

Abstract
Apoptosis occurs in many tissues, during both normal and pathogenic processes. Normally, apoptotic cells are rapidly cleared, either by neighboring or recruited phagocytes. The prompt clearance of apoptotic cells requires that the apoptotic cells announce their presence through the release of chemotactic factors, known as ‘find-me’ signals, to recruit phagocytes to the site of death, and through the exposure of so-called ‘eat-me’ signals, which are ligands for phagocytic uptake. The importance of prompt apoptotic cell clearance is revealed by findings that decreasing the efficiency of engulfment results in the persistence of apoptotic cells, which is often associated with chronic inflammation and autoimmunity. Additionally, the proper clearance of apoptotic cells is actively anti-inflammatory, which is thought to play a crucial role in immunologic tolerance. Therefore, defects associated with clearance of apoptotic cells may contribute to the pathogenesis of several inflammatory diseases, including autoimmunity and atherosclerosis. Here, we review the role of nucleotides in the apoptotic cell clearance process, and discuss their implications for disease pathogenesis.
Keywords: Apoptosis, Engulfment, Autoimmunity, Inflammation
Introduction
Apoptosis occurs in a variety of tissues during the development of an organism, tissue homeostasis, and pathogenic processes [1, 2]. During development, apoptosis plays a role in sculpting structures and getting rid of excess cells. Cell turnover is also a vital part of tissue homeostasis, as senescent cells are disposed of before they become less functional or even dysplastic. In addition to the normal processes, apoptotic cells are also observed in tumors [1], atherosclerotic plaques [3], and neurodegenerative diseases [4]. In healthy tissues, the efficient and rapid clearance of apoptotic cells occurs through a concerted effort by the apoptotic cells themselves, and the neighboring or recruited phagocytes. The quick clearance of these dying cells helps to prevent an inflammatory insult resulting from the uncontrolled release of intracellular contents. In this review, we provide an overview of the components of apoptotic cell clearance and discuss the general link between apoptotic cell clearance and disease pathogenesis. We also specifically evaluate the role of nucleotides in apoptotic cell clearance.
The clearance process
Apoptotic cell clearance can be broken down into four general steps: migrating towards the dying cell; binding/recognition of the apoptotic cell; phagocytosis/internalization of the target; and processing the ingested apoptotic cell (see Fig. 1) [5]. Here we will briefly discuss various components of how this process occurs.
Fig. 1.

Steps in the apoptotic cell clearance process. Find-me signals (such as nucleotides) recruit motile phagocytes to the site of death. Exposure of eat-me signals (such as PtdSer) on the apoptotic cell allow for binding/recognition by the engulfment receptors (such as BAI1, TIM-4) on the phagocyte. The recognition is followed by phagocytosis/internalization of the apoptotic cell. The degradation of the apoptotic cell and managing the ingested cellular components then follow.
Locating the dying cell
The clearance of apoptotic cells by so-called professional phagocytes requires that the phagocyte can properly locate the dying cell. Locating cells that are undergoing apoptosis involves the first two steps of the clearance process: migrating towards the dying cell (if there are no phagocytes already in the vicinity of the cell) and recognizing the apoptotic cell amongst its living neighbors. Cells undergoing apoptosis release factors, such as the nucleotides ATP and UTP [6-8], the chemokine fractalkine (CX3CL1) [9], and the lipids lysophosphatidylcholine (LPC) [10] and spingosine-1-phosphate (S1P) [11], to promote the recruitment of motile phagocytes. In the context of apoptotic cell clearance, these chemotactic factors are called ‘find-me’ signals for the role they play in the recruitment of phagocytes to the dying cells (a molecular ‘apoptotic cell beacon’). These factors, released by different mechanisms, set up a concentration gradient that allows phagocytes expressing their cognate receptors to migrate toward the site of death. Receptors for some of these find-me signals have been identified, including the purinergic receptor P2Y2 for ATP and UTP [6] and the chemokine receptor CX3CR1 for fractalkine (CX3CL1) [9]. The receptor G2A has been shown to mediate chemotaxis to LPC [12]. However, the original study that identified LPC as a G2A ligand has been retracted [13], which raises some questions regarding the ligand that mediates chemotaxis in these systems. We will focus on the role of nucleotides as a find-me signal in the section titled ‘Nucleotides as a find-me signal’, and in doing so will discuss some aspects of find-me signals in general (for a review of all potential find-me signals see Muñoz et al. Autoimmunity Reviews 2010 [14]).
In addition to releasing find-me signals, apoptotic cells also expose ‘eat-me’ signals on their surface, promoting their recognition by the recruited phagocyte. The most well known of these eat-me signals is phosphatidylserine (PtdSer). Normally concentrated on the inner leaflet of the plasma membrane, PtdSer loses its asymmetric distribution during apoptosis and appears on the outer leaflet of the plasma membrane [15, 16]. The exact mechanism by which PtdSer is exposed during apoptosis is still unclear. Normally, phosphatidylserine is concentrated on the inner leaflet of the plasma membrane by aminophospholipid translocase (APLT) activity. Several groups have demonstrated that APLT activity decreases during apoptosis, thereby removing the driving force that maintains PtdSer asymmetry [17, 18]. However, how does PtdSer make it to the outer leaflet? Since the headgroup of PtdSer is polar, spontaneous jumping from the inner to the outer leaflet of a bilayer occurs rather slowly [18]. Several mechanisms, which are not exclusive of each other, have been suggested to mediate the increase in PtdSer on the outer leaflet. One mechanism is that increased ‘scramblase’ activity, which catalyzes the bidirectional transbilayer movement of phospholipids, might allow for PtdSer to ‘diffuse’ down its concentration gradient to the outer leaflet during apoptosis [17, 19]. The exposure of PtdSer on the outer leaflet might also be due to fusion of vesicles with the plasma membrane [20], perhaps as part of a calcium-induced membrane repair response [21]. While the APLT and scramblase activities are thought to play a role in the exposure of PtdSer during apoptosis, the identity of the proteins mediating these activities are largely unknown and controversial [22-24].
