

The results of a European retrospective, multicenter study led by the European Musculoskeletal Oncology Society on the clinical behavior and characteristics of enchondromas in patients with Ollier disease and Maffucci syndrome are reported, with the goals of better defining the presentation and characteristics of enchondromas in these patients, estimating the cumulative probability of secondary transformation of enchondromas over a lifetime, and finding variables significantly associated with this latter outcome and mortality.
Keywords: Enchondroma, Chondrosarcoma, Ollier disease, Maffucci syndrome
Learning Objectives
After completing this course, the reader will be able to:
Describe major enchondroma distribution patterns that were identified in this study.
Identify variables that are predictive for the secondary transformation of enchondroma over the lifetime of individuals with Ollier disease or Maffucci syndrome.
This article is available for continuing medical education credit at CME.TheOncologist.com
Abstract
Background.
Enchondromatosis is characterized by the presence of multiple benign cartilage lesions in bone. While Ollier disease is typified by multiple enchondromas, in Maffucci syndrome these are associated with hemangiomas. Studies evaluating the predictive value of clinical symptoms for development of secondary chondrosarcoma and prognosis are lacking. This multi-institute study evaluates the clinical characteristics of patients, to get better insight on behavior and prognosis of these diseases.
Method.
A retrospective study was conducted using clinical data of 144 Ollier and 17 Maffucci patients from 13 European centers and one national databank supplied by members of the European Musculoskeletal Oncology Society.
Results.
Patients had multiple enchondromas in the hands and feet only (group I, 18%), in long bones including scapula and pelvis only (group II, 39%), and in both small and long/flat bones (group III, 43%), respectively. The overall incidence of chondrosarcoma thus far is 40%. In group I, only 4 patients (15%) developed chondrosarcoma, in contrast to 27 patients (43%) in group II and 26 patients (46%) in group III, respectively. The risk of developing chondrosarcoma is increased when enchondromas are located in the pelvis (odds ratio, 3.8; p = 0.00l).
Conclusions.
Overall incidence of development of chondrosarcoma is 40%, but may, due to age-dependency, increase when considered as a lifelong risk. Patients with enchondromas located in long bones or axial skeleton, especially the pelvis, have a seriously increased risk of developing chondrosarcoma, and are identified as the population that needs regular screening on early detection of malignant transformation.
Introduction
Ollier disease [1] and Maffucci syndrome [2] are both rare, nonhereditary disorders in which patients develop multiple enchondromas, which are benign cartilaginous tumors in the bone [3–5].
The diagnosis of Ollier disease, with a prevalence of one in 100,000 [6], is mainly based on clinical, radiological, and histological evaluation [7]. There is asymmetrical involvement of the extremities, with one side of the skeleton being affected with enchondromas either exclusively or predominantly [6, 8]. Throughout their lives, patients experience a variety of different kinds of symptoms. Medical problems like leg length discrepancies or bowing deformities resulting from skeletal deformities caused by the enchondromas become prominent during childhood and adolescence. These deformities, mainly developing as a consequence of the asymmetrical distribution of the enchondromas, often require surgical correction [9, 10]. If the enchondromas are located in the small tubular bones, the function of the hands and feet may be disabled to varying degrees depending on the severity of enlargement and deformities. The most severe complication is malignant transformation of enchondromas toward secondary chondrosarcomas, for which the reported incidence is highly variable, in the range of 5%–50% in the literature [11–15]. In addition, gliomas, acute myeloid leukemia, and juvenile granulosa cell tumors have been found in patients with Ollier disease [3, 16].
With Maffucci syndrome, cutaneous, soft tissue, or visceral hemangiomas are found in addition to multiple enchondromas [11, 17–19]. Deformities of the bones resulting from asymmetrical involvement of the extremities are seen, as in Ollier disease. According to the existing literature, a large number of other malignancies—particularly, pancreatic and hepatic adenocarcinoma, mesenchymal ovarian tumors, brain tumors such as glioma and astrocytoma, and various kinds of sarcomas—are observed with this disease [3, 15, 17, 20–22].
Mutations of the gene encoding parathyroid hormone receptor 1 (PTHR1) are found in a small subset (∼10%) of patients with Ollier disease [3, 23–25]. Recently, mutations in the gene encoding isocitrate dehydrogenase 1 (IDH1) and IDH2 were detected in solitary cartilaginous tumors as well is in patients with multiple enchondromas. These mutations might represent early postzygotic genetic events and account for the initiation of the disease process [16].
No specific therapy yet exists to cure these potentially disabling diseases. Thus far, surgical therapy is the only available option when complications occur, for example, pathological fractures, growth defects, or malignant transformation.
When properly diagnosed, osteochondromas do not appear in patients with Ollier disease or Maffucci syndrome. The combination of multiple enchondromas and osteochondroma-like lesions is known as metachondromatosis [3].
To gain more insight into and a better understanding of the clinical behavior and characteristics of enchondromas in patients with Ollier disease and Maffucci syndrome, this European retrospective, multicenter study aimed at better defining the presentation and characteristics of enchondromas in patients with Ollier disease and Maffucci syndrome, estimate the cumulative probability of secondary transformation of enchondroma over a lifetime, and find variables significantly associated with this latter outcome and mortality. Data were collected by the European Musculoskeletal Oncology Society (EMSOS), a multidisciplinary society with great interest in bone and soft tissue tumors.
