

Abstract
Background
Dilated cardiomyopathy (DCM) is a heritable, genetically heterogeneous disorder, typically exhibiting autosomal dominant inheritance. Genomic strategies enable discovery of novel, unsuspected molecular underpinnings of familial DCM. We performed genome-wide mapping and exome sequencing in a unique family wherein DCM segregated as an autosomal recessive (AR) trait.
Methods and Results
Echocardiography in 17 adult descendants of first cousins revealed DCM in two female siblings and idiopathic left ventricular enlargement in their brother. Genotyping and linkage analysis mapped an AR DCM locus to chromosome 7q21, which was validated and refined by high-density homozygosity mapping. Exome sequencing of the affected sisters was then employed as a complementary strategy for mutation discovery. An iterative bioinformatics process was used to filter >40,000 genetic variants, revealing a single shared homozygous missense mutation localized to the 7q21 critical region. The mutation, absent in HapMap, 1000Genomes and 474 ethnically matched controls, altered a conserved residue of GATAD1, encoding GATA zinc finger domain-containing protein 1. Thirteen relatives were heterozygous mutation-carriers with no evidence of myocardial disease, even at advanced ages. Immunohistochemistry demonstrated nuclear localization of GATAD1 in left ventricular myocytes, yet subcellular expression and nuclear morphology were aberrant in the proband.
Conclusions
Linkage analysis and exome sequencing were used as synergistic genomic strategies to identify GATAD1 as a gene for AR DCM. GATAD1 binds to a histone modification site that regulates gene expression. Consistent with murine DCM caused by genetic disruption of histone deacetylases, our data implicate an inherited basis for epigenetic dysregulation in human heart failure.
Keywords: Cardiomyopathy, Genetics, Genomics, Epigenetics, Next generation sequencing
Introduction
Idiopathic dilated cardiomyopathy (DCM) is a familial disorder in 20% to 48% of cases1-5 providing a rationale for screening echocardiography in at-risk relatives to detect presymptomatic disease.6 Moreover, recognition of DCM as a heritable condition has been the impetus for human genetic investigations to uncover the molecular bases of myopathic heart failure.7 Since 1993 pathogenic mutations in over 30 genes have been identified in patients with DCM, yet mutations in these genes are estimated to account for only a third of cases.6 The majority of DCM genes have been discovered by targeted DNA sequence analyses of candidate genes. However, a vast number of genes are expressed in the heart and hypothesis-based approaches, by definition, cannot reveal unsuspected molecular underpinnings of disease.
DCM gene mapping by genome-wide linkage analysis8 offers a powerful alternative strategy, enabling discovery of novel, unanticipated disease genes. Furthermore, pathogenicity of a mutation can be robustly validated by demonstrating co-segregation with DCM. This approach is limited to rare multigenerational families in which DCM segregates as a Mendelian, single-gene disorder. In suitable families, the ultimate success of linkage analysis largely depends on the physical size of the mapped chromosomal locus and the number of genes it comprises. To date, X-chromosome and whole-genome linkage analyses have led to the identification of 1 gene for X-linked DCM9 and 7 genes for autosomal dominant DCM,10-16 respectively. Each encoded protein has a distinct subcellular location in cardiac myocytes: DMD, membrane-associated cytoskeleton; LMNA, nuclear membrane; TNNT2, thin filament of sarcomere; SCN5A, sarcolemma; TTN, sarcomeric cytoskeleton; EYA4, nucleus; MYH7, thick filament of sarcomere; RBM20, spliceosome. Accordingly, the pathobiology of DCM as revealed by linkage studies is attributable to mutations that perturb contractile force generation/transmission, nuclear structure, ion homeostasis, and messenger RNA transcription/splicing.
Next generation genomic technologies have recently emerged, providing unprecedented opportunities to complement and overcome inherent limitations of traditional DCM gene discovery strategies. Microarray-based comparative genomic hybridization17 enables detection of aneuploidy (gains or losses of chromosomal material) with a resolution approximately 100-fold higher than a conventional chromosome study. This “molecular karyotype” has become a routine clinical diagnostic tool and recently uncovered a large deletion in BAG3, encoding a cochaperone of heat shock proteins, as a cause of DCM.18 In 2009, advances in exon capture and massively parallel high-throughput DNA sequencing enabled scanning of the protein-coding regions of virtually all genes, which comprise ~1-2% of the genome.19 Several recent reports have highlighted the success of this powerful technique in identifying both known and novel molecular genetic bases for human disease.20 In particular, exome sequencing has proved an efficient strategy for discovery of homozygous recessive mutations in unanticipated disease genes.21
Here, we identified and phenotypically characterized a consanguineous family with nonsyndromic DCM, which segregated as an autosomal recessive trait. Linkage analysis, homozygosity mapping, and whole-exome sequencing were employed as synergistic, large-scale genomic strategies to identify GATAD1 as a novel disease gene, thus implicating epigenetic perturbation as an underlying basis for human heart failure.
Methods
Study Subjects
Patients with DCM evaluated at Mayo Clinic since 1987 and their relatives were recruited for screening echocardiograms and molecular genetic investigations. We enrolled 273 unrelated probands; enriched for familial DCM (confirmed or suspected in 51%). The multigenerational family in this study is White and of northern European ancestry by self-reporting. An ethnically matched group of 474 control subjects with normal echocardiograms was randomly selected from a community-based cohort.22 Subjects provided written informed consent under a research protocol approved by the Mayo Clinic Institutional Review Board. Diagnostic criteria for DCM were: lack of an identifiable cause for disease, left ventricular diastolic and/or systolic dimensions >95th percentile indexed for body surface area,23 and left ventricular ejection fraction <50%. Idiopathic left ventricular enlargement (LVE) was considered an intermediate phenotype of DCM and individuals with LVE were classified as “affected.”1,4,24,25 Genomic deoxyribonucleic acid (DNA) was isolated from peripheral-blood white cells. Paraffin embedded cardiac biopsy tissue from the proband was available for further analysis.