Phagocytosis/corpse internalization
The recognition and subsequent engulfment of apoptotic cells by phagocytes is mediated by receptors that either directly or indirectly (via bridging molecules) bind eat-me signals. Here, we will briefly discuss several receptors and bridging molecules that bind the eat-me signal PtdSer. Bridging molecules (opsonins) are secreted proteins that bind PtdSer on the surface of apoptotic cells and are subsequently recognized by their cognate receptors on the phagocyte. MFG-E8 and Gas6 are two bridging molecules that bind the vitronectin receptor (αVβ3 integrin) and the receptor tyrosine kinase Mer respectively [25, 26]. In addition to the indirect link to PtdSer, several membrane receptors that directly bind PtdSer have been identified. BAI1 [27], Tim4 and Tim1 [28, 29], and Stabilin-2 [30] have been shown to mediate uptake of apoptotic cells by directly binding PtdSer. Related receptors such as Tim3 [31] and Stabilin-1 [32] have also been shown to play a similar role. For some of these receptors, ligation to PtdSer, either directly or indirectly, results in Rac-dependent cytoskeletal reorganization, which ultimately leads to engulfment of the apoptotic cell [5]. However, Tim-4 does not appear to signal significantly through any of the known intracellular signaling pathways for engulfment, and its cytoplasmic tail appears dispensable [33].
Activation of Rac during phagocytosis of apoptotic cells occurs through one of two delineated intracellular signaling pathways: through the mammalian intracellular signaling molecules ELMO, Dock180, and CrkII, or the adaptor molecule GULP. ELMO and Dock180 interact together to form a bipartite guanine nucleotide exchange factor (GEF) for Rac, while it is still unknown how GULP leads to Rac activation (for a more detailed overview of intracellular signaling for apoptotic cell phagocytosis see [5, 34]).
Processing the internalized cell
Once the target has been internalized, the phagosome is progressively acidified, leading to degradation of the ingested cell [35] (for a review of phagosome maturation see [35-38]). Processing of the ingested cell within the phagolysosome leads to an increased load of cellular metabolites. How the phagocyte deals with the raw materials and energy derived from this catabolic process is an interesting question that remains largely unanswered [5]. The work that has been done has addressed cholesterol homeostasis in the phagocyte. Engulfment of apoptotic cells results in increased cholesterol efflux by the phagocyte through ABCA1, in addition to incorporation of cholesterol derived from the apoptotic cell into the phagocyte’s membrane [39]. However, it appears that this response was not mediated by metabolites derived from the engulfed cell, rather from binding of PtdSer, as surrogate targets (beads which mimic PtdSer on apoptotic cells) also elicited a similar response. This suggests that simply tickling the engulfment receptors may be sufficient to trigger a homeostatic response in the phagocyte for controlling its own cellular contents (in this case, cholesterol).
Post-engulfment responses
Aside from dealing with the ingested cellular components, phagocytes also change their ‘behavior’ in several interesting ways. One post-engulfment response that has been described is the LXR nuclear receptor dependent upregulation of Mer (receptor that binds PtdSer indirectly through the bridging molecule Gas6) [40]. This finding suggests that phagocytes may become more efficient after every meal, possibly via signaling by lipid metabolites derived from degradation of the internalized apoptotic cells.
Engulfment-induced modulation of the phagocyte is not limited to metabolic processes or the phagocytic machinery. Apoptotic cell clearance is described as an immunologically ‘silent’ process, due to the fact that it does not elicit an immune response (like necrosis) [41, 42]. Therefore, immunologic consequences of engulfment have been of particular interest in the field. The clearance of apoptotic cells is not only silent, but it is also actively anti-inflammatory; engulfment promotes the secretion of the anti-inflammatory cytokines such as TGFβ by macrophages [43, 44]. This anti-inflammatory effect is even potent enough to suppress LPS-induced inflammatory cytokine release [43, 45]. Interestingly, it appears that internalization of the corpse is not required for the anti-inflammatory effect mediated by the phagocyte [46, 47], and that PtdSer recognition alone can mimic the response [44, 48].
Collectively, it is through the coordinated release of find-me signals and exposure of eat-me signals that apoptotic cells assure their proper disposal. Phagocytes also play a role in promoting the immunologically ‘quiet’ nature of clearance, by releasing anti-inflammatory cytokines and coordinating the proper disposal of the engulfed contents.
