to measure outer diameter (OD) at 4× magnification with an acquisition frequency of 1.2Hz.
Analysis of vessel mechanical properties
The viscoelastic properties of left PAs were obtained from pressure-stretch curves as well as stress-strain curves. We used the pressure-stretch curve hysteresis for the calculation of damping capacity because calculating wall stress required use of the optically measured wall thickness, which decreased precision with no gain in accuracy. Stretch was calculated as the ratio of pressure-dependent OD to OD at the baseline pressure of 10 mmHg (OD10). PA stiffness was defined by the slope of the line best-fit to the pressure-stretch loop generated by loading and unloading. PA damping capacity was calculated in two ways as shown in Fig. 1. In the first method, the stored energy (WS) and the dissipated energy (WD) were defined as area of the triangle defined by the maximum and minimum stretch in the hysteresis loop and the area within the hysteresis loop, respectively (Lakes 1999). Then, damping capacity (d) was calculated as WD/ (WD + WS). In the second method, the width of the hysteresis loop at the mid-loop pressure (Pmid = (Pmax + Pmin) / 2) and the maximal change of stretch in one dynamic cycle were measured; these distances are denoted by ‘a’ and ‘b’ in Fig. 1. Then, the phase angle (δ) between pressure and stretch was determined by δ = sin−1(a / b) and damping capacity (d’) was calculated as:
| (1) |
as in (Lakes 1999) such that
| (2) |
In the isolated vessel tests, the arteries were assumed to be homogeneous and incompressible. Assuming conservation of mass and no axial extension, the wall thickness as a function of pressure was calculated as:
| (3) |
where OD40 and h40 are the OD and h measured optically at 40 mmHg, respectively.
Fig. 1.
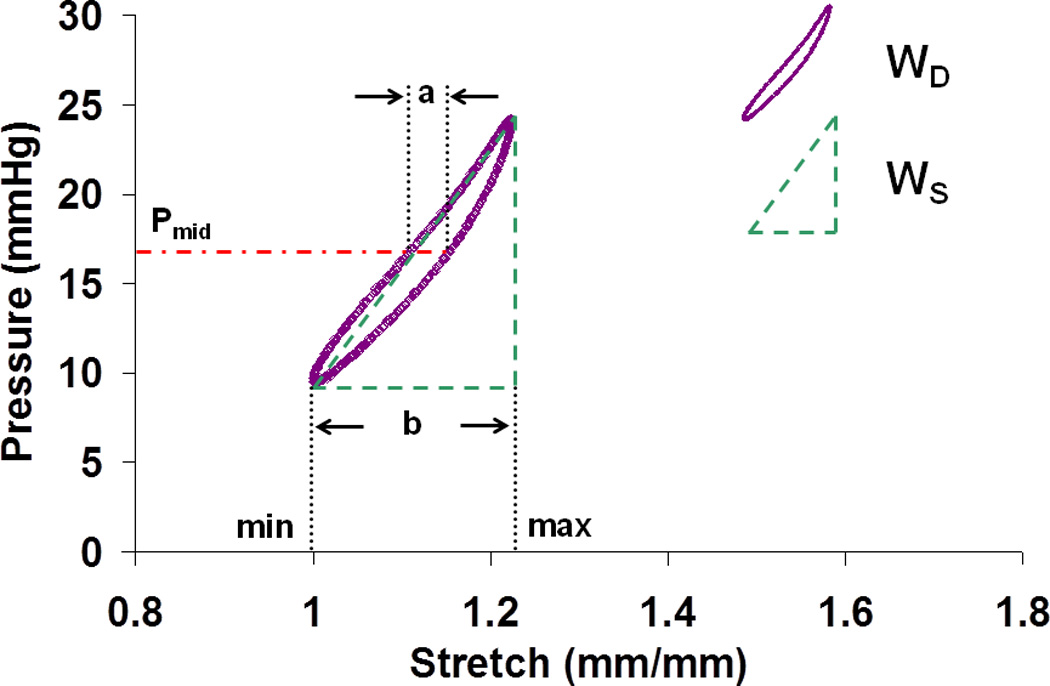
A typical clockwise pressure-stretch curve from the dynamic mechanical test. Pmid = (Pmax + Pmin) / 2. a and b mark the width of the hysteresis loop at the pressure of Pmid and the maximal change of stretch in one dynamic cycle, respectively. WD: dissipated energy; WS: stored energy.
We also analyzed PA material behavior with the stress-strain relationship to permit comparison to prior work. The midwall circumferential stress (σ) and strain (ε) were calculated using thin-walled assumption and Green’s formulation for finite deformation, respectively:
| (4) |
| (5) |
where Dm10 is the mid-wall diameter at 10 mmHg. Midwall stress and strain were chosen so that the no-load state can be used as the reference state instead of the zero-stress state (Zhao et al. 2002). Next, the transition strain at which the stress-strain curve turns into a steeper slope was examined. Briefly, two straight trend lines that best fit the low and high strain regions of the stress-strain curve over the entire pressure range were determined and the slopes of these lines (elastic moduli) were denoted as Elow and Ehigh. Then the intersection point of these two lines was used to approximate the transition strain (Tabima and Chesler 2010). Moreover, we derived the elasticity of elastin (Ee, which is the same as Elow) and collagen (Ec) from the original stress-strain curves using a technique established by Armentano et al. (Armentano et al. 1991).
Statistics
All results are presented as mean ± SD. For each strain, comparisons between exposure groups were performed by two-way (for Elow and Ehigh) or one-way (for all other parameters) analysis of variance (ANOVA) with Tukey’s multiple comparisons using R (www.r-project.org) version 2.5.1. To assess the correlative relationships between mechanical and structural properties for each mouse strain, simple linear correlations were determined for the PA stiffness or damping capacity with collagen total content or cross-linking using Microsoft Excel. R2 values are reported. P < 0.05 was considered significant (* vs. normoxia, and # vs. hypoxia. N = 5–8 per group).
Results
Biological changes in large PAs
The collagen total content (hydroxyproline; OHP) and cross-linking (pyridinoline; PYD) in large PAs were examined biochemically. As predicted, hypoxia increased large PA collagen content by 79% and 48% and cross-linking by 60% and 45% in Col1a1+/+ and Col1a1R/R mice, respectively (Fig. 2). However, these increases were only statistically significant in the Col1a1+/+ mice.
Fig. 2.