Linkage Analysis
Nineteen family members underwent genome-wide linkage analysis with the ABI PRISM Linkage Mapping Set HD5, Version 2.5 (Applied Biosystems, Foster City, CA). Polymerase chain reactions (PCR) were carried out using DNA samples and primer pairs that flanked 811 unique short tandem repeat (STR) markers. Amplified fragments were resolved on an ABI PRISM 3130xl, and genotypes were scored with GeneMapper Software (Applied Biosystems). Multipoint linkage analyses were performed with Simwalk2 Version 2.91,26 modeling recessive inheritance and specifying the following variables: phenocopy rate 0.001, equal marker allele frequencies, dichotomous liability classes (“affected” and “unaffected”). Secondary analyses were performed after reclassifying an individual with LVE as “uncertain.”
Homozygosity Mapping
The nine children of consanguineous parents were genotyped using the Omni 1M Quad SNV (single nucleotide variant) Chip (Illumina, San Diego, CA), with a mean call rate of 99.8%. A subset of 445,875 informative SNVs were analyzed with PLINK Version 1.07 software (homozyg)27 to identify genomic regions of homozygosity (≥100 adjacent SNVs) shared among three affected siblings. Secondary analysis was performed to identify homozygous intervals present only in two affected sisters. Large scale chromosomal abnormalities were screened for by analyzing B allele frequency plots as well as any areas where the Log R Ratio deviated from zero. Analysis was also performed using PennCNV28 to identify copy number variation within the genome.
Exome Sequencing and Bioinformatics Analysis
Exome sequencing and variant annotation were performed utilizing the Mayo Clinic Advanced Genomics Technology Center and Bioinformatics Core facilities. Following exome capture with the SureSelect Target Enrichment System (Version 1.0; Agilent, Santa Clara, CA), 76-base pair paired end sequencing was carried out on Illumina’s GAIIx platform. The reads were aligned to human genome 36 (hg18). For mappable reads, “Mapping and Assembly with Quality” (MAQ) was used for the detection of SNVs.29 To identify insertions and deletions (INDELs), a gapped alignment to the reference genome was tolerated by using “Burrows-Wheeler Alignment” (BWA).30 See Supplemental Material detailing quality control for mapping and variant calling. SNVs were called within MAQ while INDELs were called using the Genome Analysis Toolkit (GATK).31 Allele frequencies were generated for known variants in HapMap32 and 1000Genomes33 databases. Classification and annotation of genetic variants was accomplished using both “Sorting Intolerant From Tolerant” (SIFT)34 and SeattleSeq35 software.
Mutation Scanning and Sanger Sequencing
Gene expression profiles were assessed by searching the Gene Expression Omnibus (GEO) link on the NCBI website,36 analyzing data derived from Affymetrix GeneChip array data for 79 normal human tissues (GDS596), 61 normal mouse tissues (GDS592), and murine heart tissue at various stages of embryonic development (GDS627). Primer pairs were designed to encompass the five translated exons and flanking splice junctions of GATAD1, and 1 KB of 5’ UTR, using Oligo Primer Analysis Software, Version 6.71 (Molecular Biology Insights, Cascade, CO). PCR-amplified products were treated with ExoSAP-IT (USB Corp., Cleveland, OH) and sequenced by the dye-terminator method using an ABI PRISM 3730xl DNA Analyzer (Applied Biosystems). The DNA sequences were viewed and analyzed with Sequencher, Version 4.5 DNA analysis software (Gene Codes Corp., Ann Arbor, MI).
Each exon was evaluated for heterozygous sequence variation in our DCM cohort using denaturing high-performance liquid chromatography (DHPLC) heteroduplex analysis (WAVE DHPLC System, Transgenomic, Omaha, NE). Because PCR reagents for optimal amplification of exon 1 were not compatible with DHPLC, Sanger sequencing was performed on all samples, as well as the 1 KB 5’ UTR in one affected individual. Exon 2, containing the identified S102P variant, was also screened in 474 ethnically matched control samples. The buffer gradient and column melting temperature(s) for DHPLC were determined with Transgenomic Navigator software (Version 1.7.0 Build 25) and experimental optimization. Chromatographic elution profiles of samples yielding traces different than the wildtype homoduplex pattern were selected for Sanger sequencing.
Immunohistochemistry
Ventricular endomyocardial biopsy tissue from the proband was formalin fixed and paraffin embedded prior to sectioning at 4 μm for immunohistochemical staining. Three additional paraffin embedded specimens served as controls. Normal heart tissue was obtained at autopsy from the left ventricles of a female and male, both of whom died from noncardiac causes. In addition, left ventricular tissue was available from an individual with DCM due to a mutation in TPM1.37 Paraffin slides were deparaffinized and pretreated in a 97°C solution (1 mmol/L EDTA, pH 8.0) for 30 minutes using a PT Link (DAKO, Carpinteria, CA). Optimization of the antibody and the final staining was carried out by an automated immunohistochemistry-staining machine (DAKO Autostainer Plus, DAKO). GATAD1 protein expression was evaluated with a mouse monoclonal anti-human GATAD1 antibody recognizing amino acids 129-261 (Abcam, Cambridge, MA). The antibody was diluted 1:25 for a 30 minute incubation using ADVANCE, a highly sensitive 2-step polymer system from DAKO. A mouse monoclonal antihuman antibody against the α and γ isoforms of muscle actin (DAKO) was diluted 1:150 and incubated for 30 minutes, using DAKO Dual polymer chemistry. For both antibodies, the chromogen used for detection was diaminobenzidine (DAB) (DAKO). A negative control underwent the same procedure, but without the addition of primary antibody. All slides were counterstained with hematoxylin and cover slipped for microscopic examination.