Nucleotides as a find-me signal
It has been a long-standing puzzle in the apoptosis/engulfment field, that very few apoptotic cells are observed, even in tissues that are known to have high turnover of cells (such as the bone marrow or thymus). This led to the notion that apoptotic cells may advertise their presence at the earliest stages of apoptosis, via the release of ‘find-me’ signals; this could attract phagocytes to their proximity and thereby lead to the prompt clearance of the dying cells. Several factors have been suggested to act as find-me signals, including the nucleotides ATP and UTP [6-8], the chemokine fractalkine (CX3CL1) [9], and the lipids lysophosphatidylcholine (LPC) [10] and spingosine-1-phosphate (S1P) [11]. Among these, only the nucleotides and fractalkine have been shown to have relevance in clearing apoptotic cells in vivo [6, 9].
How do find-me signals attract the phagocytes? While receptors mediating chemotaxis to these various find-me signals have also been identified in these studies [6, 9, 12], the effective ranges of these find-me signals still need to be defined. For example, it has been shown that ATP added to the basolateral side of an endothelial cell monolayer can promote monocyte transmigration in vitro [49]. Do the find-me signals ATP and UTP permeate into the intravascular space to promote extravasation of circulating monocytes directly? This would require at least two things to occur. The nucleotides would need to cross the endothelial cell barrier somehow (transcytosis or diffusion through the endothelial cell barrier) and then the nucleotides would need to be presented on the luminal surface, in order to provide some spatial information about the location of the dying cells without being swept away in the circulation [50]. Additionally, these would need to occur while avoiding degradation by extracellular nucleotidases. These requirements could be avoided if the find-me signals were to act on endothelial cells, causing the upregulation of some stable molecular signpost on the luminal surface that promotes monocyte extravasation. Along these lines, it has been shown that ATP and UTP are able to induce Vascular Cell Adhesion Molecule-1 (VCAM-1) expression in Human Coronary Artery Endothelial Cells (HCAECs) in vitro [51]. The above ideas can be synthesized into the following model (see Fig. 2). Apoptotic cells release nucleotides, which would promote monocyte extravasation by upregulating adhesion molecule/chemokine expression by vascular endothelial cells. Once the macrophage is in the interstitium, the chemotactic gradient set up by the nucleotides would then mediate the attraction of the phagocyte to the dying cell. In the case where there are motile resident phagocytic cells (such as macrophages or microglia), the nucleotides would not need to promote extravasation of phagocytes. As an example, nucleotides have been shown to mediate microglial (resident phagocytes in the central nervous system) chemotaxis towards injured neurons [52, 53].
Fig. 2.
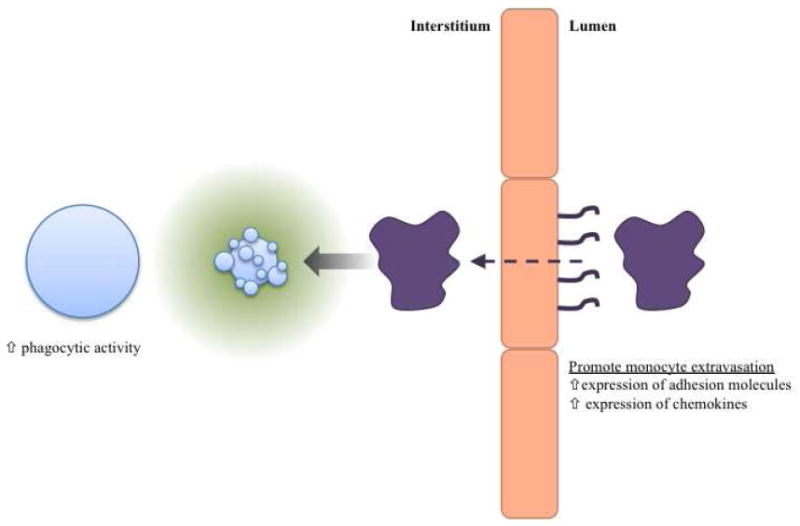
Schematic of the potential activites of find-me signals. Find-me signals may promote phagocytic activity of neighboring cells, perhaps by upregulating phagocytic machinery (receptors and intracellular signaling molecules). Find-me signals may also act on vascular endothelial cells to induce the upregulation of molecules that promote monocyte extravasation (such as adhesion molecules and chemokines). Find-me signal chemotactic gradient would then mediate migration of resident or recruited phagocytes within the interstitium.
In addition to setting up a chemotactic gradient to aid in the location of the dying cells, find-me signals might also have a role in modulating the phagocytic ability or activity of cells in the direct vicinity of the apoptotic cells (see Fig. 2). Apoptotic cells release UTP [6], which is degraded by extracellular enzymes through the removal of 5’ phosphates to produce UDP (in addition to other nucleotide metabolites). In the context of neuronal injury, UDP has been shown to promote phagocytosis by microglia [54]. While it was demonstrated that UDP promotes phagocytic activity via the P2Y6 nucleotide receptor, the mechanism of the boosted activity is unknown. This could occur by inducing the upregulation of proteins necessary for the uptake of apoptotic cells, such as secreted opsonins, receptors, or intracellular signaling molecules. In fact, ramping up of proteins involved in the engulfment of apoptotic cells has been seen in the context of increased cell death [55-57], but the mechanism of the induction in these systems is unknown. Even though it was not shown in the context of cell death, it has been demonstrated that the find-me signal fractalkine can induce production of the bridging molecule MFG-E8 by macrophages [58]. This suggests that find-me signals may indeed play a role in modulating the activity of phagocytes.