Methods
Data Collection
The objectives of this retrospective cohort study were formulated and discussed at the EMSOS annual meeting in Porto, Portugal, in 2007. A questionnaire was designed (by S.H.M.V., J.V.M.G.B., T.C.P., P.C.W.H., and A.H.M.T.) to collect clinical data on patients with Ollier disease and Maffucci syndrome. The questionnaire was digitally sent as an Excel file to 130 EMSOS members at 76 hospitals in 26 countries in Europe and the Russian Federation. The digital file was sent with an accompanying manual. The questionnaire was completed by the participating physicians using patients' clinical files, radiological test results, and, when relevant, surgical and histological reports.
Each worksheet was used to record the available information of the included patients. Requested patient characteristics included: gender, age, family history, comorbidity, leg length discrepancies, and bowing deformities. Abnormalities of the spine were scored to exclude other, rare enchondromatosis subtypes [3]. Requested radiological characteristics included: estimated number and location of enchondromas, local cortical destruction, soft tissue extension, scalloping, and fractures. Requested tumor characteristics included: site and distribution of enchondromas, development of secondary chondrosarcomas, histological grading in cases with biopsy or surgery, and location and histology of vascular lesions in cases of Maffucci syndrome. Requested surgical information included: any surgery that had been performed to correct deformities or leg length discrepancies, surgery for benign lesions, type and extent of surgery performed for malignancies, follow-up time after treatment, and prognosis with respect to metastasis, dedifferentiation, and survival.
When more than one surgery was performed for a chondrosarcoma at a specific location, this was recorded as a single tumor with local recurrence rather than as a second chondrosarcoma.
Statistical Analysis
Data were collected by different institutions using the unified Excel spreadsheets, which contained data validation and explicit definitions of all items asked. The spreadsheets were then converted and merged to one SPSS data file for analysis (SPSS, Inc., Chicago, IL).
Descriptive analyses consisted of tabular overviews of means, medians, percentiles, and standard deviations. Bivariate associations of discrete variables were tested in crosstabulations using the likelihood ratio test or Fisher's exact test (in the case of low counts).
Estimates related to the occurrence in time of a specific event, for example, death, were obtained in a survival analysis framework. The primary approach was Kaplan–Meier estimation. The primary outcome of interest was patient survival, defined as the time from birth to the time of death from any cause.
A logistic regression framework was used to estimate the probability of “being diagnosed with chondrosarcoma” as a function of the occurrence of enchondromas at various locations in the body. To this end, binary variables were constructed as indicators of the presence of enchondromas on plain x-rays in the scapula, humerus, ulna, radius, carpus, metacarpus, phalanges of the hand, pelvis, femur, tibia, fibula, tarsus, metatarsus, and phalanges of the feet. They were entered into the model and a backward elimination of multivariately nonsignificant predictors was performed.
A discriminant analysis was used for the same purpose, but only as verification using another statistical method.
To calculate the cumulative incidence of enchondromas in combination with chondrosarcomas over time, a competing risk framework was used. The competing risk in this case was death resulting from any cause. The starting point of these cumulative incidence curve estimates was the date of birth of the patient. Because of the construction of this dataset, the probability estimates should never be interpreted as “life-long probabilities” since birth. This is not a cohort study following patients from birth but a study population highly selected on the occurrence of disease, and hence all probabilities (or proportions) have only a descriptive meaning conditional on the disease having been diagnosed.
Results
In total, 14 bone tumor referral centers and centralized national databanks in nine different European countries contributed patient data for the study, resulting in 161 patients. Apart from nonresponse, the primary reason for nonparticipation was a lack of patients who clinically fit the study's profile.
General Characteristics
In total, 144 patients with Ollier disease and 17 patients with Maffucci syndrome were included in the study. The mean age at the time of this study was 32 years (range, 5–72 years). Information regarding comorbidities, the development of other malignancies, and family history was provided for <3% of the patients, and therefore no evaluation of these data was performed. No positive family history or aberrations of the spine were reported [3] (Table 1).
Table 1.
Characteristics of patients with Ollier disease and Maffucci syndrome

Overview of characteristics of patients with Ollier disease and Maffucci syndrome. The mean ages at first surgery were, respectively, 33 years and 30 years; noting that the mean age at the time of the study was 32 years, the incidence of chondrosarcoma in our patient cohort is expected to increase in the future.
Age at Diagnosis of Ollier Disease or Maffucci Syndrome
The mean age of patients at the time of diagnosis of Ollier disease in this series was 13 years (range, 0–59 years; data from 105 of 141 patients with Ollier disease). For Maffucci syndrome, the mean age was 12 years (range, 1–65 years; data completed for 11 of 17 patients with Maffucci syndrome). Seventy-five percent of the patients were diagnosed before the age of 20 years, both for Maffucci syndrome and for Ollier disease (Fig. 1).
Figure 1.