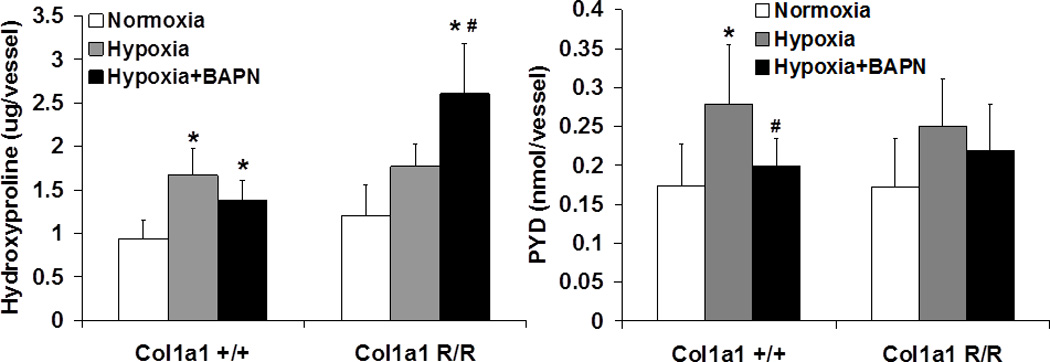
Total amount of hydroxyproline (Left, N = 6–8 per group) and pyridinoline (PYD, Right, N = 5–8 per group) per vessel measured from large PAs in normoxia, hypoxia, and hypoxia + BAPN treatment groups. (p < 0.05 for * hypoxia or hypoxia+BAPN vs. normoxia, and # hypoxia+BAPN vs. hypoxia).
BAPN treatment during hypoxia led to different effects on collagen accumulation in Col1a1+/+ and Col1a1R/R mice. As expected, because BAPN prevented cross-link formation in newly synthesized collagen, the total amount of cross-linking was less in the BAPN-treated group compared to the untreated group after hypoxia in both mouse strains. Again, this difference only reached statistical significance in the Col1a1+/+ mice. However, notably, whereas in the Col1a1+/+ mice BAPN treatment slightly decreased collagen total content (by 18% from the untreated hypoxia group), in the Col1a1R/R mice BAPN treatment significantly increased the collagen total content by 47% of the untreated hypoxia group.
Mechanical changes in large PAs
Representative pressure-stretch curves for each group are shown in Fig. 3. For all arteries tested, hypoxia induced a left-ward shift of the curve, which reflects stiffening of the artery. This change was more dramatic in Col1a1+/+ mice compared to Col1a1R/R mice. BAPN treatment during hypoxia abolished this left-ward shift in Col1a1+/+ mice and had only a minimal effect in Col1a1R/R mice.
Fig. 3.
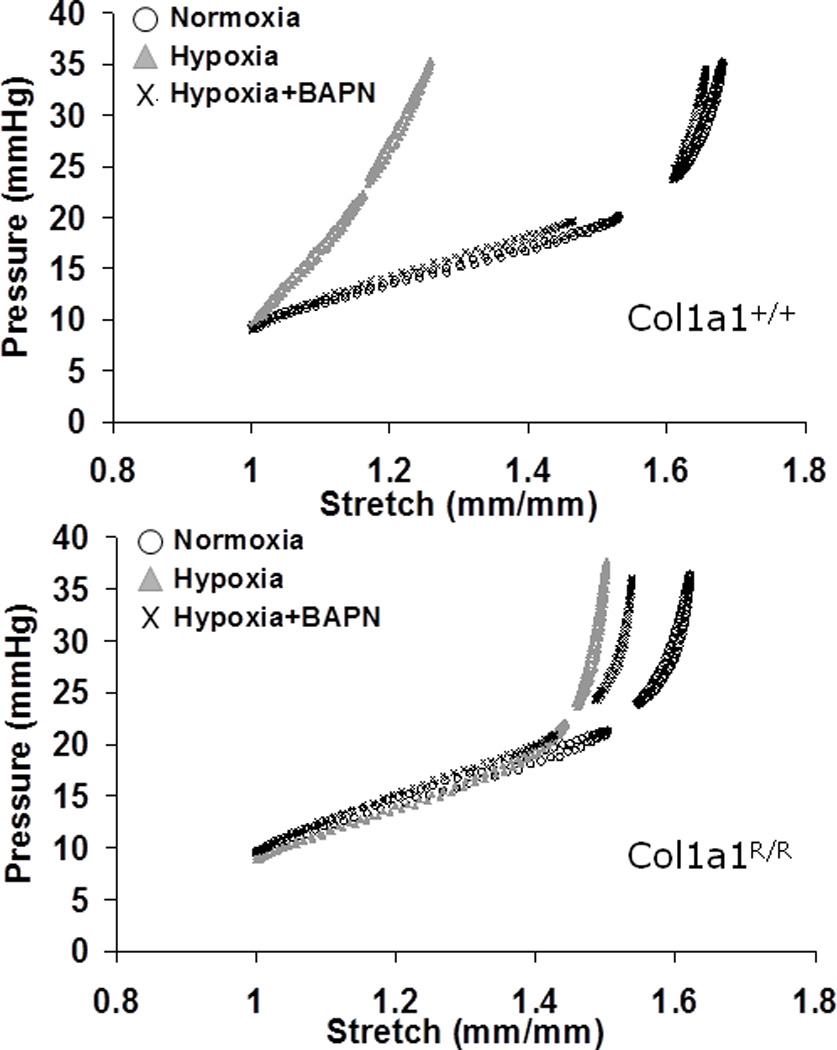
Representative pressure-stretch curves from large PAs (Upper: Col1a1+/+ mice; Lower: Col1a1R/R mice) in normoxia, hypoxia, and hypoxia + BAPN treatment groups.
Within the entire pressure range tested (10 – 40 mmHg), a single parameter that represents mechanical behavior such as stiffness cannot be easily extracted. Moreover, the different conditions (normoxia, hypoxia, hypoxia+BAPN) resulted in different stretch or strain ranges for the same testing pressures (Fig. 3). Therefore, instead of comparing mechanical behavior at the same pressure but different stretch or strain, we selected a fixed range of stretch of 1 – 1.5, which corresponds to the testing pressure of 10 – 25 mmHg, to examine the viscoelastic properties of the PA. This stretch range is equivalent to Green strain of 0 – 0.625, which includes the recruitment of collagen fibers (Armentano et al. 1991; Zulliger et al. 2004; Lammers et al. 2008) and thus enables us to observe the effects of collagen on the passive mechanical properties. The stiffness and damping capacity of the PAs in the pressure range of 10 – 25 mmHg were then quantified as described in the Methods.
As expected, the stiffness and damping capacity of large PAs in Col1a1+/+ mice increased with hypoxia, and BAPN treatment during hypoxia prevented these increases (Fig. 4). The changes in damping capacity d were caused by changes in WS with essentially no changes in WD (Table 1). In the Col1a1R/R mice, hypoxia induced a smaller but significant increase in the stiffness; and to our surprise, hypoxia decreased damping capacity, which is the opposite of the result for Col1a1+/+ mice. A decrease in WD with no change in WS was responsible for the decrease in d (Table 1). BAPN treatment did not result in any significant changes in the stiffness or damping capacity compared to the untreated hypoxic group in the Col1a1R/R mice. The measurement of damping capacity by the length method (d’) showed the same trends but differences did not reach statistical significance (Table 2). This method is advantageous for irregularly shaped hysteresis curves typical of non-linear materials; since the pressure-stretch curves here were reasonably elliptical, the length method provided no new insight.