Even though ATP has been typically thought of as a danger signal [59], apoptotic cell supernatants appear to preferentially recruit monocytes over neutrophils in vivo [6]. Furthermore, apoptotic cell clearance is usually anti-inflammatory and immunologically silent, as phagocytes release anti-inflammatory mediators (such as TGFβ, IL-10 and Prostaglandin E2) after ingestion of apoptotic cells [43, 45]. Therefore, the recent identification of nucleotides as a find-me signal raises the following question: How can apoptotic cell clearance be immunologically silent if the apoptotic cells release factors that are considered inflammatory molecules, or danger signals?
There are perhaps several differences between the release of nucleotides during cell death via cytolysis and apoptosis (see Fig. 3). First and foremost, we will raise the issue of quantity. The nucleotide release seen during apoptosis appears to represent a rather small quantity of the total cellular ATP content (< 2%) [6]. Therefore, the regulated release of nucleotides during apoptosis is significantly less than that seen during cell lysis, or damage-induced loss of membrane integrity. ATP’s reputation as an inflammatory molecule is based largely on its ability to activate the ionotropic nucleotide receptor P2X7, which in turn results in activation of the inflammasome and release of pro-inflammatory cytokines [60, 61]. Along these lines, ATP derived from necrotic cells has been shown to result in sterile inflammation via inflammasome activation [62]. However, the concentrations necessary for activation of P2X7 (EC50 > 100μM) are much higher than those necessary for activation of the receptors mediating chemotaxis (such as P2Y2; EC50 < 1μM) [59, 60]. Intriguingly, it has been shown that lower concentrations of ATP may actually have an anti-inflammatory effect by suppressing the secretion of inflammatory cytokines, while promoting the release of anti-inflammatory cytokines [61, 63-65]. Therefore, the concept of ATP as a universal danger signal might be too simplistic.
Fig. 3.
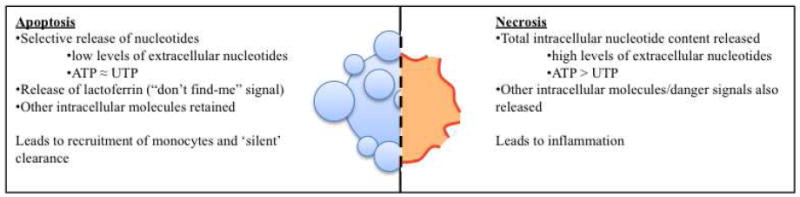
Differential release of molecules from apoptotic versus necrotic cells results in different responses.
In addition to differences between quantities of ATP released by apoptotic cells versus necrotic cells, there are also other cellular factors that are differentially released [66]. While necrotic cells theoretically release all of their intracellular contents, apoptotic cells become selectively permeable, retaining most of their intracellular contents [66]. HMGB-1 is an example of a molecule that is released by necrotic cells, but not apoptotic cells, that is capable of inciting inflammation [67]. Interestingly, it appears that the lack of HMGB-1 release by apoptotic cells is not simply because of selective membrane permeability. Apoptotic cells actively retain HMGB-1 by deacetylating histones, which increases the affinity of HMGB-1 for the chromatin [67]. Apoptotic cells also release factors that are not released by necrotic cells to modulate the inflammatory signature of ATP. It was recently demonstrated that apoptotic cells release lactoferrin, which acts as a “don’t find-me” signal for neutrophils [68]. Perhaps lactoferrin plays a role in dampening the attraction of neutrophils to ATP released by apoptotic cells. Yet another potential distinguishing factor between nucleotide release during apoptosis versus necrosis is the relative quantity of UTP that is released with ATP. Apoptotic cells release roughly similar quantities of the two nucleotides (even though ATP levels in the cell are several fold higher than UTP), whereas necrotic cells release the nucleotides proportionally to their intracellular levels [6]. Therefore, differential activation of various nucleotide receptors (which have different affinities for the various nucleotides) may also play a role in the ability to distinguish necrosis from apoptosis.
Relevance of apoptotic cell clearance to disease
It has been suggested that the efficient clearance of apoptotic cells is important for the maintenance of self tolerance, and that deficiencies in this process may lead to the development of autoimmunity. Reducing the efficiency of the clearance process, at any of the steps preceding engulfment, results in the persistence of apoptotic cells [6, 31, 40, 57, 69-73]. Early apoptotic cells are relatively intact (i.e., plasma membrane is selectively permeable, allowing regulated release of small molecules [6]), but if they are not cleared in a timely manner, they eventually become secondarily necrotic. It is thought that secondary necrosis may promote an immune response to intracellular antigens, since the antigens are exposed in an inflammatory environment, caused by the release of various danger signals. Mounting an immune response to normally hidden intracellular proteins does not seem to be simply a product exposure to these antigens due to cytolysis. During apoptosis, some antigens undergo post-translational modifications [74], and are also concentrated in membrane blebs on the surface of cells during apoptosis [75-77]. Post-translational modifications and altered localization may contribute to bypassing tolerance in various autoimmune diseases [74, 78]. Regardless of the mechanism by which autoantigens are revealed to the immune system, it appears that persistence of apoptotic cells plays a role in the development of autoantibodies, and perhaps autoimmune disease. Interestingly, intravenous administration of apoptotic cells have been shown to result in the development of autoantibodies, presumably due to excessive apoptotic cells that cannot be efficiently cleared, [79]. Hindering clearance in vivo has also been shown to induce the development of autoantibodies, and in some cases autoimmune-disease-like phenotypes [28, 31, 40, 69, 70, 72, 73, 80]. Complicating the interpretation of some of these results, several of the receptors (such as Tim4 and the vitronectin receptor) have other roles in immune tolerance that might be responsible for the phenotype seen [81]. In fact, it is not clear what components are necessary to induce autoimmunity, as reducing clearance does not always result in the development of autoantibodies [82] and development of autoantibodies does not always result in autoimmunity (for a broader review of the link between apoptotic cell clearance, autoimmunity and other disease processes, please see Elliott and Ravichandran, The Journal of Cell Biology 2010 [83]).