Results
Family Phenotype
The proband (III.12, Figure 1 and Table 1) presented at age 50 years with heart failure and cardiomegaly. Her medical history was negative for hypertension, diabetes, and tobacco or alcohol use. Echocardiography was diagnostic for DCM and angiography ruled out coronary artery disease. Left ventricular endomyocardial biopsy revealed moderate myocyte hypertrophy, mild focal interstitial fibrosis, and mild diffuse endocardial fibrosis, consistent with a chronic cardiomyopathy. Her medical therapy included digoxin, furosemide, carvedilol, enalapril, spironolactone, and warfarin (for apical thrombus). She has had no history of arrhythmia and, 24 years after her initial presentation, is in NYHA class II heart failure with moderate-severe left ventricular enlargement and ejection fraction of 25%.
Figure 1.

Pedigree structure and chromosome 7q21 haplotypes for family with autosomal recessive dilated cardiomyopathy. * = proband; square = male; circle = female; solid = DCM; half symbol = idiopathic left ventricular enlargement; open = unaffected; gray = uncertain; slash through the symbol = deceased with age at death indicated for generations I and II. Markers are listed from centromere to q telomere, with map locations according to the National Center for Biotechnology Information website (36.3 Build) and given in megabases (MB) and centimorgans (cM). Haplotypes for marker genotypes are shown in columns below pedigree symbols and are unambiguously inferred for individuals without DNA samples (II.1, II.2, III.7). Consistent with homozygous recessive inheritance, identical disease-associated haplotypes (boxed) are inherited by the three affected family members from their first-cousin parents. Recombination events account for homozygous inheritance of portions of the haplotype in an unaffected (III.4) and affected (III.6) individual, defining D7S669 as the upper flanking marker. The lower flanking marker, D7S515, is defined by heterozygous genotypes in all three affected individuals (III.6, III.10, and III.12). GATAD1 maps within this 24 MB DCM locus. Presence of the identified GATAD1-S102P mutation is indicated by a plus symbol and its absence indicated by a minus symbol. Clinically affected individuals are homozygous for the mutation.
Table 1.
Phenotypic and Genetic Data for Family with Autosomal Recessive Dilated Cardiomyopathy
| Pedigree Number | Age at Diagnosis/ Evaluation, y | LVDD, mm* | LVSD, mm* | EF, % | Coronary Angiography | Status | Cardiac Phenotype | S102P Mutation in GATAD1 |
|---|---|---|---|---|---|---|---|---|
| III.1 | -/69 | Normal | Normal | 55 | - | Alive 91 y | Unaffected | Normal |
| III.2 | -/81 | 43 (52) | 28 (35) | 62 | - | Alive 87 y | Unaffected | Heterozygous |
| III.3 | -/64** | 50 (53) | 32 (35) | 64 | - | Alive 86 y | Unaffected | Heterozygous |
| III.4 | -/79 | 47 (51) | 33 (34) | 60 | - | Alive 83 y | Unaffected | Heterozygous |
| III.5 | -/60 | Normal | Normal | 55 | - | Died 70 y, PD | Unaffected | Heterozygous |
| III.6 | 57/60 | 59 (55) | 40 (36) | 54 | 30% RCA | Died 73 y, CA | LVE | Homozygous |
| III.8 | -/55 | 46 (52) | 30 (34) | 60 | - | Alive 77 y | Unaffected | Heterozygous |
| III.10 | 53/53 | 53 (52) | 40 (34) | 43 | 10% RCA | Alive 76 y, NYHA II, AF | DCM | Homozygous |
| III.12 | 50/50 | 60 (53) | 53 (35) | 21 | No CAD | Alive 74 y, NYHA II | DCM | Homozygous |
| IV.1 | -/29 | 47 (52) | 30 (35) | 64 | - | Alive 51 y | Unaffected | Heterozygous |
| IV.2 | -/26 | 48 (52) | 31 (35) | 62 | - | Alive 47 y | Unaffected | Heterozygous |
| IV.3 | -/25 | 44 (53) | 29 (35) | 57 | - | Alive 43 y | Unaffected | Heterozygous |
| IV.4 | -/39 | 53 (56) | 36 (37) | 56 | - | Alive 49 y | Unaffected | Heterozygous |
| IV.5 | -/28 | 50 (53) | 35 (35) | 56 | - | Alive 48 y | Unaffected | Heterozygous |
| IV.6 | -/24 | 54 (56) | 37 (37) | 54 | - | Alive 46 y | Unaffected | Heterozygous |
| IV.7 | -/22 | 55 (55) | 37 (36) | 55 | - | Alive 44 y | Unaffected | Heterozygous |
| IV.8 | -/17 | 53 (54) | 36 (37) | 58 | - | Alive 39 y | Unaffected | Heterozygous |
LVDD, left ventricular diastolic dimension; LVSD, left ventricular systolic dimension; EF, left ventricular ejection fraction; RCA, right coronary artery occlusion; CAD, coronary artery disease; PD, Parkinson’s disease; CA, cancer; NYHA, New York Heart Association; AF, atrial fibrillation; LVE, idiopathic left ventricular enlargement; DCM, idiopathic dilated cardiomyopathy.
The 95th percentiles for left ventricular dimensions, based on body surface area and age, are indicated in parentheses. Abnormal measurements are in bold font.
EF was 51% on Technetium Sestamibi scan at age 79
The extended family is of Norwegian ancestry with initial history negative for cardiomyopathy, heart failure, and sudden death. However, the parents of the proband were first cousins (II.1 and II.2) who died at advanced ages without apparent heart disease, suggesting a recessive form of DCM. Screening echocardiograms were performed in 16 of her relatives, and genomic DNA samples were procured. Two of eight asymptomatic siblings of the proband were found to have cardiomyopathy in the absence of established risk factors for coronary and myopathic heart disease. A sister (III.10) was diagnosed with DCM at 53 years of age. Her treatment included furosemide, metoprolol, losartan, spironolactone, warfarin, and AV node ablation with pacemaker implantation for persistent atrial fibrillation. She has remained in NYHA class II heart failure 23 years after diagnosis with mild left ventricular enlargement and ejection fraction of 40%. A 57-year-old brother (III.6) was diagnosed with idiopathic left ventricular enlargement, a known intermediate phenotype of DCM,1,4,24,25 confirmed by a follow up echocardiogram 3 years later. He was treated with furosemide, metoprolol, and captopril. At age 68 years, he had cardiomegaly on chest radiography and a borderline ejection fraction of 50% on left ventricular angiography. He died of cancer at age 73 years. The remaining six siblings (III.1-5, 8), ages 55 to 81 years, had completely normal echocardiograms and none developed heart failure over 4 to 22 years of follow up. DCM was also excluded in all 8 offspring of the 3 affected siblings, and none have developed heart failure over 20 years of follow up. While male-male transmission was not observed, X-linked recessive inheritance was effectively ruled out based on the milder cardiomyopathy phenotype in the brother compared to his affected sisters. In summary, the family history and clinical evaluations indicated that nonsyndromic DCM was inherited as an autosomal recessive trait.
