

Abstract
Histone deacetylases (HDACs) are enzymes involved in many important biological functions. They have been linked to a variety of cancers, psychiatric disorders, and other diseases. Since small molecules can serve as probes to study the relevant biological roles of HDACs, novel scaffolds are necessary to develop more efficient, selective drug candidates. Screening libraries of molecules may yield structurally diverse probes that bind these enzymes and modulate their functions in cells. Here we report a small molecule with a novel hydroxy-pyrimidine scaffold that inhibits multiple HDAC enzymes and modulates acetylation levels in cells. Analogs were synthesized in an effort to evaluate structure-activity relationships.
Keywords: Histone deacetylase, non-selective inhibitor, hydroxy-pyrimidine, SAR studies
Post-translational modifications on histones are essential for regulating gene expression.1 A wide variety of chemical modifications, such as methylation, phosphorylation, and acetylation, can alter chromatin structure, influencing levels of gene expression.2 One important modification to histones is N-ε-lysine acetylation. The positive charge of the lysine residues on histone tails allows the negatively charged DNA to tightly coil around histones, contributing to inhibition of transcription. An increase in lysine side chain acetylation has the opposite effect, neutralizing the positive charge and relaxing the DNA from the histone proteins, causing an increase in the amount of transcriptional activity. This tight balance of hyperacetylation and hypoacetylation is maintained by two counteracting family of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively.
Since the initial discovery of the first mammalian HDAC,3 17 additional HDACs have been identified. They are divided into several classes based on sequence homology to their respective yeast orthologs.2 The classical zinc-dependent HDACs are divided into three subclasses, class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), and class IV (HDAC11). Class III HDACs, also known as sirtuins, are zinc-independent and instead require a NAD+ cofactor for deacetylase activity.4 HDACs are present in a variety of organisms and participate in a myriad of biological functions, including post-translational modification of various non-histone substrates. The aberrant activity of HDACs has been connected to numerous diseases, such as inflammation5 and neurosis,6 as well as regulation of tumor suppressor genes and oncogenes, contributing to cancer pathogenesis.2, 7
Using small molecules to inhibit HDACs has provided promising clinical results in different models of cancer, such as cutaneous T-cell lymphoma and neuroblastoma.4, 7–8 Among HDAC inhibitors (HDACi) identified so far, four major classes of structures are frequently studied, including hydoxamates (1), benzamides (2), aliphatic acids (3), and cyclic peptides (4a–b) (Fig. 1).7, 9 Most HDACi follow the pharmacophore model of a capping region that interacts with the surface of the protein, a metal-binding region that chelates zinc to disrupt the enzymatic activity, and a linker domain consisting of a hydrophobic spacer that interacts with the hydrophobic-binding channel (Fig. 1).6b, 10
Figure 1.

Pharmacophore model and representative structures of HDACi. Hydroxamic acid: suberoylanilide hydroxamic acid (SAHA) (1), benzamide: MS-275 (2), aliphatic acid: valproic acid (3), and cyclic peptides: largazole (4a) and trapoxin (4b).
A key motivation for investigating compounds that inhibit HDACs is to find a compound that selectivity targets individual HDACs or a subclass of HDACs. A few compounds have been found to selectivity target HDACs; however progress has been limited.11 Screening diverse small molecules allows for discovery of structurally novel HDACi that may modulate the proteins through different mechanisms or with previously unobserved patterns of selectivity.12 Here we report a structurally novel HDACi, BRD-2577 (7) that was discovered in a binding assay involving a small-molecule microarray (SMM) (Fig. 2a). It is interesting to note that the structure of BRD-2577 is a hydroxy-pyrimidine scaffold, and does not conform to the four main structural classes of HDACi. We further explored the structure-activity-relationships (SAR) and cellular effects of this molecule. Recently, Donald and coworkers evaluated the pyrimidine hydroxamic acid moiety and found that their compounds were able to inhibit HDACs, demonstrating the potential importance of pyrimidines in inhibition of HDACs.13
Figure 2.