Systemic Lupus Erythematosus (SLE) is an autoimmune disease characterized by a variety of symptoms, which are thought to be largely due to the presence of antibodies directed against self antigens (autoantibodies) [84]. Interestingly, patients with systemic lupus erythematosus (SLE) have higher amounts of apoptotic cells in their circulation [85] and in lymph nodes [86]. Macrophages from SLE patients have also been shown to have less phagocytic capacity [87], suggesting that the accumulation of apoptotic cells is due to a defect in clearance. Whether defects in apoptotic cell clearance cause SLE or are a byproduct of the inflammation is still unclear (for a more in-depth discussion of the role of apoptotic cell clearance in autoimmunity see Muñoz et al. Nature Reviews Rheumatology 2010 [88]).
Inefficient clearance of apoptotic cells also appears to be a factor in the pathogenesis of atherosclerosis [89]. Defective clearance may contribute to the development of a necrotic core in an atheroma, as uncleared apoptotic cells become secondarily necrotic. Accumulation of secondarily necrotic cells in an atherosclerotic plaque is thought to play a role in the establishment of an inflammatory environment in the plaque and also contributes to its structural instability. This inflammatory plaque, laden with necrotic cells is the late stage plaque that precedes plaque rupture and thrombosis. Knocking out molecules (such as the bridging molecules C1q and MFG-E8 or the receptor tyrosine kinase Mer) that mediate phagocytic removal of apoptotic cells in atherosclerosis-prone mice (LDLR-/- or ApoE-/-), results in accumulation of apoptotic cells and acceleration of atherosclerosis [90-93]. One group demonstrated that functionally knocking out Mer in bone marrow derived cells alone is sufficient to see the increase in apoptotic cells (presumably due to decreased clearance) and acceleration of atherosclerosis [93]. However, it must be noted that these molecules have other roles in the immune system, and therefore the effect cannot be conclusively attributed solely to deficiencies in clearance.
Two post-engulfment responses are of particular interest to the field of atherosclerosis research: the upregulation of ABCA1 expression [39] and the release of anti-inflammatory cytokines [43] by phagocytes. Lipid-laden macrophages (foam cells) play a crucial role in the atherogenesis [94]. Since ABCA1 expression has been shown to be inversely related to atherogenesis [94], perhaps apoptotic cell induced ABCA1 upregulation plays a role in protection against atherosclerosis by preventing the accumulation of lipids within macrophages. The release of anti-inflammatory cytokines by phagocytes is also relevant to atherosclerosis for at least two reasons. First, atherosclerosis is an inflammatory disease [95, 96], and therefore quenching inflammation through the release of cytokines could have an effect in promoting disease resolution. Second, TGFβ, which is one of the anti-inflammatory cytokines released after engulfment, has been shown to promote fibrous cap development in atherosclerotic plaques, protecting against thrombotic events due to plaque rupture [97-100]. Furthermore, TGFβ expression correlates with plaque stability in humans [101]. Therefore, it is feasible that defects in phagocytosis of apoptotic cells may exacerbate atherosclerosis, since the post-engulfment responses (cholesterol efflux and anti-inflammatory cytokine release) are generally atheroprotective.
Apoptotic cell clearance is not only relevant to disease from the standpoint of deficiencies leading to inflammatory milieus, but also for its potential role in perpetuating other disease processes. The presence of macrophages plays a critical role in the progression of some disease processes, including atherosclerosis [94] and cancer [102]. A number of chemokines have been implicated in the recruitment of monocytes to tumors [103] and atherosclerotic plaques [104]. However, since apoptotic cells are present in both atherosclerotic plaques [3, 105] and tumors [1], perhaps apoptosis plays a role in the initial recruitment of monocytes in these disease processes. This recruitment of monocytes does not need to be independent of chemokines, since as addressed above, find-me signals may play a role in inducing the production of chemokines by neighboring cells. It has been suggested that lipid deposition in the sub-endothelial space results in macrophage accumulation in atherosclerosis [94]. However, it is not known how lipid deposition results in the recruitment of monocytes. Perhaps lipids promote apoptosis, which leads to monocyte recruitment. Aside from fractalkine release from Burkitt Lymphoma cells [9], the role of find-me signals in the initial recruitment of macrophages to tumors has not been investigated. However, elevated levels of ATP have been detected in tumors [106], suggesting that nucleotide find-me signals might be released by apoptotic tumor cells.