Fig. 4.
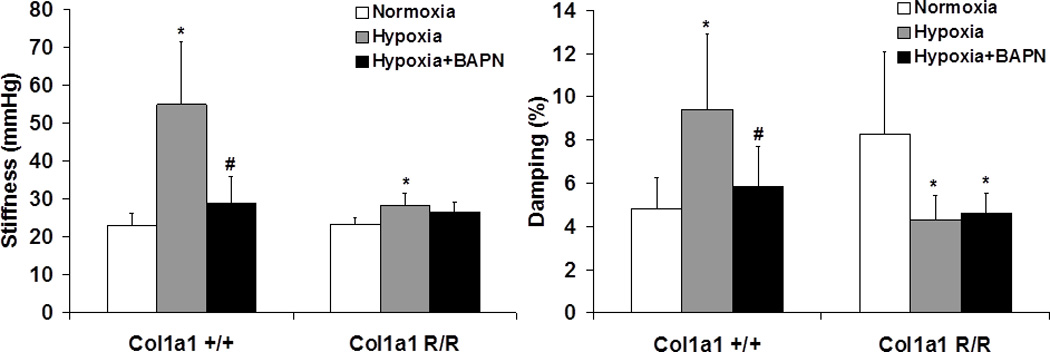
PA stiffness (Left) and damping capacity calculated using the area method (Right) measured from pressure-stretch curves (N = 5–7 per group, p < 0.05 for * hypoxia or hypoxia+BAPN vs. normoxia, and # hypoxia+BAPN vs. hypoxia).
Table 1.
Dissipated energy (WD) and stored energy (WS) measured from pressure-stretch curves using the area method for each experimental group
| Mouse Strain | WD (mmHg) | WS (mmHg) | |
|---|---|---|---|
| Col1a1+/+ | Normoxia | 0.16±0.02 | 3.31±0.80 |
| Hypoxia | 0.18±0.08 | 1.91±0.92* | |
| Hypoxia+BAPN | 0.14±0.05 | 2.35±0.27 | |
| Col1a1R/R | Normoxia | 0.31±0.08 | 3.77±1.26 |
| Hypoxia | 0.16±0.04* | 3.67±0.68 | |
| Hypoxia+BAPN | 0.13±0.02* | 2.64±0.34 |
(N = 5–7 per group, *p<0.05 hypoxia or hypoxia+BAPN for vs. normoxia).
Table 2.
Damping capacity measured from pressure-stretch curves using the length method (d’) and comparison to damping capacity measured from pressure-stretch curves using the area method (d)
| Mouse Strain | d' (%) | d (%) | |
|---|---|---|---|
| Col1a1+/+ | Normoxia | 5.3±2.5 | 4.8±1.4 |
| Hypoxia | 9.4±3.9 | 9.4±3.5* | |
| Hypoxia+BAPN | 6.3±2.4 | 5.8±1.8# | |
| Col1a1R/R | Normoxia | 8.2±3.8 | 8.3±3.8 |
| Hypoxia | 4.8±1.8 | 4.3±1.1* | |
| Hypoxia+BAPN | 5.4±1.2 | 4.6±0.9* |
(N = 5–7 per group, *p<0.05 hypoxia or hypoxia+BAPN for vs. normoxia, #p<0.05 hypoxia+BAPN for vs. hypoxia).
We also examined the transition strain (Fig. 5) for each group from the stress-strain curves (not shown). Consistent with our findings in C57BL6/J mice (Tabima and Chesler 2010), in the Col1a1+/+ mice, hypoxia decreased the transition strain. This suggests that collagen fibers were engaged earlier (at lower strains) after hypoxia-induced remodeling. BAPN treatment prevented the decrease in transition strain in Col1a1+/+ mice. However, in the Col1a1R/R mice, no changes were observed either after hypoxia alone or after hypoxia with BAPN treatment.
Fig. 5.
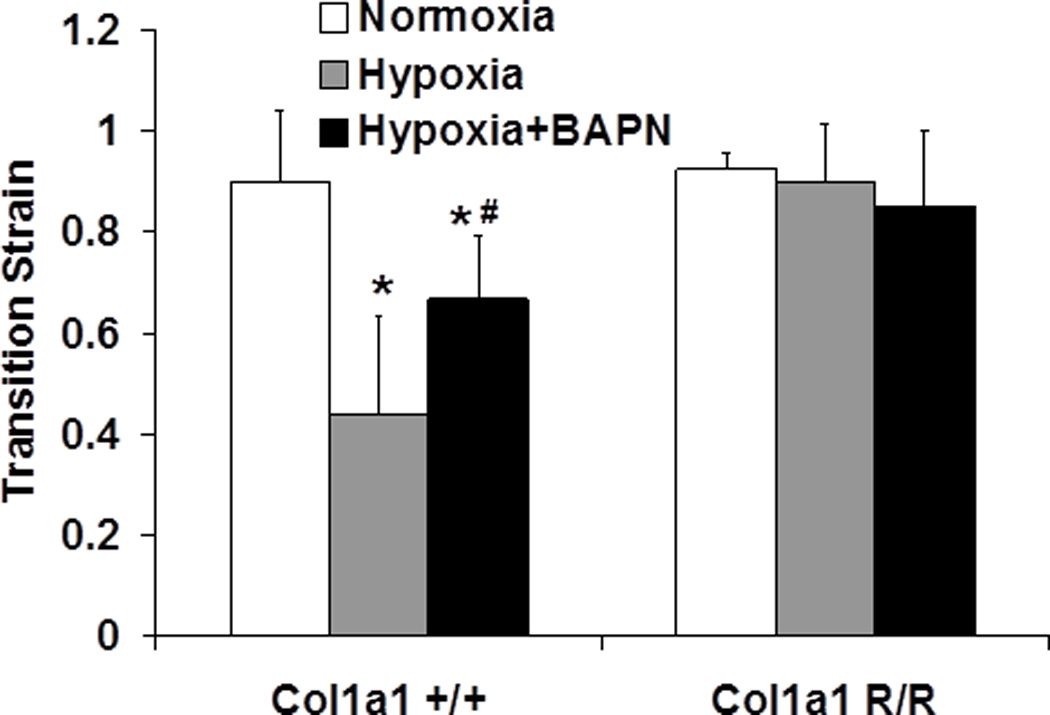
Transition strain measured from stress-strain curves in normoxia, hypoxia, and hypoxia + BAPN treatment groups (N = 5–7 per group, p < 0.05 for * hypoxia or hypoxia+BAPN vs. normoxia, and # hypoxia+BAPN vs. hypoxia).
The results for elastic moduli at low and high strain regions (Elow and Ehigh) as well as for collagen (Ec) are presented in Table 3. As expected, we did not observe significant changes in elasticity of elastin or Elow. However, we did observe significant decreases in Ehigh and Ec in the Col1a1+/+ mice with hypoxia (Table 3).
Table 3.