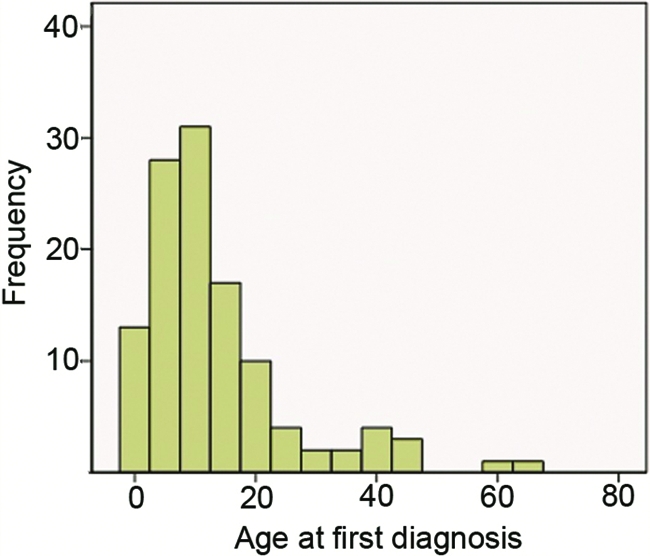
Age when disorder was first discovered and a diagnosis of Ollier disease or Maffucci syndrome was made (n = 116). Twenty-three percent of the patients were diagnosed between age 0 and age 5 years and 45% were diagnosed before the age of 10 years. At 20 years of age, 75% of all patients had been diagnosed. Mean age at diagnosis, 13.38 years; standard deviation, 12.579 years.
Location and Distribution of Enchondromas
Eighty-nine patients (55%) had cartilaginous lesions on one side of the body and 68 patients (42%) had bilateral disease (four patients had missing data). Enchondromas were predominantly found in the femur (affected in 59% of patients), tibia (affected in 47% of patients), humerus (affected in 32% of patients), fibula (affected in 27% of patients), and pelvis (affected in 25% of patients). The small tubular bones of the hands were more often affected with enchondromas (carpal bones, 11%; metacarpals, 35%; phalanges of the hands, 45%) than the small tubular bones of the feet (tarsal bones, 10%; metatarsals, 19%; phalanges of the feet, 21%).
We distinguished three patterns of distribution of enchondromas. In 18% of cases, only the hands and/or feet were affected (designated as group I). Forty percent of patients had enchondromas in the long tubular and/or flat bones (group II). In 42% of patients, both the long and the flat bones as well as the small tubular bones of the hands and/or feet were affected (group III) (Table 2).
Table 2.
Distribution patterns of enchondroma

The local distribution pattern of enchondromas correlated with the risk for developing chondrosarcoma.
aTotal deaths: eleven. DOD (death of disease; these patients died as a result of the disease): eight patients, pulmonary metastasis secondary to chondrosarcoma; three patients, nondisease related (glioma, n = 2; hepatic carcinoma, n = 1).
Skeletal Deformities
Forty-four patients with Ollier disease (31%) and three patients with Maffucci syndrome (18%) had bowing or leg length deformities, particularly as a result of asymmetric distribution of enchondromas in the metaphysis and diaphysis of the long bones. The types of surgery performed included mainly lengthening procedures in the case of length discrepancies, osteotomies, and local surgery like debulking or amputation to correct disabling enlargement of the fingers and toes. With respect to the site of surgery, 80% involved the long bones of the lower extremities, 13% involved the long bones of the upper extremities, and 7% of the procedures were related to the metacarpals or phalanges of the hands.
Radiology
The radiological features of enchondromas and chondrosarcomas were compared on conventional radiographs (95 patients versus 66 patients). When both cortical destruction and soft tissue extension were present, the chance of dealing with a chondrosarcoma instead of an enchondroma was greater by a factor of 2.3 (95% confidence interval [CI], 1.28–4.8; p = .019).
Hemangiomas in Patients with Maffucci Syndrome
One or more skin lesions were present in 12 of 17 patients with Maffucci syndrome. The location was mainly in the upper extremities (forearm, n = 4; hands, n = 6) and lower extremities (leg, n = 3; lower leg, n = 2; foot, n = 2).
Excision of the lesion was performed in eight patients. Histological analysis identified spindle cell hemangioma in all cases.
Development of Chondrosarcomas
Sixty-six patients (41%) developed one or more secondary chondrosarcomas (Ollier disease, n = 57; Maffucci syndrome, n = 9). The mean age at which they first underwent surgery for chondrosarcoma was 33 years for patients with Ollier disease (range, 10–59 years) and 30 years for patients with Maffucci syndrome (range, 14–51 years) (Fig. 2).
Figure 2.

Distribution of age at first surgery for chondrosarcoma over time. Only 50% of the patients had their first event before the age of 35 years. Mean age at first event, 33.0 years; standard deviation, 13.2 years.
Of these 66 patients, 48 developed one chondrosarcoma whereas 18 developed two to four chondrosarcomas. Of these 18 patients, 33% had synchronous and 56% had metachronous chondrosarcomas (unknown, n = 2 [11%]) (Table 1).
Altogether, 89 chondrosarcomas were diagnosed in the 66 patients. The primary locations affected in the long bones were the humerus (n = 10), femur (n = 18), and tibia (n = 10). Nineteen chondrosarcomas were found in the flat bones (scapula, n = 8; pelvis, n = 11). Of the small tubular bones, the metacarpals and metatarsals were less often involved than the phalanges of the hands and feet (n = 9 and n = 14, respectively), which contradicts a nonsyndromal distribution [26].
Chondrosarcomas developed in 45% and 46% of patients in group II and group III, respectively (Table 2). In contrast, in group I wherein enchondromas were restricted to the small tubular bones of the hands and feet, the risk for developing chondrosarcoma was lower, at 15%. In patients with Maffucci syndrome, both the short and the long tubular bones were affected more often with enchondromas (group III) (Table 2).