Summary
Apoptotic cell clearance is an important process that helps to maintain homeostasis in healthy tissue. We have outlined the various steps in the clearance process and discussed how they are related to pathogenesis of disease. We have also discussed the nucleotide find-me signals and how they may play the two seemingly disparate roles as a find-me signal and a ‘danger’ molecule. There have been many contributions to this field recently, including the identification of several receptors for phosphatidylserine, which is exposed on the surface of apoptotic cells, and new find-me signals released by cells undergoing apoptosis. What remains to be determined is the relative contribution of each of these players to clearance in vivo, and how impairment of the molecules or the pathways they regulate could result in disease. A potential future direction that needs to be explored is how to ‘improve’ or restore engulfment in certain disease states and whether such manipulations could be therapeutically beneficial. The recent advances in the field and the pace of discoveries portend a bright future and possibilities for therapies based on targeting the engulfment machinery.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health. F.B.C. was supported by a Pharmacological Sciences Training Grant (National Institute of General Medical Studies) and a F30 pre-doctoral fellowship (National Heart, Lung and Blood Institute). This work was supported by funding (to K.S.R.) from the National Institutes of Health, American Asthma Foundation and The Goldhirsh Foundation. K.S.R. is a William Benter Senior Fellow of the American Asthma Foundation.
Footnotes
The authors declare no conflict of interests related to this study.
References
- 1.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 2.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 3.Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res. 2000;45:736–746. doi: 10.1016/s0008-6363(99)00235-7.S0008-6363(99)00235-7 [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 5.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–974. doi: 10.1038/nri2214.nri2214 [DOI] [PubMed] [Google Scholar]
- 6.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296.nature08296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028.nm.2028 [DOI] [PubMed] [Google Scholar]
- 8.Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, Smyth MJ, Zitvogel L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 70:855–858. doi: 10.1158/0008-5472.CAN-09-3566.0008-5472.CAN-09-3566 [DOI] [PubMed] [Google Scholar]
- 9.Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404.blood-2008-06-162404 [DOI] [PubMed] [Google Scholar]
- 10.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7.S0092867403004227 [DOI] [PubMed] [Google Scholar]
- 11.Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–2638. doi: 10.1096/fj.08-107169.fj.08-107169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, Wesselborg S, Lauber K. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. 2008;283:5296–5305. doi: 10.1074/jbc.M706586200.M706586200 [DOI] [PubMed] [Google Scholar]
- 13.Witte ON, Kabarowski JH, Xu Y, Le LQ, Zhu K. Retraction. Science. 2005;307:206. doi: 10.1126/science.307.5707.206b.307/5707/206b [DOI] [PubMed] [Google Scholar]
- 14.Munoz LE, Peter C, Herrmann M, Wesselborg S, Lauber K. Scent of dying cells: the role of attraction signals in the clearance of apoptotic cells and its immunological consequences. Autoimmun Rev. 9:425–430. doi: 10.1016/j.autrev.2009.11.016.S1568-9972(09)00194-3 [DOI] [PubMed] [Google Scholar]
- 15.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 16.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhoven B, Schlegel RA, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272:26159–26165. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- 19.Williamson P, Kulick A, Zachowski A, Schlegel RA, Devaux PF. Ca2+ induces transbilayer redistribution of all major phospholipids in human erythrocytes. Biochemistry. 1992;31:6355–6360. doi: 10.1021/bi00142a027. [DOI] [PubMed] [Google Scholar]
- 20.Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem. 1989;264:17049–17057. [PubMed] [Google Scholar]
- 21.Mirnikjoo B, Balasubramanian K, Schroit AJ. Suicidal membrane repair regulates phosphatidylserine externalization during apoptosis. J Biol Chem. 2009;284:22512–22516. doi: 10.1074/jbc.C109.022913.C109.022913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlegel RA, Williamson P. P.S to PS (phosphatidylserine)--pertinent proteins in apoptotic cell clearance. Sci STKE 2007. 2007:pe57. doi: 10.1126/stke.4082007pe57.stke.4082007pe57 [DOI] [PubMed] [Google Scholar]
- 23.Zullig S, Neukomm LJ, Jovanovic M, Charette SJ, Lyssenko NN, Halleck MS, Reutelingsperger CP, Schlegel RA, Hengartner MO. Aminophospholipid translocase TAT-1 promotes phosphatidylserine exposure during C. elegans apoptosis. Curr Biol. 2007;17:994–999. doi: 10.1016/j.cub.2007.05.024.S0960-9822(07)01403-0 [DOI] [PubMed] [Google Scholar]
- 24.Darland-Ransom M, Wang X, Sun CL, Mapes J, Gengyo-Ando K, Mitani S, Xue D. Role of C elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science. 2008;320:528–531. doi: 10.1126/science.1155847.320/5875/528 [DOI] [PubMed] [Google Scholar]
- 25.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a.417182a [DOI] [PubMed] [Google Scholar]
- 26.Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- 27.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329.nature06329 [DOI] [PubMed] [Google Scholar]
- 28.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307.nature06307 [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011.S1074-7613(07)00546-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242.4402242 [DOI] [PubMed] [Google Scholar]
- 31.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–3830. doi: 10.1182/blood-2008-10-185884.blood-2008-10-185884 [DOI] [PubMed] [Google Scholar]