Elastic moduli derived from stress-strain curves
| Mouse Strain | Elow (Ee) (kPa) | Ehigh (kPa) | Ec (kPa) | |
|---|---|---|---|---|
| Col1a1+/+ | Normoxia | 27±14 | 335±183 | 305±169 |
| Hypoxia | 37±16 | 190±71* | 153±70* | |
| Hypoxia+BAPN | 27±3 | 281±44 | 254±43 | |
| Col1a1R/R | Normoxia | 28±13 | 258±121 | 230±109 |
| Hypoxia | 16±2 | 138±42 | 123±43 | |
| Hypoxia+BAPN | 17±4 | 185±117 | 169±114 |
(N = 5–7 per group, *p<0.05 for hypoxia vs. normoxia).
Correlations between collagen accumulation and mechanical changes of large PAs
Next, we performed vessel-specific correlations between the structural (i.e., collagen content and cross-linking) and functional (i.e., stiffness and damping capacity) properties of the tested PAs. Fig. 6 presents the linear correlations between PA stiffness and collagen content without and with BAPN treatment in Col1a1+/+ and Col1a1R/R mice. In Col1a1+/+ mice, PA stiffness was positively correlated with the collagen content, and BAPN treatment tended to weaken the relationship. In Col1a1R/R mice, there was a similar but more moderate correlation between PA stiffness and collagen content, and BAPN treatment again weakened the correlation. We did not observe any correlations between PA stiffness and collagen cross-linking (not shown).
Fig. 6.
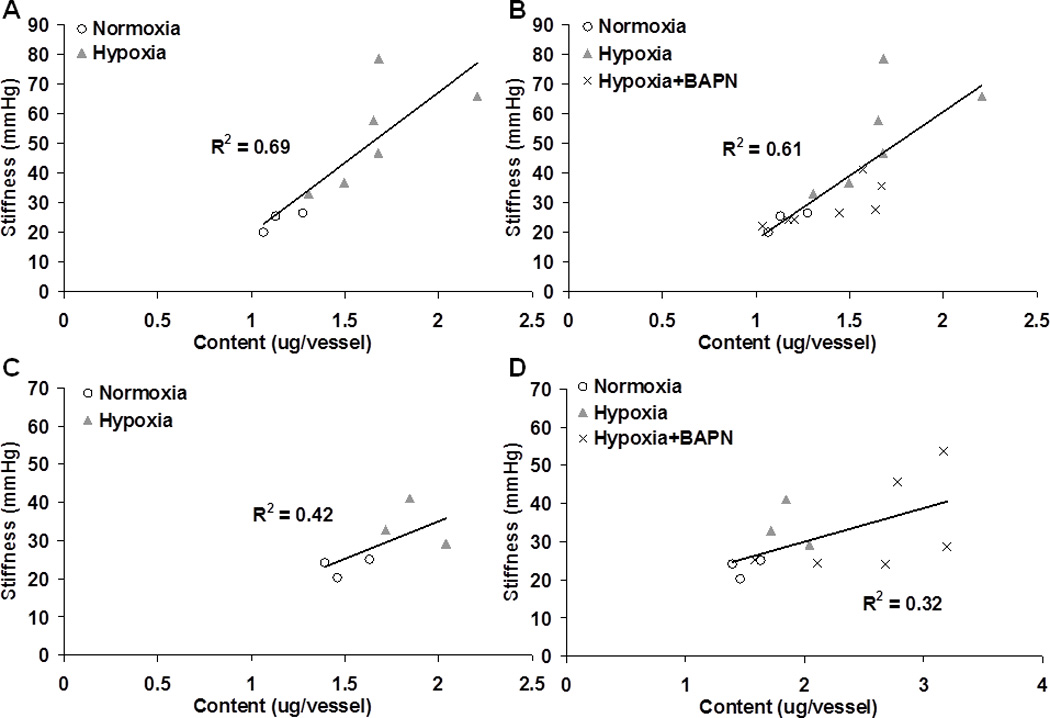
Linear correlations between PA stiffness and collagen total content in the groups of Col1a1+/+ mice without (A) and with (B) BAPN treatment and in the groups of Col1a1R/R mice without (C) and with (D) BAPN treatment.
Similarly, we performed linear correlations between PA damping capacity and collagen content without and with BAPN treatment in Col1a1+/+ and Col1a1R/R mice (Fig. 7). In Col1a1+/+ mice, we observed a positive correlation between PA damping capacity and collagen content. In Col1a1R/R mice, a negative correlation was observed. In both mouse strains, BAPN treatment substantially weakened the correlations. We did not observe any correlations between PA damping capacity and collagen cross-linking in Col1a1+/+ mice, and there was only a moderate, negative correlation (R2=0.59) in the non-BAPN treated groups in Col1a1R/R mice between PA damping capacity and collagen cross-linking (not shown).
Fig. 7.
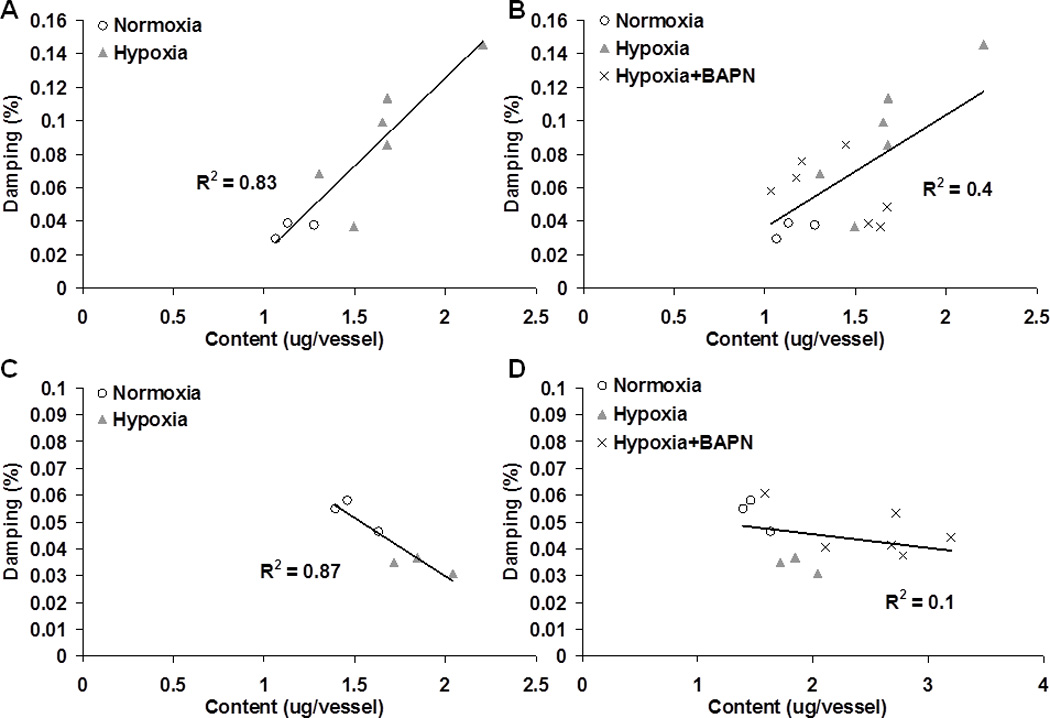
Linear correlations between PA damping capacity and collagen total content in the groups of Col1a1+/+ mice without (A) and with (B) BAPN treatment and in the groups of Col1a1R/R mice without (C) and with (D) BAPN treatment.