Using a logistic regression model for estimation of the probability of having chondrosarcoma as a function of the location of enchondromas, the only indicator remaining was the presence of an enchondroma in the pelvis. The other locations of enchondroma did not contribute significantly to the outcome. The odds ratio associated with enchondroma of the pelvis was 3.8, with a 95% CI of 1.8–8.0 (p = .001).
In the previously mentioned group of 47 patients with surgery for skeletal deformities, 19 also developed one or more chondrosarcomas in the course of their disease. In three patients with Ollier disease and one patient with Maffucci syndrome, chondrosarcomas developed at the site of previous surgery. All four demonstrated unilateral disease and had been operated on for deformities of the femur.
Histology of Chondrosarcomas
Histological data were provided for 90% of the chondrosarcomas. Fifty-two percent were found to be grade I, 32% were grade II, 6% were grade III, and 10% were of unknown grade (Table 3).
Table 3.
Histological grade of chondrosarcoma

Based on the histological grade of chondrosarcomas as diagnosed in the center of origin. Eighty-seven chondrosarcomas developed in 66 patients. Fifty percent of the lesions were low-grade chondrosarcoma.
Surgery for Chondrosarcomas
Eighty-nine surgeries were performed for chondrosarcomas in 66 patients. In five cases with grade I chondrosarcomas, no surgery was performed after the biopsy. In 24 cases, intralesional curettage was performed (grade I, n = 17; grade II, n = 4; unknown grade, n = 3). In eight of these cases (33%), local adjuvant therapy was used with intralesional curettage (phenol/ethanol, n = 4; cryosurgery, n = 3; radiofrequency ablation and phenol, n = 1). Resection was performed in 46 cases (grade I, n = 17; grade II, n = 20; grade III, n = 3; unknown grade, n = 6). In two patients, surgery was followed by radiation therapy (intralesional curettage of grade II lesion of the skull, n = 1; resection of grade II lesion of the humerus, n = 1).
Amputation was performed in 13 cases. Sites of amputation were the phalanges of the hands and feet (eight patients; grade I, n = 5; grade II, n = 1; unknown grade, n = 1), metacarpals (grade II, n = 1), tarsal bone (grade II, n = 1), femur (two patients; grade II, n = 1; unknown grade, n = 1), and humerus (grade III, n = 1).
Mortality
The disease-related mortality rate of the 161 patients included in this series was 6.8% (11 patients). Three patients died as a result of malignancies other than chondrosarcoma (Table 2, and Fig. 4). Seven of the eight disease-related deceased patients were diagnosed with Ollier disease and died as a result of pulmonary metastases secondary to chondrosarcomas. The chondrosarcomas were primarily located in the humerus (n = 1), radius (n = 1), pelvis (n = 1), femur (n = 3), and tibia (n = 1).
Figure 4.
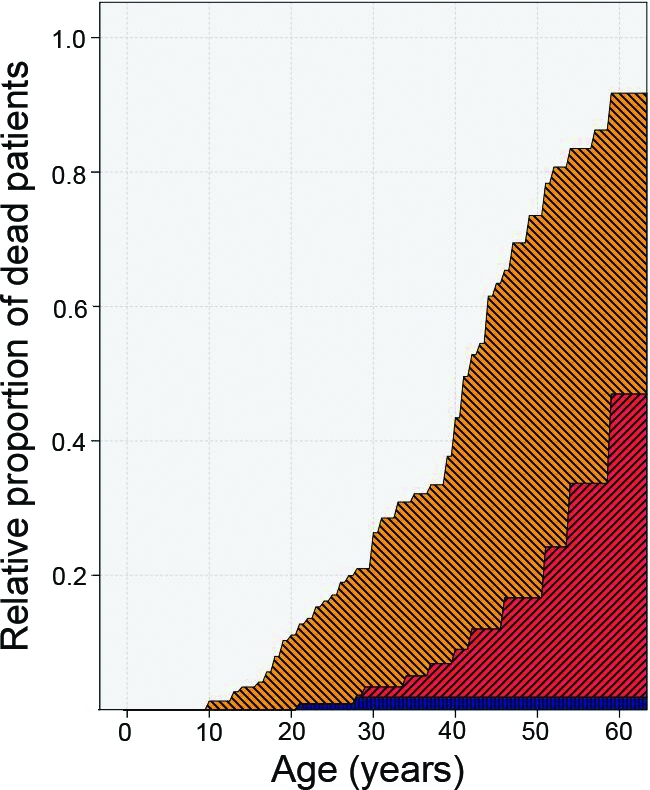
Survival analysis using a competing risk model. This analysis only aims at describing relative proportions (the composition of the study population) for specific patient ages. It therefore only pretends to give a (an unbiased) graphical description for each possible number of years since birth of how many patients at that moment were actually already dead or alive with enchondroma (EC) or chondrosarcoma (CHS) or alive without detection of EC or CHS. More precisely, at each point in time (patient age), it shows the relative proportion of study patients already dead without EC or CHS (the blue pattern); the proportion already dead after detection of EC or CHS (the red or inclining pattern); the proportion at that age alive with EC or CHS (the yellow or declining pattern); and the proportion alive at that age without EC or CHS. Note that the separation between the two shaded areas (red and orange) is in fact the overall survival duration of this group of patients; the border between the silver and the orange area is the survival interval free from EC and CHS. Again, the interpretation is a description of the age at onset of EC or CHS death among these specific patients; it is useful as a description of the population and gives insight into the interplay of the competing risks (dying versus acquiring EC or CHS). It is not a predictive model per se because this is not a cohort followed from birth but a set of patients defined retrospectively on the basis of the occurrence of disease.