- 32.Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, Kzhyshkowska J, Kim IS. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci. 2009;122:3365–3373. doi: 10.1242/jcs.049569.122/18/3365 [DOI] [PubMed] [Google Scholar]
- 33.Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19:346–351. doi: 10.1016/j.cub.2009.01.042.S0960-9822(09)00614-9 [DOI] [PubMed] [Google Scholar]
- 34.Kinchen JM. A model to die for: signaling to apoptotic cell removal in worm, fly and mouse. Apoptosis. doi: 10.1007/s10495-010-0509-5. [DOI] [PubMed] [Google Scholar]
- 35.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515.nrm2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184.4402184 [DOI] [PubMed] [Google Scholar]
- 37.Zhou Z, Yu X. Phagosome maturation during the removal of apoptotic cells: receptors lead the way. Trends Cell Biol. 2008;18:474–485. doi: 10.1016/j.tcb.2008.08.002.S0962-8924(08)00211-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029.ni1006-1029 [DOI] [PubMed] [Google Scholar]
- 39.Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16:2252–2258. doi: 10.1016/j.cub.2006.09.043.S0960-9822(06)02275-5 [DOI] [PubMed] [Google Scholar]
- 40.N AG, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018.S1074-7613(09)00318-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–1181. doi: 10.1038/ni1205-1179.ni1205-1179 [DOI] [PubMed] [Google Scholar]
- 42.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 43.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 46.Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M, Silverstein R, Savill J, Lacy-Hulbert A. Requirements for apoptotic cell contact in regulation of macrophage responses. J Immunol. 2006;177:4047–4054. doi: 10.4049/jimmunol.177.6.4047.177/6/4047 [DOI] [PubMed] [Google Scholar]
- 47.Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–889. doi: 10.4049/jimmunol.172.2.880. [DOI] [PubMed] [Google Scholar]
- 48.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009.S1074761304003000 [DOI] [PubMed] [Google Scholar]
- 49.Goepfert C, Sundberg C, Sevigny J, Enjyoji K, Hoshi T, Csizmadia E, Robson S. Disordered cellular migration and angiogenesis in cd39-null mice. Circulation. 2001;104:3109–3115. doi: 10.1161/hc5001.100663. [DOI] [PubMed] [Google Scholar]
- 50.Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853.100/12/3853 [DOI] [PubMed] [Google Scholar]
- 51.Seye CI, Yu N, Jain R, Kong Q, Minor T, Newton J, Erb L, Gonzalez FA, Weisman GA. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem. 2003;278:24960–24965. doi: 10.1074/jbc.M301439200. [DOI] [PubMed] [Google Scholar]
- 52.Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001.21/6/1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472.nn1472 [DOI] [PubMed] [Google Scholar]
- 54.Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704.nature05704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakatani H, Aoki N, Nakagawa Y, Jin-No S, Aoyama K, Oshima K, Ohira S, Sato C, Nadano D, Matsuda T. Weaning-induced expression of a milk-fat globule protein, MFG-E8, in mouse mammary glands, as demonstrated by the analyses of its mRNA, protein and phosphatidylserine-binding activity. Biochem J. 2006;395:21–30. doi: 10.1042/BJ20051459.BJ20051459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028.S0896-6273(06)00319-9 [DOI] [PubMed] [Google Scholar]
- 57.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 112:733–743. doi: 10.1111/j.1471-4159.2009.06494.x.JNC6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miksa M, Amin D, Wu R, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med. 2007;13:553–560. doi: 10.2119/2007-00019.Miksa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal. 2009;2:pe6. doi: 10.1126/scisignal.256pe6.scisignal.256pe6 [DOI] [PubMed] [Google Scholar]
- 60.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013.S0163-7258(06)00066-0 [DOI] [PubMed] [Google Scholar]
- 61.Di Virgilio F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci. 2007;28:465–472. doi: 10.1016/j.tips.2007.07.002.S0165-6147(07)00184-8 [DOI] [PubMed] [Google Scholar]
- 62.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106.0908698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hasko G, Kuhel DG, Salzman AL, Szabo C. ATP suppression of interleukin-12 and tumour necrosis factor-alpha release from macrophages. Br J Pharmacol. 2000;129:909–914. doi: 10.1038/sj.bjp.0703134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.la Sala A, Ferrari D, Corinti S, Cavani A, Di Virgilio F, Girolomoni G. Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. J Immunol. 2001;166:1611–1617. doi: 10.4049/jimmunol.166.3.1611. [DOI] [PubMed] [Google Scholar]
- 65.Wilkin F, Stordeur P, Goldman M, Boeynaems JM, Robaye B. Extracellular adenine nucleotides modulate cytokine production by human monocyte-derived dendritic cells: dual effect on IL-12 and stimulation of IL-10. Eur J Immunol. 2002;32:2409–2417. doi: 10.1002/1521-4141(200209)32:9<2409∷AID-IMMU2409>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 66.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215.nri2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 68.Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, Rossi AG, Gregory CD. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest. 2009;119:20–32. doi: 10.1172/JCI36226.36226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359.304/5674/1147 [DOI] [PubMed] [Google Scholar]
- 70.Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 71.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603.35075603 [DOI] [PubMed] [Google Scholar]