DCM Locus Mapping
Genome-wide linkage analysis specifying a homozygous recessive mode of inheritance identified a DCM locus on chromosome 7q21 with a peak multipoint logarithm of the odds (LOD) score of 3.1 between markers D7S669 and D7S515 (Figure 2). Construction of haplotypes revealed homozygosity for 2 adjacent STR markers in the 3 affected siblings but none of the 6 unaffected siblings. Flanking markers defined a region of 24 MB that encompassed 258 genes (Figure 1). A more conservative approach, where the phenotype of III.6 was adjusted to “uncertain,” resulted in a peak multipoint LOD score of 1.9 at the same 7q21 locus (data not shown). In separate linkage analyses specifying a dominant mode of inheritance; 60%, 80%, or 100% penetrance; and classification of subject III.6 as “affected” or “uncertain,” no locus with a multipoint LOD score ≥1.3 (P-value ≤0.05) was identified.
Figure 2.
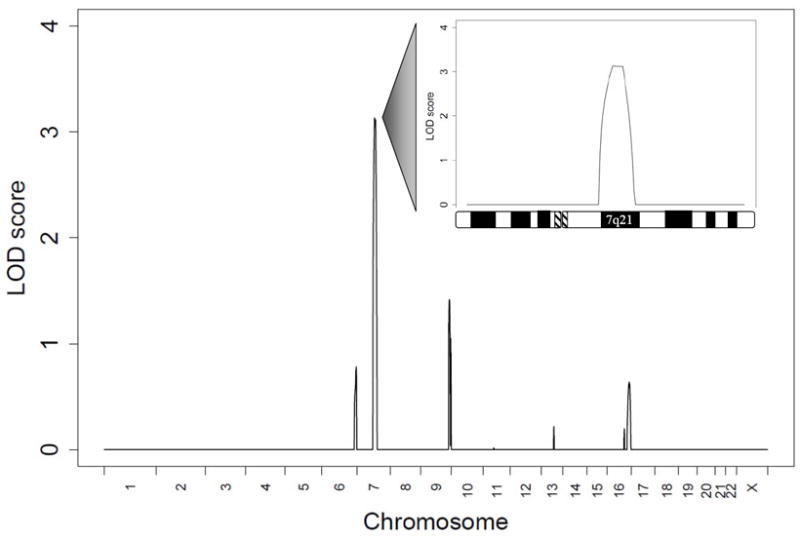
Genome-wide linkage analysis revealed a locus for autosomal recessive DCM on chromosome 7, with a peak multipoint LOD score of 3.1 (>1,000:1 odds). The graph illustrates the peak multipoint LOD scores for individual chromosomes with the lower threshold set at zero. The inset details the LOD score plot for chromosome 7, highlighting the linked 7q21 region flanked by markers D7S669 (78 MB) and D7S515 (102 MB).
Homozygosity Mapping
High-density homozygosity mapping revealed a contiguous block of homozygous SNVs at the 7q21 locus, verifying that this region harbored an AR DCM gene and significantly narrowing the size of the critical region (Figure 3). An identical block of 1900 homozygous SNVs spanning 15.9 MB was identified at 7q21 in the three affected siblings. An unaffected individual (III.4) was homozygous for the identical haplotype over an 8.6 Mb portion of this region (1011 contiguous SNVs), thus refining the centromeric boundary of the critical region. Among the five other unaffected siblings, four were heterozygous for the DCM-associated haplotype and one inherited no copies. The 7q21 locus was the only region of shared homozygosity among the three affected individuals. The more conservative approach, in which only the two affected sisters were analyzed, revealed five additional homozygous blocks on four chromosomes. However, each haplotype was shared by at least one unaffected sibling (Supplemental Table 1). Homozygosity mapping enabled a three-fold reduction in the size of the 7q21 locus, a 7.3 MB critical region comprised of 61 gene candidates. Further analysis of the SNV chip data revealed no instances of copy number variants, i.e sub-chromosomal deletions or duplications, shared among the three affected individuals.
Figure 3.
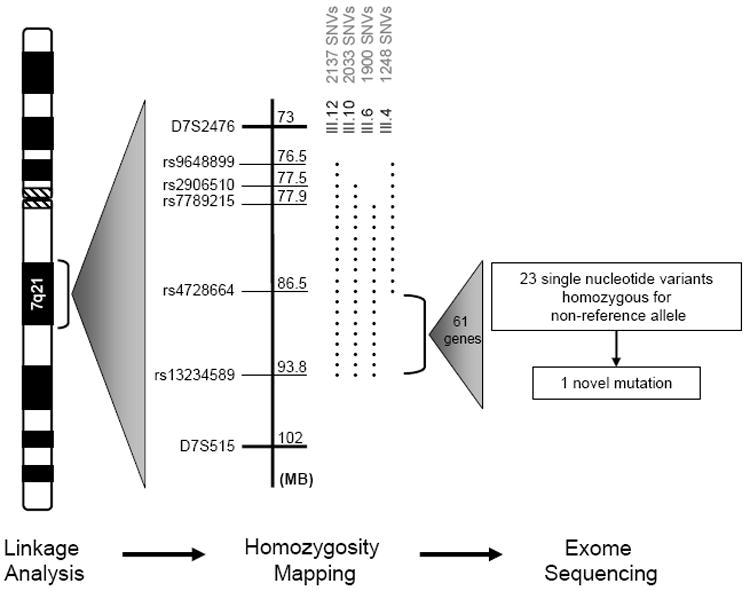
Genome-wide homozygosity mapping revealed a single region of homozygosity on chromosome 7q21 in the three affected siblings. Adjacent to the chromosome ideogram is a depiction of the SNVs for these individuals (III.12, III.10, and III.6) and an unaffected individual (III.4), which enabled fine mapping of the critical region. The dotted lines represent blocks of contiguous, homozygous SNVs identified in each family member. The unaffected individual (III.4) shared a portion of the homozygous block with her affected siblings, thus establishing the upper flank for the locus. The lower flank was defined by the telomeric ends of the homozygous blocks in the three affected individuals. A block of 889 contiguous, homozygous SNVs defined the 7.3 MB critical region encompassing 61 genes. Subsequent analysis of exome sequencing data within this region identified a total of 23 SNVs homozygous for the non-reference allele. All but one SNV were present in HapMap or 1000Genomes and several of our 16 other exomes.