Discussion
In this study, we performed isolated vessel dynamic mechanical tests on the large PAs of mice exposed to chronic hypoxia, and examined structural (collagen content and cross-linking) and mechanical (stiffness and damping capacity) changes as well as their correlations. By treating Col1a1 mice with BAPN, we were able to decouple the relative contributions of collagen content and cross-linking in HPH-induced remodeling in large PAs. Our results suggest that large PA viscoelasticity in a passive state is more dependent on collagen content than the cross-linking. These results support a growing body of literature implicating collagen accumulation in extralobar PA stiffening, which is potentially clinically relevant to outcomes in pulmonary hypertension.
Large conduit arterial stiffening is well known to occur in human pulmonary hypertension (Mahapatra et al. 2006; Gan et al. 2007) and has been confirmed in animal studies in a variety of species (Tozzi et al. 1994; Kobs et al. 2005; Lammers et al. 2008; Drexler 2008). The mechanical changes in large PAs during stiffening have been linked with increases in both collagen and elastin (Tozzi et al. 1994; Kobs et al. 2005; Kobs and Chesler 2006; Lammers et al. 2008; Drexler 2008). Our recent study using the same mouse strain as used here (Col1a1) suggested that collagen accumulation is a key factor in the extralobar PA stiffening during HPH development (Ooi et al. 2010). Therefore, the focus of the present study was to investigate how exactly collagen accumulation affects vascular biomechanics. As expected, we observed large PA stiffening in response to the hypoxia exposure (Figs. 3 and 4) from the dynamic pressure–outer diameter measurements. This stiffening, via the vessel-specific correlations, was found to be related to collagen content rather than the cross-linking (Fig. 6).
The increase in PA stiffness with hypoxia in Col1a1R/R mice was not as significant as in Col1a1+/+ mice, which was not unexpected. In our prior study, Col1a1R/R mice exposed to chronic hypoxia had a smaller increase in elastic modulus than Col1a1+/+ mice despite a greater relative increase in mean PA pressure (Ooi et al. 2010). We previously attributed the discrepancy to impaired collagen turnover due to the collagen degradation resistance mutation, which likely limits remodeling. This speculation is consistent with the OHP results (Fig. 2), in which the relative increase in collagen content in hypoxic Col1a1R/R PAs tended to be less than in hypoxic Col1a1+/+ PAs. However, we also observed a less significant correlation (R2 = 0.42) between PA stiffness and collagen content in Col1a1R/R mice compared to Col1a1+/+ mice (R2 = 0.69) (Fig. 6), which suggests that changes in collagen fiber orientation, organization and interactions, i.e., overall architecture, are also a consequence of the defect in collagen metabolism. The decrease in correlation coefficients between structural and functional parameters when the BAPN treated groups are included supports this suggestion that factors other than content and cross-linking are critical to PA stiffness. We will further discuss the potential impacts of abnormal collagen metabolism in the Col1a1R/R strain below.
The decrease in transition strain with HPH-induced remodeling in the Col1a1+/+ mice (Fig. 5) has been reported in mouse pulmonary hypertensive arteries (Tabima and Chesler 2010) and systemic hypertensive arteries (Intengan et al. 1999; Danpinid et al. 2010). This decrease is typically interpreted as recruitment of collagen at lower strains, which suggests an increasing mechanical load on the collagen fibers in diseased conditions. In hypertensive aortas, a reduced transition strain was associated with the degradation of elastin and formation of collagen after angiotensin-II administration (Danpinid et al. 2010), which led to a decreased ratio of elastin to collagen in the vessel wall. The same mechanism may exist in the Col1a1+/+ mice. Our prior study on Col1a1 mouse pulmonary arteries found no significant changes in elastin content after hypoxia (Ooi et al. 2010). Thus, the ratio of elastin to collagen was reduced in hypoxic Col1a1+/+ PAs. We speculate that, with normal collagen metabolism, collagen engagement depends on the ratio of elastin to collagen in the vessel wall, with a lower ratio indicating a smaller transition strain. The ability of BAPN treatment to impede the reduction of transition strain with hypertension-induced remodeling is a novel finding.
In the Col1a1R/R mice, which have abnormal collagen accumulation, collagen total content increased with hypoxia and increased further with hypoxia+BAPN treatment despite impaired cross-linking formation (Fig. 2), but these changes had no effect on transition strain (Fig. 5). Because collagen degradation is required for normal turnover during wall growth and remodeling (Strauss et al. 1996; Fratzl 2008; Humphrey 2008b), we speculate that in the mutant Col1a1R/R mice, the mechanical loading of collagen fibers does not occur normally after hypoxia. For example, a mix of old, un-degraded and new, differently- or un-organized collagen fibers might have reduced efficiency in cell-matrix or matrix-matrix interactions or these fibers may be at sub-optimal lengths and stretch ratios within the artery wall to engage and bear stress with increased strain (Humphrey 1999). Thus, we propose that the recruitment of collagen fibers is not only dependent on the elastin to collagen ratio but also on the collagen fiber architecture. The parameter transition strain appears sensitive to this change in architecture and, although less well recognized, may be of clinical relevance.
We did not observe significant changes in elasticity of elastin (or Elow), which suggests a limited role of elastin in the hypertensive remodeling process. We did observe a significant decrease in the elasticity of collagen (and Ehigh) in the Col1a1+/+ mice with hypoxia (Table 3). But, it is important to note that this decrease in modulus occurred not because the artery became more compliant with hypoxia but instead because the modulus was measured in a lower strain range in the hypoxic arteries. As mentioned before, the hypoxic condition led to a smaller stretch range for the same pressure compared to the normoxic condition, which is why we chose a fixed stretch range instead of a fixed pressure range to compare the viscoelastic behavior of large PAs.