Three of these eight deceased patients with Ollier disease also developed a second chondrosarcoma located in the fibula (grade I), tarsus (grade II), and phalanges of the foot (grade I).
One patient with Maffucci syndrome died as a result of metastases of a chondrosarcoma located in the humerus. On average, there were 57 months between the first surgery for chondrosarcoma and the time of death. The mean age at the time of death in the deceased patients was 44.5 years (range, 29.2–58.9 years) (Fig. 3).
Figure 3.

Kaplan–Meier overall survival curves for group I (blue), group II (green), and group III (black) demonstrating excellent prognosis when enchondromas are restricted to the small bones of the hands and feet (group I). However, as soon as enchondromas are located in the long and flat bones, with or without disease in the small bones as well (group II and group III, respectively), the overall survival time is shorter. The difference between the curves is borderline significant (log-rank trend test, p = .08). Note that there are only zero, four, and seven events in the three groups, respectively, which explains the rather low power of the test. Point estimates (95% confidence intervals) at time = 40 years were: group II, 88% (75%–100%); group III, 85% (70%–100%).
Three nonchondrosarcoma-related deaths concerned patients with Ollier disease who died at an average age of 29 years (range, 21–37 years) from hepatic carcinoma and glioma (n = 2). The patient who died from hepatic carcinoma had previously undergone a surgical resection of a grade I chondrosarcoma in the pelvis; no evidence of local recurrence or distant metastasis was found after 91 months of follow-up (Table 1).
Discussion
Enchondromatosis, of which Ollier disease and Maffucci syndrome are the most common subtypes, is a rare disorder, and descriptive clinical studies are sparse [3]. This study was performed to gain more insight into and a better understanding of the clinical behavior of these disabling diseases.
The reported incidence in previous studies of malignant transformation in enchondromas is variable, and it is estimated to occur in 5%–50% of cases. The cause of the wide variation in the incidence of secondary chondrosarcomas and other related tumors, especially in patients with Maffucci syndrome, is the small numbers of patients in the series published so far [11–15]. The present study recorded the development of one or more chondrosarcomas in 40% of patients. The fact that our data were mainly collected from referral centers for musculoskeletal oncology may have led to a selection bias and the true incidence of malignancy may be slightly lower. It is also unclear whether patients were only referred to these reference centers because they had malignancy suspected or because of the underlying condition. On the other hand, this is the selected group of patients that we deal with in a specialized center, and this study represents what actually happens once the patient visits the hospital.
As a result of the fact that we did not perform a cohort study in which all patients are followed until death, and because a substantial percentage of patients had their first surgery before the actual mean age in our study population, combined with the fact that we did observe malignancies among those patients substantially older than the mean age (32 years), we may expect the young patients in our study group to survive a substantial number of years and thus indeed develop additional malignancies, the probability of which is clearly not negligible.
Fiorenza et al. [27] performed multivariate analysis on independent risk factors for rate of survival in patients with solitary chondrosarcomas of bone. Extracompartmental spread, the development of local recurrence, and high histological grade were defined. Cumulative rates of death in 153 patients at 10 years and 15 years were 30% and 37%, respectively. With respect to mortality in that study, eight patients (5%) died as a result of chondrosarcoma with high-grade malignancy. Compared with the above-mentioned study, the percentage in our series is relative low. Considering the age at time time of the study, however, a higher number of deaths can be expected in the future.
The difference in skeletal deformities of 31% for patients with Ollier disease and 18% for patients with Maffucci syndrome is not statistically significant (Fisher's exact test, p = .40). The odds ratio showed a factor of two difference (95% CI, 0.56–7.5). The variability in the estimates of the percentage of deformities is so large that a difference of both a factor of two lower and a factor of seven higher are compatible with the data. Hence, the difference between 31% and 18% is well within the change fluctuation.
In this study, we tried to define characteristics of enchondromas in patients with Ollier disease and Maffucci syndrome. We discovered that the distinction of a solitary enchondroma from a solitary low-grade central chondrosarcoma is notoriously difficult when analyzing conventional radiographs [28]. Normally, no cortical destruction and soft tissue extension are seen with enchondromas on conventional radiographs. This study shows, however, that, in the case of Ollier disease and Maffucci syndrome, the behavior of the enchondroma is locally more aggressive, and cortical destruction and/or soft tissue extension are seen in 44% of cases. In addition, the aforementioned distinction is also difficult at a histological level [29] and is, as is histological grading, subject to a high level of interobserver variability [30]. In the case of Ollier disease or Maffucci syndrome, the distinction is even more difficult because objective criteria for determining the occurrence of these diseases are lacking and, therefore, in general, more worrisome histological features are tolerated within this context.
This study is hampered by the fact that no central review of radiographs and histology was performed. Comparable studies carried out previously within EMSOS have shown that, in practice, it is too difficult to try to perform this in a retrospective, multicenter study because of, among other reasons, different national regulations regarding tissue handling [31, 32].