Exome Sequencing and Bioinformatics Analysis
Genomic DNA samples from two of the affected individuals (III.10 and III.12) were submitted for whole-exome sequencing following targeted exome capture. Each sample was sequenced in two lanes of the same flow cell, yielding sixty-five million pairs of nucleotide sequences from paired end, 76-base pair reads. Data analysis determined that 86% of the sequence mapped to the genome and 50% was found within targeted regions. Representation of the exome was the same in both individuals, with 73% and 92% coverage of the targeted regions demonstrating a minimal read depth of 20-fold and 5-fold, respectively.
Detected SNVs and INDELs were annotated and loaded onto a spreadsheet, enabling an iterative filtering process to identify the pathogenic mutation (Figure 4). Largely due to the homozygous recessive mode of inheritance, extensive unbiased filtering was possible without taking into account the chromosome 7q21 critical region or predicting the functional impact of nonsynonymous variants. We first excluded SNVs reported in HapMap, 1000Genomes, or 16 exomes of individuals sequenced in our core facility. We then filtered the remaining novel variants by removing synonymous SNVs, intergenic variants, and intronic variants not localized to splice sites. Non-silent SNVs were thus defined as missense, nonsense, and INDEL coding variants, and variants residing within UTRs or canonical splice donor/acceptor sites. Based on the recessive mode of inheritance, the next filter excluded heterozygous SNVs. We subsequently narrowed the list of remaining homozygous SNVs to those shared by the affected sisters. Our filtering process culminated in a restricted list of only 3 homozygous missense variants, each of which was verified by Sanger sequencing and absent from the Human Gene Mutation Database. Variants in SETD1A and MYLK3 were each localized to a region of chromosome 16 identified by affected sisters-only homozygosity mapping but, in both cases, a phenotypically normal elderly sibling (III.3) was homozygous for the variant (Supplemental Figure 1). These variants were thus ruled out as having a primary, causative role in AR DCM, although a disease modifying effect can not be excluded.
Figure 4.
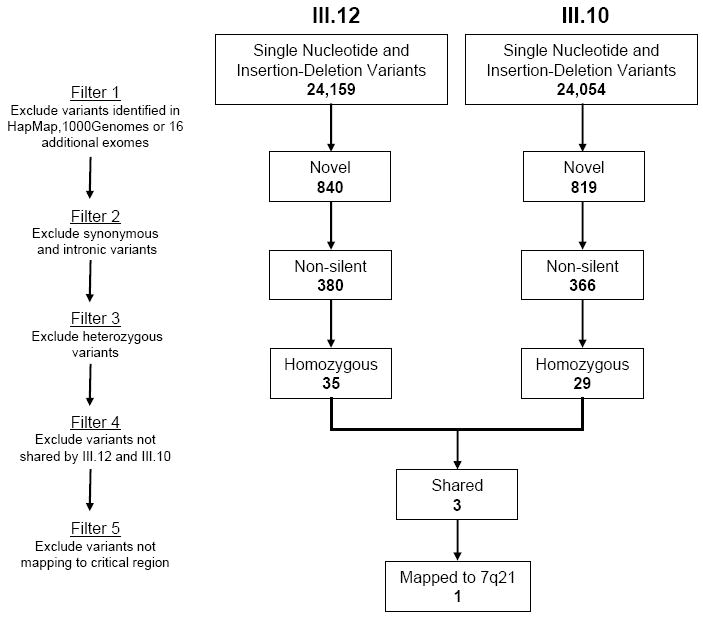
To identify a pathogenic mutation that underlies autosomal recessive DCM, an iterative filtering process was applied to SNVs and INDELs identified in III.12 and III.10 by exome sequencing. The list of potential candidate genes was narrowed to three, each harboring a novel, non-silent, homozygous SNV shared by the affected sisters. Only one of these – GATAD1-S102P - mapped to the critical region on chromosome 7q21.
A solitary mutation in GATAD1 remained, for which the three affected siblings were homozygous and which mapped to the 7q21 critical region. The six unaffected siblings were either heterozygous or homozygous for the normal allele. The eight unaffected offspring of affected individuals were obligate heterozygous carriers of the mutation. The identified missense mutation was a c.304 T>C nucleotide transition in exon 2 of GATAD1, encoding GATA zinc finger domain-containing protein 1 (Figure 5). The resultant S102P substitution replaces a polar hydroxyl serine with a nonpolar cyclic proline. No additional mutations in GATAD1 were identified in the coding region or 5’ UTR of the proband. In addition to being absent from the large variant databases used in the filtering process, the mutation was not present in 474 healthy control samples (948 chromosomes) of similar ancestry. To determine if heterozygous mutations in GATAD1 underlie autosomal dominant or sporadic DCM, the 5 exons of this gene were screened in 273 unrelated subjects. Except for two reported polymorphisms in exons 1 and 5, (G54S and R233W), there were no instances of unreported nonsynonymous variants within GATAD1, indicating highly conserved nucleotide and protein sequences. Together with control samples, the S102P mutation was absent in a total of 1,558 chromosomes. Furthermore, examination of the exome sequencing data did not reveal homozygous mutations in more than 30 genes previously associated with DCM.6
Figure 5.