Arterial viscosity, the capacity to damp pulsatile pressure and flow oscillations, contributes to circulatory energy losses (Armentano et al. 2007), affects the magnitude of arterial wave reflections and modulates ventricular function via impaired ventricular-vascular coupling (Grignola et al. 2007). This arterial wall property is changed during aging and with atherosclerosis and systemic hypertension (Learoyd and Taylor 1966; Armentano et al. 1995; Armentano et al. 1998). However, despite its clinical consequences, arterial viscosity has received less attention than stiffness. In the present study, we observed an increase in the damping capacity of the large PAs in Col1a1+/+ mice after chronic hypoxia (Fig. 4), which is consistent with our previous findings from extralobar mouse PAs in passive mechanical tests (Kobs et al. 2005; Kobs and Chesler 2006). Traditionally, smooth muscle cells (SMCs) are considered to be the primary source of arterial viscosity (Boutouyrie et al. 1998; Armentano et al. 2007) but our results suggest that SMCs may not be the only determinant of arterial viscosity. In the passive mechanical tests from our current and prior studies, SMC activity was eliminated by depletion of calcium and magnesium ions in the medium. Therefore, the mechanical properties were almost entirely determined by the extracellular matrix proteins. In tendons, which also have no active elements (e.g., SMC), slippage of collagen fibrils is an important contributor to viscous energy dissipation (Silver et al. 2001). In arteries, the relationships between micro-structural arrangement of vascular wall components and their energy dissipating function are not established yet. But collagen and elastin make up about 50% of the dry weight of a vessel, so measurable energy dissipation may occur through cell-matrix interactions and sliding between the collagen fibers (Silver et al. 2001; Silver et al. 2003). With HPH-induced remodeling in large PAs, this fibrillar slippage may occur more frequently or to a greater degree with excess collagen deposition.
In Col1a1R/R mice, we observed a decrease in damping capacity in response to hypoxia, and this change persisted with BAPN treatment (Fig. 4). The opposite contributions of collagen content to the damping capacity between Col1a1+/+ and Col1a1R/R mice (Fig. 7) supports our suggestion above that the abnormal collagen metabolism in Col1a1R/R mice alters the mechanical function of synthesized collagen. To date, there is no report on the abnormal vascular remodeling in this transgenic mouse (Col1a1R/R) except by our group. We suspect that the defect in Col1a1R/R mice leads to poorly organized collagen fibers with modified mechanical function. In normal conditions, vascular fibrillar collagen functions by effective cross-link formation and attachment at certain angles of orientation to the smooth muscle cells via integrins (Silver et al. 2003; Fratzl 2008). We did not examine the microscopic distribution of collagen fibers in Col1a1R/R PAs in the present study, but we speculate that the mix of old, un-degraded and new, un-organized collagen fibers impairs the normal structure-function relationship of collagen. Therefore, the accumulated dysfunctional collagen may not only prevent normal collagen recruitment as strain increases, but also impair the effective interactions and slippage between collagen fibers as in normal conditions to reduce energy dissipation. This may explain why, despite a preserved linear correlation between collagen content and damping capacity, the relationship changed from positive to negative. In addition, a blood vessel is a complicated composite material, and other arterial wall components that generate friction between extracellular molecules during cyclic wall motion may be critical in determining arterial viscosity, which requires further investigation. Again, the decrease in correlation coefficients when the BAPN treated groups are included supports the suggestion that factors other than content and cross-linking are critical to PA damping capacity.
Collagen accumulation is invariably observed in HPH-induced remodeling in different species, both in the media and adventitia of the pulmonary arteries (Poiani et al. 1990; Kobs et al. 2005; Stenmark et al. 2006a). Unlike elastin, which is produced and organized primarily during the perinatal period and is stable for decades (Mithieux and Weiss 2005; Humphrey et al. 2009), collagen turnover can be significant during arterial remodeling and collagen deposition is a major event in hypertensive vessels (Fratzl 2008). In the current study, we specifically examined the effects of collagen content vs. cross-linking in the passive mechanical behavior during HPH development. From the vessel-specific correlations performed in each mouse strain, we found that the functional changes were correlated with the collagen content but not cross-linking, except that in Col1a1R/R mice the damping capacity showed a moderate, negative correlation (R2=0.59) with collagen cross-linking.
Cross-link formation is a critical step in the maturation of collagen fibers to provide tensile strength, and it is reasonable to assume cross-links contribute to the elasticity of the vessel. However, we did not observe correlations between cross-linking and PA mechanics in this study. There are two major groups of cross-links: those initiated by the enzyme lysyl oxidase and those derived from nonenzymatically sugar glycosylation (Last et al. 1990); the latter form often during aging or diabetes (Aronson 2003). The measure of cross-linking (PYD) used in the current study quantifies non-reducible (lysyl oxidase) enzyme-initiated cross-links between adjacent mature collagen molecules, which are formed by a sequence of post-translational modifications and can be inhibited by BAPN (Pfeiffer et al. 2005). A previous study in rats showed that inhibition of collagen cross-linking by BAPN results in reduced aortic stiffness (Bruel et al. 1998). However, in that case, the cross-linking (PYD) was presented as an amount relative to the total collagen content (OHP). In our study, because of the small volume of mouse PA tissue, we could not perform OHP and PYD assays on the same artery and only measured the total amount of cross-linking. Therefore, although we observed an overall increase in PYD in right PAs after hypoxia (Fig. 2), its density or percentage in the vessel wall may remain at control levels, which may explain the lack of correlation between cross-linking and vessel biomechanics.
The effect of BAPN treatment on HPH progression has been shown previously in rats. Via attenuated collagen deposition, BAPN limited the development of pulmonary hypertension and RV hypertrophy, although the mechanical changes in large PAs were not explored (Kerr et al. 1984). While BAPN may directly impair distal pulmonary arterial remodeling in response to chronic hypoxia, we speculate that the therapeutic effects of BAPN originate from its ability to limit large PA stiffening, as shown here. That is, PA stiffening is thought to be key to pulmonary hypertension progression through two mechanisms: increased distal arterial cyclic strain damage (Li et al. 2009), which promotes SMC proliferation, and proximal wave reflections (Milnor et al. 1969), which increase RV afterload. Indeed, conduit PA stiffening accounts for over a third of the RV workload increase with pulmonary hypertension (Stenmark et al. 2006b). The destabilizing influence of BAPN on new collagen fibrils in proximal PAs may reduce HPH progression through both of these mechanisms. However, the impact of BAPN on distal pulmonary arterial remodeling remains unknown; thus the primary effect of BAPN may be to reduce pulmonary vascular resistance by preventing distal arterial remodeling with the impact on the proximal PAs a secondary effect. Future studies are required to distinguish between these potential mechanisms of action. Finally, as noted above, BAPN treatment may affect conduit PA collagen fiber architecture independent of content and cross-linking, as evidenced by the consistently decreased correlation coefficients when the BAPN treated groups are included (Figs. 6 and 7).
Limitations
In the present study, the dynamic mechanical tests were performed at sub-physiological frequency (0.01Hz) and in a passive state. We selected these conditions to allow a comparison with our prior experimental findings (Kobs et al. 2005; Kobs and Chesler 2006). Also, we used OD at 10 mmHg as the reference for the no-load state instead of 5 mmHg. We estimate this will lead to approximately 12% underestimation of the stretch (OD/ODRef) compared to our previous studies (Kobs et al. 2005; Ooi et al. 2010; Tabima and Chesler 2010). Moreover, the stress-strain curves calculation involved the measurement of wall thickness, which was obtained optically. Hence we did not use stress-strain curves but pressure-stretch curves to analyze PA damping capacity.