To estimate the cumulative probability of secondary transformation over a lifetime, it is important to distinguish the distribution patterns of enchondroma. Patients with enchondromas restricted to the small bones of their hands and/or feet have a relatively low chance (14%, group I) of developing malignancies. In contrast, when enchondromas are found in the long bones or axial skeleton, there is a higher risk (44%–50%) for developing chondrosarcomas (group II and III). The only variable that was significantly associated with a higher risk for developing chondrosarcoma was the occurrence of enchondromas in the pelvis. Patients who have enchondromas located in the pelvis had a 3.8× higher risk for developing chondrosarcoma anywhere in their skeleton. Most importantly, disease-related mortality only occurred in patients with chondrosarcoma of the long or flat bones.
In the literature, various other malignancies have been reported in patients with Maffucci syndrome, such as pancreatic and hepatic adenocarcinoma, mesenchymal ovarian tumors, brain tumors (glioma and astrocytoma), acute myeloid leukemia [16], and various kinds of sarcomas (reviewed by Pansuriya et al. [3]). Only one case has been described in which autopsy-based molecular genetic tests were performed on a 34-year-old man with Ollier disease [33]. Therefore, several authors have advocated an abdominal computed tomography (CT) scan upon the diagnosis of Maffucci syndrome [34]. For Ollier disease, the spectrum of associated malignancies is much smaller, mainly consisting of gliomas and juvenile granulosa cell tumors [3]. Brain tumors in patients with Ollier disease are almost exclusively of glial origin, and patients are almost 10 years younger than patients with Maffucci syndrome when developing brain tumors [21, 35]. Our results are in line with this; two patients (1.2%) developed and died from gliomas and one patient died as a result of hepatic carcinoma. In our series, in 17 patients with Maffucci syndrome, no other malignancies were reported. Therefore, minimal neurological complaints or abdominal symptoms should warrant a cerebral or abdominal CT.
This study found that metastases mainly arose in the lungs, which is in line with conventional chondrosarcoma and should be a guidance for follow-up.
Following the results of this study and summarizing data from the literature, we would like to recommend our opinion in the grading and follow-up of patients with multiple enchondromas. In cases when two or more enchondromas are detected in a patient, the patient should be staged by a technetium scan. X-rays of every single enchondroma should be performed to have a point of departure for the future. If any hemangioma is detected, the patient is diagnosed with Maffucci syndrome, otherwise the patient has Ollier disease. According to the locations of the enchondromas, patients can be divided into one of the above-mentioned groups to assess the risk for developing chondrosarcoma in the future.
In follow up, random periodical x-rays of enchondromas usually give little information. In cases in which patients have dozens or hundreds of enchondromas, in particular, local situations can change at any moment. Patients with enchondromas of the long and/or flat bones, and especially those with enchondromas of the pelvis, should be screened more carefully radiologically using plain x-rays when any complaints of pain, swelling, or neurological disorders appear or increase, whereas for patients with only enchondromas of the short tubular bones of the hands and feet, longer intervals can be used. When cortical and/or soft tissue extension on a plain radiograph is new or increases, a gadolinium (Gd) magnetic resonance imaging (MRI) scan should be performed [28, 36].
When malignant transformation is suspected, a biopsy should be completed. To prevent sampling error resulting from tumor heterogeneity, Gd-MRI can be helpful to increase tissue characterization [36–39]. Depending on the number of suspected lesions, the location of the lesions, and the histology of the biopsy, additional surgical therapy should be carried out. As a result of the fact that the patients with Ollier disease or Maffucci syndrome reported in literature had a higher risk for other malignant tumors, an additional CT scan of the brain or abdomen should always be considered when patients have symptoms [16].
Acknowledgments
The authors would like to thank Leida Rozeman, Hans Vogelaar, Ron Wolterbeek, and Erik van der Zwet for their help with the Excel software and data analysis. This study was financially supported by The Netherlands Organization for Scientific Research (917-76-315 to J.V.M.G.B. and T.C.P.) and EuroBoNet, a European Commission sponsored by the Network of Excellence for studying Bone Tumours (S.H.M.V., J.V.M.G.B., T.C.P., R.J.G., A.L., F.M.K., P.C.W.H, R.B., A.H.M.T.).