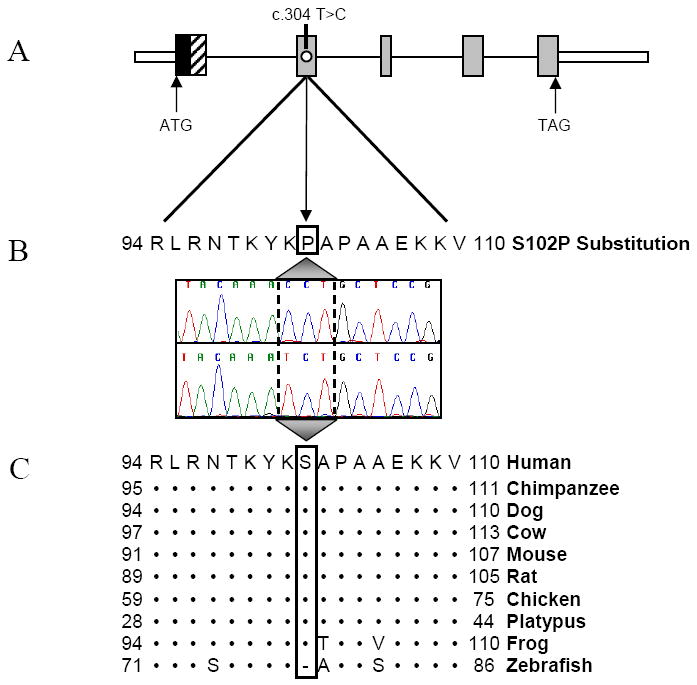
(A) The gene topology of GATAD1, including functional GATA-type Zinc Finger (black) and Glycine-Rich (diagonal lines) domains and the location of the homozygous mutation c.304 T>C in exon 2. ATG, start codon; TAG, stop codon. (B) Sanger sequencing verified the mutation, which results in an S102P substitution. (C) Conservation of this residue is illustrated whereby (•) indicates identical residues and (-) indicates a gap. Overall, the zebrafish and human homologues share only 73% protein identity and the N-terminus of the zebrafish homologue lacks the glycine-rich domain, implicated in protein-protein interactions and present in all other species.
GATAD1 Protein Localization
According to GEO array data, GATAD1 is ubiquitously expressed among various tissue types and in embryonic mouse hearts at all stages of development. To qualitatively verify cardiac expression, determine subcellular localization, and explore the mutation’s potential effect on histology, we performed immunohistochemistry using a monoclonal antibody against GATAD1. Within healthy left ventricular tissue, GATAD1 localized to the nucleus and cytoplasm with a homogenous, striated staining pattern (Figure 6A-B). A similar staining pattern was seen in the left ventricular myocardium of a patient with genetically defined DCM37 (Figure 6C). In contrast, homozygosity for the S102P in GATD1 resulted in a marked disturbance in its extranuclear distribution in the proband (Figure 6D). Actin staining of an adjacent tissue section (Figure 6E) was indistinguishable from control tissues and complete absence of staining in the negative control lent further support to the specificity of the antibody (data not shown). Additionally, myocyte nuclei in the proband had a unique globular morphology, contrasting with the elongated spindle shape observed in the controls.
Figure 6.
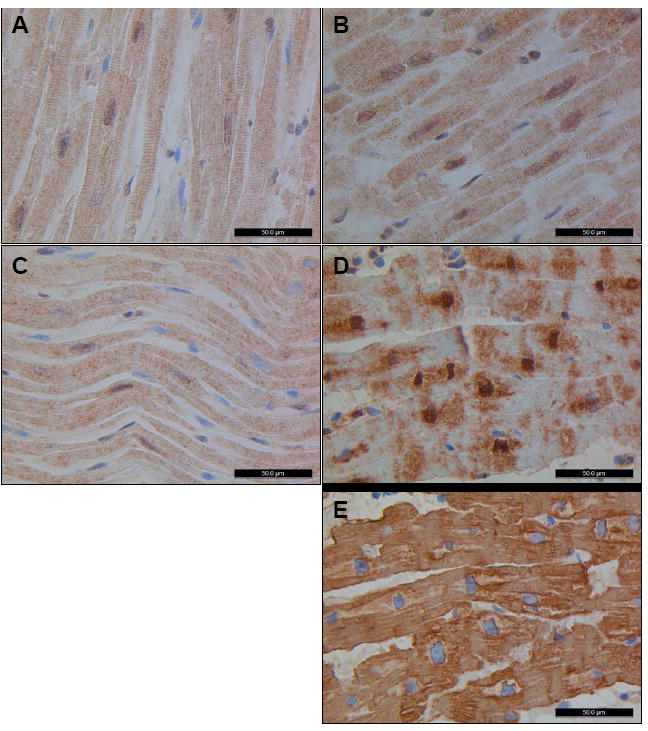
Immunohistochemistry was performed with a monoclonal antibody against GATAD1 on left ventricular tissue from four individuals. Normal tissue from a male (A) and a female (B) display nuclear staining of GATAD1 with a homogeneous striated pattern in the extranuclear space, similar to what was noted in tissue from an individual with DCM due to a mutation in TPM1 (C). In contrast, GATAD1 in biopsy tissue from the proband shows an abnormal staining pattern (D). While there is still nuclear localization, extranuclear distribution of GATAD1 is perturbed whereas actin staining of an adjacent section (E) was indistinguishable from normal controls (data not shown). In addition, the globular morphology of nuclei is unique from the spindle shape observed in control tissue.
Discussion
DCM is characterized by marked genetic and clinical heterogeneity.6 Knowledge of the molecular basis of this disorder continues to evolve with the identification of mutations in genes involved in distinct biological pathways. Here, we employed synergistic genomic strategies for hypothesis-free disease gene discovery in a family with autosomal recessive DCM, leading to identification of a pathogenic homozygous mutation in GATAD1, encoding GATA zinc finger domain-containing 1.