Because of the non-linear behavior of PAs over entire testing range (10–40 mmHg), and because of the different stretch/strain ranges that resulted from different conditions (normoxia, hypoxia, hypoxia+BAPN) under the same testing pressures, especially in the testing range of 25–40 mmHg, we selected a fixed range of stretch to compare mechanical properties. This range includes the recruitment of collagen but not necessary all collagen fibers in the PA. In addition, the strain range for mechanical characterization includes contributions from both elastin and collagen, but we did not explicitly examine the changes in elastin in this study. In our prior study using Col1a1 mice, we did not observe significant changes in total elastin content of large PAs (Ooi et al. 2010). Here, we also did not include controls with saline injections. Since we did not observe systemic effects of the injections (i.e., weight loss) in the BAPN-treated groups, we assumed the local effects of injection alone (i.e., on the PA biomechanics) to be negligible.
Due to the small size of the mouse extralobar pulmonary artery, all the available tissues were used for collagen content and cross-linking measurements and we did not examine the changes in content of collagen subtypes (e.g. type I vs. III). However, since type I collagen is the major collagen subtype in PAs (Diez 2007), a defect in type I collagen should translate into a defect in total collagen content. A final limitation worth noting is that we did not measure in vivo hemodynamics, pulmonary vascular resistance or distal pulmonary artery remodeling. While these data are important for elucidating the therapeutic impact of BAPN, differences in pulmonary artery pressure between groups could either be a cause or a consequence of the differences in collagen content and cross-linking in the Col1a1R/R and BAPN-treated mice.
In summary, the current study examined the relative contributions of collagen content (hydroxyproline) and cross-linking (pyridinoline) in the passive mechanical properties of large PAs after exposure to chronic hypoxia. In concert with collagen accumulation, the stiffness and damping capacity increased with normal collagen metabolism and these changes were correlated with the total content rather than cross-linking in the vessel wall. BAPN treatment that prevents lysyl oxidase-initiated cross-linking formation impeded PA stiffening during HPH and weakened the relationships between collagen content and PA mechanics. The linked relationship between structural and functional changes in large PAs suggests collagen total content is critical to the extralobar PA stiffening, which has potentially significant implications for the progression of pulmonary hypertension and subsequent right ventricular dysfunction.
Acknowledgements
We thank Dr. R.S. Lakes for suggestions in the characterization of arterial viscoelastic properties. This study is supported by NIH R01 HL086939 (NCC) and AHA Midwest Affiliate Postdoctoral Fellowship 10POST2640148 (ZW).
Reference
- Armentano R, Megnien JL, Simon A, Bellenfant F, Barra J, Levenson J. Effects of hypertension on viscoelasticity of carotid and femoral arteries in humans. Hypertension. 1995;26(1):48–54. doi: 10.1161/01.hyp.26.1.48. [DOI] [PubMed] [Google Scholar]
- Armentano RL, Barra JG, Pessana FM, Craiem DO, Graf S, Santana DB, Sanchez RA. Smart smooth muscle spring-dampers. Smooth muscle smart filtering helps to more efficiently protect the arterial wall. IEEE Eng Med Biol Mag. 2007;26(1):62–70. doi: 10.1109/memb.2007.289123. [DOI] [PubMed] [Google Scholar]
- Armentano RL, Graf S, Barra JG, Velikovsky G, Baglivo H, Sanchez R, Simon A, Pichel RH, Levenson J. Carotid wall viscosity increase is related to intima-media thickening in hypertensive patients. Hypertension. 1998;31(1 Pt 2):534–539. doi: 10.1161/01.hyp.31.1.534. [DOI] [PubMed] [Google Scholar]
- Armentano RL, Levenson J, Barra JG, Fischer EI, Breitbart GJ, Pichel RH, Simon A. Assessment of elastin and collagen contribution to aortic elasticity in conscious dogs. Am J Physiol. 1991;260(6 Pt 2):H1870–H1877. doi: 10.1152/ajpheart.1991.260.6.H1870. [DOI] [PubMed] [Google Scholar]
- Aronson D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens. 2003;21(1):3–12. doi: 10.1097/00004872-200301000-00002. [DOI] [PubMed] [Google Scholar]
- Barbera JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21(5):892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- Boutouyrie P, Boumaza S, Challande P, Lacolley P, Laurent S. Smooth muscle tone and arterial wall viscosity: an in vivo/in vitro study. Hypertension. 1998;32(2):360–364. doi: 10.1161/01.hyp.32.2.360. [DOI] [PubMed] [Google Scholar]
- Boutten B, Brazier M, Morche N, Morel A, Vendeuvre JL. Effects of animal and muscle characteristics on collagen and consequences for ham production. Meat Science. 2000;55:233–238. doi: 10.1016/s0309-1740(99)00148-5. [DOI] [PubMed] [Google Scholar]
- Bruel A, Ortoft G, Oxlund H. Inhibition of cross-links in collagen is associated with reduced stiffness of the aorta in young rats. Atherosclerosis. 1998;140(1):135–145. doi: 10.1016/s0021-9150(98)00130-0. [DOI] [PubMed] [Google Scholar]
- Danpinid A, Luo J, Vappou J, Terdtoon P, Konofagou EE. In vivo characterization of the aortic wall stress-strain relationship. Ultrasonics. 2010;50(7):654–665. doi: 10.1016/j.ultras.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez J. Arterial stiffness and extracellular matrix. Adv Cardiol. 2007;44:76–95. doi: 10.1159/000096722. [DOI] [PubMed] [Google Scholar]
- Drexler ES, Bischoff JE, Slifka AJ, McCowan CN, Quinn TP, Shandas R, Ivy DD, Stenmark KR. Stiffening of the extrapulmonary arteries from rats in chronic hypoxic pulmonary hypertension. Journal of Research of the National Institute of Standards and Technology. 2008;113(4):239–249. doi: 10.6028/jres.113.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser KL, Tullis DE, Sasson Z, Hyland RH, Thornley KS, Hanly PJ. Pulmonary hypertension and cardiac function in adult cystic fibrosis: role of hypoxemia. Chest. 1999;115(5):1321–1328. doi: 10.1378/chest.115.5.1321. [DOI] [PubMed] [Google Scholar]
- Fratzl P, editor. Collagen structure and mechancis. New York, NY: Springer; 2008. [Google Scholar]
- Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest. 2007;132(6):1906–1912. doi: 10.1378/chest.07-1246. [DOI] [PubMed] [Google Scholar]