Footnotes
- (C/A)
- Consulting/advisory relationship
- (RF)
- Research funding
- (E)
- Employment
- (H)
- Honoraria received
- (OI)
- Ownership interests
- (IP)
- Intellectual property rights/inventor/patent holder
Author Contributions
Conception/Design: Suzan H.M. Verdegaal, Judith V.M.G. Bovée, Pancras C.W. Hogendoorn, Ronald Brand, Antonie H.M. Taminiau
Provision of study material or patients: Judith V.M.G. Bovée, Twinkal C. Pansuriya, Robert J. Grimer, Ronald Brand
Collection and/or assembly of data: Suzan H.M. Verdegaal, Twinkal C. Pansuriya, Robert J. Grimer, Harzem Ozger, Paul C. Jutte, Mikel San Julian, David J. Biau, Ingrid C.M. van der Geest, Andreas Leithner, Arne Streitbürger, Frank M. Klenke, Francois G. Gouin, Domenico A. Campanacci, Perrine Marec-Berard, Antonie H.M. Taminiau
Data analysis and interpretation: Suzan H.M. Verdegaal, Judith V.M.G. Bovée, Pancras C.W. Hogendoorn, Ronald Brand, Antonie H.M. Taminiau
Manuscript writing: Suzan H.M. Verdegaal, Twinkal C. Pansuriya
Final approval of manuscript: Suzan H.M. Verdegaal, Judith V.M.G. Bovée, Twinkal C. Pansuriya, Robert J. Grimer, Harzem Ozger, Paul C. Jutte, Mikel San Julian, David J. Biau, Ingrid C.M. van der Geest, Andreas Leithner, Arne Streitbürger, Frank M. Klenke, Francois G. Gouin, Domenico A. Campanacci, Perrine Marec-Berard, Pancras C.W. Hogendoorn, Ronald Brand, Antonie H.M. Taminiau
References
- 1.Ollier M. Dyschondroplasie. Lyon Med. 1900;93:23–25. [Google Scholar]
- 2.Maffucci A. Di un caso encondroma ed angioma multiplo. Mov Med Chir (Napoli) 1881;3:399–412. 565–575. [Google Scholar]
- 3.Pansuriya TC, Kroon HM, Bovée JV. Enchondromatosis: Insights on the different subtypes. Int J Clin Exp Pathol. 2010;3:557–569. [PMC free article] [PubMed] [Google Scholar]
- 4.Bovée JV, Hogendoorn PC, Wunder JS, et al. Cartilage tumours and bone development: Molecular pathology and possible therapeutic targets. Nat Rev Cancer. 2010;10:481–488. doi: 10.1038/nrc2869. [DOI] [PubMed] [Google Scholar]
- 5.Bovée JV, Cleton-Jansen AM, Taminiau AH, et al. Emerging pathways in the development of chondrosarcoma of bone and implications for targeted treatment. Lancet Oncol. 2005;6:599–607. doi: 10.1016/S1470-2045(05)70282-5. [DOI] [PubMed] [Google Scholar]
- 6.Silve C, Jp¨pner H. Ollier disease. Orphanet J Rare Dis. 2006;1:37. doi: 10.1186/1750-1172-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unni KK. Cartilaginous lesions of bone. J Orthop Sci. 2001;6:457–472. doi: 10.1007/s007760170015. [DOI] [PubMed] [Google Scholar]
- 8.Lucas DR, Bridge JA. Chondromas: Enchondroma, periosteal chondroma, and enchondromatosis. World Health Organization Classification of Tumours. In: Fletcher CDM, Unni KK, Mertens F, editors. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. pp. 237–240. [Google Scholar]
- 9.Baumgart R, Burklein D, Hinterwimmer S, et al. The management of leg-length discrepancy in Ollier's disease with a fully implantable lengthening nail. J Bone Joint Surg Br. 2005;87:1000–1004. doi: 10.1302/0301-620X.87B7.16365. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro F. Ollier's disease. An assessment of angular deformity, shortening, and pathological fracture in twenty-one patients. J Bone Joint Surg Am. 1982;64:95–103. [PubMed] [Google Scholar]
- 11.World Health Organization Classification of Tumours. Lyon, France: IARC Press; 2002. Pathology and Genetics of Tumours of Soft Tissue and Bone; pp. 234–257. [Google Scholar]
- 12.Liu J, Hudkins PG, Swee RG, et al. Bone sarcomas associated with Ollier's disease. Cancer. 1987;59:1376–1385. doi: 10.1002/1097-0142(19870401)59:7<1376::aid-cncr2820590725>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 13.Rozeman LB, Hogendoorn PC, Bovée JV. Diagnosis and prognosis of chondrosarcoma of bone. Expert Rev Mol Diagn. 2002;2:461–472. doi: 10.1586/14737159.2.5.461. [DOI] [PubMed] [Google Scholar]
- 14.Schaison F, Anract P, Coste F, et al. [Chondrosarcoma secondary to multiple cartilage diseases. Study of 29 clinical cases and review of the literature] Rev Chir Orthop Reparatrice Appar Mot. 1999;85:834–845. [PubMed] [Google Scholar]
- 15.Schwartz HS, Zimmerman NB, Simon MA, et al. The malignant potential of enchondromatosis. J Bone Joint Surg Am. 1987;69:269–274. [PubMed] [Google Scholar]
- 16.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours 1. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 17.Lewis RJ, Ketcham AS. Maffucci's syndrome: Functional and neoplastic significance. Case report and review of the literature. J Bone Joint Surg Am. 1973;55:1465–1479. [PubMed] [Google Scholar]
- 18.Mertens F, Unni KK. Enchondromatosis: Ollier disease and Maffucci syndrome. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and bone. Lyon, France: IARC Press; 2002. pp. 356–357. [Google Scholar]
- 19.Pansuriya TC, Bovée JVMG. Enchondromatosis. Atlas Genet Cytogenet Oncol Haematol. 2008. [accessed August 26, 2011]. Available at http://AtlasGeneticsOncology.org/Kprones/EnchondromatosisID10151.html.