GATAD1 is a Reader of the Histone Code
Histones are the fundamental building blocks of chromatin, proteins that package and condense DNA into structural units known as nucleosomes. Histone tails serve as targets for multiple post-translational modifications, such as methylation, acetylation and phosphorylation, which modulate epigenetic gene expression. Over the past 10 years, increased knowledge of the histone code has revealed the biochemical complexity of gene regulation by multiple protein readers known to interact with individual modifications.38 GATAD1, a 269 amino acid protein with ubiquitous tissue expression, was recently shown to assemble with proteins of a chromatin complex that specifically interact with a prototypic epigenetic mark - H3K4Me3 (trimethylation of the fourth residue (lysine) on histone 3).39 Moreover, a role for H3K4Me3 in heart failure has been implicated based on a unique epigenetic profile in left ventricular myocardium of patients with DCM compared to normal heart tissue.40 While the specific effects of GATAD1-S102P on nucleosome protein interactions and downstream gene expression are unknown, the mutation leads to a heart-specific phenotype and implicates epigenetic dysregulation as a novel basis for human DCM. Functional studies will be required to gain further insight into the mechanisms by which the GATAD1 mutation causes DCM. GATAD1 mutations appear to be a rare cause for DCM, yet defects within other nucleosome interacting proteins may prove to be important in disease pathogenesis.
Epigenetics and Dilated Cardiomyopathy
The link between epigenetics and myopathic heart failure has been well established in genetically modified mouse models, best exemplified by dysregulation of histone deacetylases.41 In particular, conditional knockout of both Hdac1 and Hdac2 in the murine heart results in DCM.42 Modified conditional knockouts which leave one allele of either Hdac1 or Hdac2 intact result in phenotypically normal mice, demonstrating a capacity to compensate for haploinsufficiency. Remarkably, human HDAC1 and HDAC2 are binding partners with GATAD1,39 lending further support to perturbed nucleosome protein interactions in the pathogenesis of DCM.
Genotype-Phenotype Correlation
Unlike early-onset, clinically aggressive heart failure reported in rare families with proven43 or suspected44,45 autosomal recessive DCM, the phenotype in our family was similar to patients with autosomal dominant or sporadic forms of disease. In fact, the only clues for recessive inheritance were consanguinity, lack of heart failure in the ancestors of affected family members by history, and lack of DCM in offspring of affected family members by echocardiographic screening. Variable disease expression is a well-recognized phenomenon in familial DCM, attributable to modifier genes, environmental variables, or epigenetic factors. An intriguing clinical observation within our family is the overt DCM phenotype in the affected sisters versus the intermediate phenotype of left ventricular enlargement with preserved ejection fraction in the male sibling. This may be partially explained by differential cardiac expression of GATAD1 observed in men and women. Indeed, GATAD1 was one of few autosomal genes found to be more highly expressed in females than males with DCM and new-onset heart failure (>1.6-fold change).46 We have clearly shown that the GATAD1-S102P mutation is recessive, as 13 heterozygous carriers had normal cardiac phenotypes. Like Hdac1 and Hdac2, this indicates that GATAD1 can carry out its regulatory role with only one wildtype allele.
Disrupted Localization of GATAD1-S102P
Our immunohistochemistry experimentation confirmed that GATAD1 is expressed in left ventricular myocytes. Moreover, the GATAD1-S102P mutant protein was detected in the proband’s myocardium, ruling out deficiency of GATAD1 as a consequence of the underlying homozygous mutation. The similarity of GATAD1 staining patterns and nuclear morphology in TPM1-DCM and normal control tissues, distinct from the proband, indicates that GATAD1-S102P has a mutation-specific effect. On the other hand, abnormal nuclear morphology is a hallmark of DCM caused by knockout of Lmna, a gene that encodes a structural protein of the nuclear membrane.47 Previous work,36,39,40,46 together with our immunohistochemistry findings, strongly supports a role for GATAD1 in nuclei of cardiac myocytes. Extranuclear staining raises the further possibility of a broader role for GATAD1 in the heart.
Synergy of Locus Mapping and Exome Sequencing
While genome-wide linkage analysis and homozygosity mapping identified a single locus for AR DCM on chromosome 7q21, 61 positional candidate genes localized to the critical region. GATAD1, functionally distinct from known DCM genes, was not readily apparent as a compelling candidate. Exome sequencing of 2 affected family members and our iterative filtering process enabled us to analyze the entire exome and narrow the number of potential candidates to only 3 genes, prior to taking into account the mapping information. Filtering was significantly aided by the family’s homozygous recessive mode of inheritance, allowing a 10-fold reduction in candidate variants in step 3 (Figure 4). Successful identification of a pathogenic mutation in our family by whole-exome sequencing thus proved to be minimally reliant on positional information. However, the mapping data provided independent verification that the disease gene resided within the 7q21 locus. Moreover, extensive phenotypic characterization of 17 family members enabled unambiguous determination of an autosomal recessive mode of inheritance and robustly demonstrated co-segregation of homozygosity for the GATAD1 mutation with DCM.
Exome sequencing is poised to have a transformative impact on novel DCM gene discovery, yet a major challenge is the large number of novel variants present in individual exomes. In families with autosomal dominant DCM attributable to unidentified heterozygous mutations, additional filtering criteria would likely be required to identify disease-causing mutations, e.g. exclusion of variants in genes with low cardiac expression levels and exclusion of gene variants predicted to have non-damaging effects on protein structure and function.19 However, the most robust genetic evidence for causality would be derived from comparative analyses of exome sequencing data for several affected family members and/or complementary whole-genome mapping data to narrow the list of candidate variants to a discrete chromosomal locus.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the family and patients who participated in this study and the physicians who referred them. We thank Adele Goodloe for her critical review of the paper and Dr. Ahmet Dogan for his assistance with immunohistochemistry.