- Grignola JC, Gines F, Bia D, Armentano R. Improved right ventricular-vascular coupling during active pulmonary hypertension. Int J Cardiol. 2007;115(2):171–182. doi: 10.1016/j.ijcard.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Hiestand D, Phillips B. The overlap syndrome: chronic obstructive pulmonary disease and obstructive sleep apnea. Crit Care Clin. 2008;24(3):551–563. doi: 10.1016/j.ccc.2008.02.005. vii. [DOI] [PubMed] [Google Scholar]
- Humphrey JD. Remodeling of a collagenous tissue at fixed lengths. J Biomech Eng. 1999;121(6):591–597. doi: 10.1115/1.2800858. [DOI] [PubMed] [Google Scholar]
- Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intramural stress. Hypertension. 2008a;52(2):195–200. doi: 10.1161/HYPERTENSIONAHA.107.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD. Vascular adaptation and mechanical homeostasis at tissue, cellular, and sub-cellular levels. Cell Biochem Biophys. 2008b;50(2):53–78. doi: 10.1007/s12013-007-9002-3. [DOI] [PubMed] [Google Scholar]
- Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech. 2009;42(1):1–8. doi: 10.1016/j.jbiomech.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intengan HD, Thibault G, Li JS, Schiffrin EL. Resistance artery mechanics, structure, and extracellular components in spontaneously hypertensive rats : effects of angiotensin receptor antagonism and converting enzyme inhibition. Circulation. 1999;100(22):2267–2275. doi: 10.1161/01.cir.100.22.2267. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Riley DJ, Frank MM, Trelstad RL, Frankel HM. Reduction of chronic hypoxic pulmonary hypertension in the rat by beta-aminopropionitrile. J Appl Physiol. 1984;57(6):1760–1766. doi: 10.1152/jappl.1984.57.6.1760. [DOI] [PubMed] [Google Scholar]
- Kobs RW, Chesler NC. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech Model Mechanobiol. 2006;5(4):217–225. doi: 10.1007/s10237-006-0018-1. [DOI] [PubMed] [Google Scholar]
- Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2005;288(3):H1209–H1217. doi: 10.1152/ajpheart.01129.2003. [DOI] [PubMed] [Google Scholar]
- Lakes RS. Viscoelastic Solids. Boca Raton, Florida: CRC Press LLC; 1999. [Google Scholar]
- Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, Hofmeister S, Mecham R, Stenmark KR, Shandas R. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol. 2008;295(4):H1451–H1459. doi: 10.1152/ajpheart.00127.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Last JA, Armstrong LG, Reiser KM. Biosynthesis of collagen crosslinks. Int J Biochem. 1990;22(6):559–564. doi: 10.1016/0020-711x(90)90031-w. [DOI] [PubMed] [Google Scholar]
- Learoyd BM, Taylor MG. Alterations with age in the viscoelastic properties of human arterial walls. Circ Res. 1966;18(3):278–292. doi: 10.1161/01.res.18.3.278. [DOI] [PubMed] [Google Scholar]
- Li M, Stenmark KR, Shandas R, Tan W. Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J Vasc Res. 2009;46(6):561–571. doi: 10.1159/000226224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, Jeffrey J, Krane S, Jaenisch R. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J Cell Biol. 1995;130(1):227–237. doi: 10.1083/jcb.130.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra S, Nishimura RA, Sorajja P, Cha S, McGoon MD. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Coll Cardiol. 2006;47(4):799–803. doi: 10.1016/j.jacc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- Merklinger SL, Wagner RA, Spiekerkoetter E, Hinek A, Knutsen RH, Kabir MG, Desai K, Hacker S, Wang L, Cann GM, Ambartsumian NS, Lukanidin E, Bernstein D, Husain M, Mecham RP, Starcher B, Yanagisawa H, Rabinovitch M. Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary hypertension. Circ Res. 2005;97(6):596–604. doi: 10.1161/01.RES.0000182425.49768.8a. [DOI] [PubMed] [Google Scholar]
- Milnor WR, Conti CR, Lewis KB, O'Rourke MF. Pulmonary arterial pulse wave velocity and impedance in man. Circ Res. 1969;25(6):637–649. doi: 10.1161/01.res.25.6.637. [DOI] [PubMed] [Google Scholar]
- Mithieux SM, Weiss AS. Elastin. Adv Protein Chem. 2005;70:437–461. doi: 10.1016/S0065-3233(05)70013-9. [DOI] [PubMed] [Google Scholar]
- Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010 doi: 10.1152/ajpheart.00493.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BJ, Franklin CL, Hsieh FH, Bank RA, Phillips CL. Alpha 2(I) collagen deficient oim mice have altered biomechanical integrity, collagen content, and collagen crosslinking of their thoracic aorta. Matrix Biol. 2005;24(7):451–458. doi: 10.1016/j.matbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Poiani GJ, Tozzi CA, Yohn SE, Pierce RA, Belsky SA, Berg RA, Yu SY, Deak SB, Riley DJ. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ Res. 1990;66(4):968–978. doi: 10.1161/01.res.66.4.968. [DOI] [PubMed] [Google Scholar]
- Silver FH, Horvath I, Foran DJ. Viscoelasticity of the vessel wall: the role of collagen and elastic fibers. Crit Rev Biomed Eng. 2001;29(3):279–301. doi: 10.1615/critrevbiomedeng.v29.i3.10. [DOI] [PubMed] [Google Scholar]
- Silver FH, Snowhill PB, Foran DJ. Mechanical behavior of vessel wall: a comparative study of aorta, vena cava, and carotid artery. Ann Biomed Eng. 2003;31(7):793–803. doi: 10.1114/1.1581287. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006a;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006b;99(7):675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- Strauss BH, Robinson R, Batchelor WB, Chisholm RJ, Ravi G, Natarajan MK, Logan RA, Mehta SR, Levy DE, Ezrin AM, Keeley FW. In vivo collagen turnover following experimental balloon angioplasty injury and the role of matrix metalloproteinases. Circ Res. 1996;79(3):541–550. doi: 10.1161/01.res.79.3.541. [DOI] [PubMed] [Google Scholar]
- Tabima DM, Chesler NC. The effects of vasoactivity and hypoxic pulmonary hypertension on extralobar pulmonary artery biomechanics. J Biomech. 2010;43(10):1864–1869. doi: 10.1016/j.jbiomech.2010.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am J Respir Crit Care Med. 1994;149(5):1317–1326. doi: 10.1164/ajrccm.149.5.8173773. [DOI] [PubMed] [Google Scholar]
- Zhao J, Day J, Yuan ZF, Gregersen H. Regional arterial stress-strain distributions referenced to the zero-stress state in the rat. Am J Physiol Heart Circ Physiol. 2002;282(2):H622–H629. doi: 10.1152/ajpheart.00620.2000. [DOI] [PubMed] [Google Scholar]
- Zhao W, Byrne MH, Boyce BF, Krane SM. Bone resorption induced by parathyroid hormone is strikingly diminished in collagenase-resistant mutant mice. J Clin Invest. 1999;103(4):517–524. doi: 10.1172/JCI5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulliger MA, Fridez P, Hayashi K, Stergiopulos N. A strain energy function for arteries accounting for wall composition and structure. J Biomech. 2004;37(7):989–1000. doi: 10.1016/j.jbiomech.2003.11.026. [DOI] [PubMed] [Google Scholar]