- 73.Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, Roes JT, Savill JS, Hynes RO. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci U S A. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104.0707421104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Utz PJ, Anderson P. Posttranslational protein modifications, apoptosis, and the bypass of tolerance to autoantigens. Arthritis Rheum. 1998;41:1152–1160. doi: 10.1002/1529-0131(199807)41:7<1152∷AID-ART3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 75.LeFeber WP, Norris DA, Ryan SR, Huff JC, Lee LA, Kubo M, Boyce ST, Kotzin BL, Weston WL. Ultraviolet light induces binding of antibodies to selected nuclear antigens on cultured human keratinocytes. J Clin Invest. 1984;74:1545–1551. doi: 10.1172/JCI111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golan TD, Elkon KB, Gharavi AE, Krueger JG. Enhanced membrane binding of autoantibodies to cultured keratinocytes of systemic lupus erythematosus patients after ultraviolet B/ultraviolet A irradiation. J Clin Invest. 1992;90:1067–1076. doi: 10.1172/JCI115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suber T, Rosen A. Apoptotic cell blebs: repositories of autoantigens and contributors to immune context. Arthritis Rheum. 2009;60:2216–2219. doi: 10.1002/art.24715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, Tanaka M. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med. 2004;200:459–467. doi: 10.1084/jem.20040342.jem.20040342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Savill J, Gregory C. Apoptotic PS to phagocyte TIM-4: eat me. Immunity. 2007;27:830–832. doi: 10.1016/j.immuni.2007.12.002.S1074-7613(07)00553-5 [DOI] [PubMed] [Google Scholar]
- 82.Devitt A, Parker KG, Ogden CA, Oldreive C, Clay MF, Melville LA, Bellamy CO, Lacy-Hulbert A, Gangloff SC, Goyert SM, Gregory CD. Persistence of apoptotic cells without autoimmune disease or inflammation in CD14-/- mice. J Cell Biol. 2004;167:1161–1170. doi: 10.1083/jcb.200410057.jcb.200410057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Bio. 2010 doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sawalha AH, Harley JB. Antinuclear autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 2004;16:534–540. doi: 10.1097/01.bor.0000135452.62800.8f.00002281-200409000-00008 [DOI] [PubMed] [Google Scholar]
- 85.Perniok A, Wedekind F, Herrmann M, Specker C, Schneider M. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus. 1998;7:113–118. doi: 10.1191/096120398678919804. [DOI] [PubMed] [Google Scholar]
- 86.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191∷AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 87.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241∷AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 88.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 6:280–289. doi: 10.1038/nrrheum.2010.46.nrrheum.2010.46 [DOI] [PubMed] [Google Scholar]
- 89.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 10:36–46. doi: 10.1038/nri2675.nri2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, Esposito B, Teissier E, Daemen MJ, Leseche G, Boulanger C, Tedgui A, Mallat Z. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–2177. doi: 10.1161/CIRCULATIONAHA.106.662080.CIRCULATIONAHA.106.662080 [DOI] [PubMed] [Google Scholar]
- 91.Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, Boyle JJ, Haskard DO. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170:416–426. doi: 10.2353/ajpath.2007.060406.170/1/416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197.ATVBAHA.108.167197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, Offenstadt G, Leseche G, Cohen PL, Tedgui A, Mallat Z. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1429–1431. doi: 10.1161/ATVBAHA.108.169078.ATVBAHA.108.169078 [DOI] [PubMed] [Google Scholar]
- 94.Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2:309–315. doi: 10.1038/ncpcardio0195.ncpcardio0195 [DOI] [PubMed] [Google Scholar]
- 95.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 96.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. doi: 10.1038/nrcardio.2009.55.nrcardio.2009.55 [DOI] [PubMed] [Google Scholar]
- 97.Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb. 1991;11:1223–1230. doi: 10.1161/01.atv.11.5.1223. [DOI] [PubMed] [Google Scholar]
- 98.Nabel EG, Shum L, Pompili VJ, Yang ZY, San H, Shu HB, Liptay S, Gold L, Gordon D, Derynck R, et al. Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc Natl Acad Sci U S A. 1993;90:10759–10763. doi: 10.1073/pnas.90.22.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, Fradelizi D, Tedgui A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89:930–934. doi: 10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 100.Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, Koteliansky VE, Daemen MJ. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002;22:975–982. doi: 10.1161/01.atv.0000019729.39500.2f. [DOI] [PubMed] [Google Scholar]
- 101.Cipollone F, Fazia M, Mincione G, Iezzi A, Pini B, Cuccurullo C, Ucchino S, Spigonardo F, Di Nisio M, Cuccurullo F, Mezzetti A, Porreca E. Increased expression of transforming growth factor-beta1 as a stabilizing factor in human atherosclerotic plaques. Stroke. 2004;35:2253–2257. doi: 10.1161/01.STR.0000140739.45472.9c.01.STR.0000140739.45472.9c [DOI] [PubMed] [Google Scholar]
- 102.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007.S0092-8674(06)00055-9 [DOI] [PubMed] [Google Scholar]
- 103.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-11092004-03-1109. [DOI] [PubMed] [Google Scholar]
- 104.Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004.S1050-1738(08)00127-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Best PJ, Hasdai D, Sangiorgi G, Schwartz RS, Holmes DR, Jr, Simari RD, Lerman A. Apoptosis Basic concepts and implications in coronary artery disease. Arterioscler Thromb Vasc Biol. 1999;19:14–22. doi: 10.1161/01.atv.19.1.14. [DOI] [PubMed] [Google Scholar]
- 106.Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008;3:e2599. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]