Funding Sources: This work was supported by NIH grants T32 HL007111 (JT), R01 HL36879 (VM), R01 HL071225 (TO), the Mayo Clinic Center for Individualized Medicine, a generous gift from James and Donna Barksdale, and a generous gift from the Marriott Family.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 2.Mestroni L, Krajinovic M, Severini GM, Pinamonti B, Di Lenarda A, Giacca M, Falaschi A, Camerini F. Familial dilated cardiomyopathy. Br Heart J. 1994;72:S35–41. doi: 10.1136/hrt.72.6_suppl.s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keeling PJ, Gang Y, Smith G, Seo H, Bent SE, Murday V, Caforio AL, McKenna WJ. Familial dilated cardiomyopathy in the United Kingdom. Br Heart J. 1995;73:417–421. doi: 10.1136/hrt.73.5.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. doi: 10.1016/s0735-1097(97)00433-6. [DOI] [PubMed] [Google Scholar]
- 5.Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 6.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12:655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson TM. Monogenic Dilated Cardiomyopathy. In: Walsh RA, editor. Molecular Mechanisms of Cardiac Hypertrophy and Failure. 1. Boca Raton: Taylor & Francis; 2005. pp. 525–540. [Google Scholar]
- 8.Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 10.Kass S, MacRae C, Graber HL, Sparks EA, McNamara D, Boudoulas H, Basson CT, Baker PB, Cody RJ, Fishman MC, Cox N, Kong A, Wooley CF, Seidman JG, Seidman CE. A gene defect that causes conduction system disease and dilated cardiomyopathy maps to chromosome 1p1-1q1. Nat Genet. 1994;7:546–551. doi: 10.1038/ng0894-546. [DOI] [PubMed] [Google Scholar]
- 11.Durand J-B, Bachinski LL, Bieling LC, Czernuszewicz GZ, Abchee AB, Yu QT, Tapscott T, Hill R, Ifegwu J, Marian AJ, Brugada R, Daiger S, Gregoritch JM, Anderson JL, Quiñones M, Towbin JA, Roberts R. Localization of a gene responsible for familial dilated cardiomyopathy to chromosome 1q32. Circulation. 1995;92:3387–3389. doi: 10.1161/01.cir.92.12.3387. [DOI] [PubMed] [Google Scholar]
- 12.Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996;97:528–532. doi: 10.1172/JCI118445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siu BL, Niimura H, Osborne JA, Fatkin D, MacRae C, Solomon S, Benson DW, Seidman JG, Seidman CE. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation. 1999;99:1022–1026. doi: 10.1161/01.cir.99.8.1022. [DOI] [PubMed] [Google Scholar]
- 14.Schönberger J, Levy H, Grünig E, Sangwatanaroj S, Fatkin D, MacRae C, Stäcker H, Halpin C, Eavey R, Philbin EF, Katus H, Seidman JG, Seidman CE. Dilated cardiomyopathy and sensorineural hearing loss: a heritable syndrome that maps to 6q23-24. Circulation. 2000;101:1812–1818. doi: 10.1161/01.cir.101.15.1812. [DOI] [PubMed] [Google Scholar]
- 15.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 16.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 18.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Züchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng SB, Nickerson DA, Bamshad MJ, Shendure J. Massively parallel sequencing and rare disease. Hum Mol Genet. 2010;19:R119–124. doi: 10.1093/hmg/ddq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redfield MM, Jacobsen SJ, Burnett JC, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community. Appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194–202. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]
- 23.Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62:1054–1061. doi: 10.1161/01.cir.62.5.1054. [DOI] [PubMed] [Google Scholar]
- 24.Michels VV, Olson TM, Miller FA, Ballman KV, Rosales AG, Driscoll DJ. Frequency of development of idiopathic dilated cardiomyopathy among relatives of patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 2003;91:1389–1392. doi: 10.1016/s0002-9149(03)00341-2. [DOI] [PubMed] [Google Scholar]
- 25.Fatkin D, Yeoh T, Hayward CS, Benson V, Sheu A, Richmond Z, Feneley MP, Keogh AM, Macdonald PS. Evaluation of left ventricular enlargement as a marker of early disease in familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:342–348. doi: 10.1161/CIRCGENETICS.110.958918. [DOI] [PubMed] [Google Scholar]
- 26.Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant S, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GATK. http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit.
- 32.HapMap. http://hapmap.ncbi.nlm.nih.gov.
- 33.1000 Genomes. http://www.1000genomes.org. Released 08/04/2011.
- 34.SIFT. http://sift.jcvi.org.
- 35.Seattle Seq. http://gvs.gs.washington.edu/SeattleSeq.Annotation/
- 36.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37(Database issue):D885–890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol. 2001;33:723–732. doi: 10.1006/jmcc.2000.1339. [DOI] [PubMed] [Google Scholar]
- 38.Gardner KE, Allis CD, Strahl BD. OPERating ON Chromatin, a Colorful Language where Context Matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, Misawa Y, Isomura T, Shimada K, Mano H. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14:69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 41.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 42.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy RT, Mogensen J, Shaw A, Kubo T, Hughes S, McKenna WJ. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet. 2004;363:371–372. doi: 10.1016/S0140-6736(04)15468-8. [DOI] [PubMed] [Google Scholar]
- 44.Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, McKenna WJ, Falaschi A, Giacca M, Camerini Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 45.Seliem MA, Mansara KB, Palileo M, Ye X, Zhang Z, Benson DW. Evidence for autosomal recessive inheritance of infantile dilated cardiomyopathy: studies from the Eastern Province of Saudi Arabia. Pediatr Res. 2000;48:770–775. doi: 10.1203/00006450-200012000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Heidecker B, Lamirault G, Kasper EK, Wittstein IS, Champion HC, Breton E, Russell SD, Hall J, Kittleson MM, Baughman KL, Hare JM. The gene expression profile of patients with new-onset heart failure reveals important gender-specific differences. Eur Heart J. 2010;31:1188–1196. doi: 10.1093/eurheartj/ehp549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.