- 20.Sun TC, Swee RG, Shives TC, et al. Chondrosarcoma in Maffucci's syndrome. J Bone Joint Surg Am. 1985;67A:1214–1219. [PubMed] [Google Scholar]
- 21.Ranger A, Szymczak A. Do intracranial neoplasms differ in Ollier disease and Maffucci syndrome? An in-depth analysis of the literature. Neurosurgery. 2009;65:1106–1113. doi: 10.1227/01.NEU.0000356984.92242.D5. discussion 1113–1115. [DOI] [PubMed] [Google Scholar]
- 22.McDermott AL, Dutt SN, Chavda SV, et al. Maffucci's syndrome: Clinical and radiological features of a rare condition. J Laryngol Otol. 2001;115:845–847. doi: 10.1258/0022215011909152. [DOI] [PubMed] [Google Scholar]
- 23.Couvineau A, Wouters V, Bertrand G, et al. PTHR1 mutations associated with Ollier disease result in receptor loss of function. Hum Mol Genet. 2008;17:2766–2775. doi: 10.1093/hmg/ddn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopyan S, Gokgoz N, Poon R, et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet. 2002;30:306–310. doi: 10.1038/ng844. [DOI] [PubMed] [Google Scholar]
- 25.Rozeman LB, Sangiorgi L, Bruijn IH, et al. Enchondromatosis (Ollier disease, Maffucci syndrome) is not caused by the PTHR1 mutation p.R150C. Hum Mutat. 2004;24:466–473. doi: 10.1002/humu.20095. [DOI] [PubMed] [Google Scholar]
- 26.Bovée JV, Van der Heul RO, Taminiau AH, et al. Chondrosarcoma of the phalanx: A locally aggressive lesion with minimal metastatic potential: A report of 35 cases and a review of the literature. Cancer. 1999;86:1724–1732. doi: 10.1002/(sici)1097-0142(19991101)86:9<1724::aid-cncr14>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 27.Fiorenza F, Abudu A, Grimer RJ, et al. Risk factors for survival and local control in chondrosarcoma of bone. J Bone Joint Surg Br. 2002;84:93–99. doi: 10.1302/0301-620x.84b1.11942. [DOI] [PubMed] [Google Scholar]
- 28.Geirnaerdt MJ, Hermans J, Bloem JL, et al. Usefulness of radiography in differentiating enchondroma from central grade 1 chondrosarcoma. AJR Am J Roentgenol. 1997;169:1097–1104. doi: 10.2214/ajr.169.4.9308471. [DOI] [PubMed] [Google Scholar]
- 29.Mirra JM, Gold R, Downs J, et al. A new histologic approach to the differentiation of enchondroma and chondrosarcoma of the bones. A clinicopathologic analysis of 51 cases. Clin Orthop Relat Res. 1985;(201):214–237. [PubMed] [Google Scholar]
- 30.Eefting D, Schrage YM, Geirnaerdt MJ, et al. Assessment of interobserver variability and histologic parameters to improve reliability in classification and grading of central cartilaginous tumors. Am J Surg Pathol. 2009;33:50–57. doi: 10.1097/PAS.0b013e31817eec2b. [DOI] [PubMed] [Google Scholar]
- 31.Surgical Subcommittee of the European Osteosarcoma Intergroup. Grimer RJ, Taminiau AM, Cannon SR. Surgical outcomes in osteosarcoma. J Bone Joint Surg Br. 2002;84:395–400. doi: 10.1302/0301-620x.84b3.12019. [DOI] [PubMed] [Google Scholar]
- 32.Grimer RJ, Gosheger G, Taminiau A, et al. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur J Cancer. 2007;43:2060–2065. doi: 10.1016/j.ejca.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Bovée JV, van Roggen JF, Cleton-Jansen AM, et al. Malignant progression in multiple enchondromatosis (Ollier's disease): An autopsy-based molecular genetic study. Hum Pathol. 2000;31:1299–1303. doi: 10.1053/hupa.2000.19308. [DOI] [PubMed] [Google Scholar]
- 34.Morgan DR, Mylankal K, el Barghouti N, et al. Small bowel haemangioma with local lymph node involvement presenting as intussusception. J Clin Pathol. 2000;53:552–553. doi: 10.1136/jcp.53.7.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ranger A, Szymczak A. The association between intracranial tumours and multiple dyschondroplasia (Ollier's disease or Maffucci's syndrome): Do children and adults differ? J Neurooncol. 2009;95:165–173. doi: 10.1007/s11060-009-9924-2. [DOI] [PubMed] [Google Scholar]
- 36.Geirnaerdt MJ, Bloem JL, Eulderink F, et al. Cartilaginous tumors: Correlation of gadolinium-enhanced MR imaging and histopathologic findings. Radiology. 1993;186:813–817. doi: 10.1148/radiology.186.3.8430192. [DOI] [PubMed] [Google Scholar]
- 37.De Beuckeleer LH, De Schepper AM, Ramon F. Magnetic resonance imaging of cartilaginous tumors: Is it useful or necessary? Skeletal Radiol. 1996;25:137–141. doi: 10.1007/s002560050050. [DOI] [PubMed] [Google Scholar]
- 38.Geirnaerdt MJ, Hogendoorn PC, Bloem JL, et al. Cartilaginous tumors: Fast contrast-enhanced MR imaging. Radiology. 2000;214:539–546. doi: 10.1148/radiology.214.2.r00fe12539. [DOI] [PubMed] [Google Scholar]
- 39.Verstraete KL. Gent, Belgium: University of Gent; 1994. Dynamic contrast-enhanced magnetic resonance imaging of tumor and tumor-like lesions of the musculoskeletal system. Thesis. [Google Scholar]