(A) Synthesis of BRD-2577 (7). (B) Inhibition profiling of BRD-2577 in triplicate against HDAC1-9. (C) Lineweaver-Burk plot of BRD-2577 at 100 μM (●), 10 μM (■), and 1 μM (▲) for HDAC2.
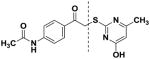
BRD-2577 was originally identified as a binder to purified HDAC8 using SMMs.12 To determine whether the binder inhibits the deacetylase activity of the enzyme, we resynthesized this compound using a nucleophilic substitution reaction between α-chloroketone (5) and mercapto-methylpyrimidine (6) (Fig. 2a) and evaluated it for HDAC inhibitory effects using a trypsin-coupled in vitro biochemical assay (Fig. 2b).14 This biochemical assay employs an acetylated lysine tripeptide substrate amide linked to aminocoumarin (AMC). When HDACs deacetylate the lysine residue, trypsin is able to cleave the AMC from the substrate, thus increasing the fluorescent signal.14–15 Small-molecule HDAC inhibition prevents the deacetylation of the lysine, preventing the generation of fluorescent signal. The effects of various concentrations of BRD-2577 were evaluated, and the data was normalized against a DMSO control. IC50 values were measured by fitting the data to a non-linear regression model. The IC50 values for BRD-2577 against HDAC1-9 ranged from ~11 μM to 74 μM, suggesting it is a non-selective inhibitor of HDACs with modest potency. A counter screen demonstrated that this compound inhibits HDAC activity and does not interfere with the proteolytic activity of trypsin (Supplementary data). The mode of inhibition was also evaluated by analyzing various doses of compound with concentrations of substrate 10 times above and below the Km of HDAC2. The Lineweaver-Burk plot for BRD-2577 and HDAC2 is shown in Figure 2c. The y-intercept does not vary across concentrations of substrate, suggesting that BRD-2577 is a competitive inhibitor, and that the inhibitor is binding to the active site of enzyme.
The structural novelty of BRD-2577 as compared to other HDACi motivated further studies to gain insight into the molecular features that may be responsible for target inhibition. Towards this end, we investigated whether two major substructures of BRD-2577, 5 and 6, were independently capable of inhibition. Neither the α-chloroketone group (5) nor the pyrimidine ring (6) exhibited inhibition in the biochemical assay. This may indicate that activity is not derived from a single biasing element but rather by the whole skeleton.
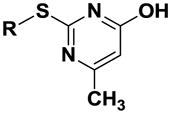
Using simple reactions, we synthesized a set of analogs to investigate the effects of chemical modifications at different positions (Table 1). The initial SAR study focused on the pyrimidine group and its substituent groups. The hydroxyl group was necessary for activity (8–10). The replacement of pyrimidine by a phenyl group resulted in loss of activity (11). Interestingly, the activity was retained when the sulfur atom was substituted by oxygen (12), albeit at a reduced potency and with an altered selectivity pattern. Additionally, removing nitrogen at the 1-position from 12 (13) led to loss of activity, demonstrating that both nitrogen atoms are essential for binding.
Table 1.
SAR on the pyrimidine ring and respective IC50 values.
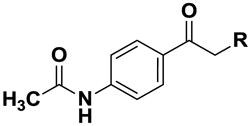 |
HDAC IC50 (μM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 7 |
|
16.4 (±1.08) | 18.9 (±1.10) | 15.2 (±1.05) | 32.5 (±1.07) | 27.9 (±1.05) | 11.2 (±1.09) | 24.4 (±1.07) | 23.9 (±1.07) | 74.1 (±1.10) |
|
| ||||||||||
| 8 |
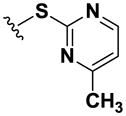
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 9 |
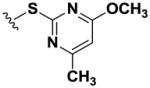
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 10 |
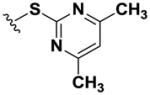
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 11 |
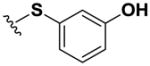
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 12 |
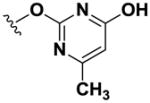
|
>100 | >100 | >100 | 54.2 | 16.7 | 88.0 | 31.9 | >100 | 11.7 |
| 13 |
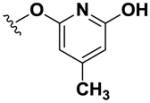
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
Values are means of >2 experiments
Next, we investigated the consequences of chemical modification of the opposing end of the structure of BRD-2577 (Table 2). The removal or the reduction of the carbonyl group (14 and 15, respectively) resulted in a loss of potency, suggesting the carbonyl function is indispensable. Similarly, the modifications and the replacement of the acetamide group with various functionalities (16–22) resulted in decreased potencies for most of the HDACs, while the chloride analog (21) retained some inhibition of HDAC1, 2, 4 and 6. The dihydroxy analog (23) was found only to inhibit HDAC6. Protection of the dihydroxy group via a dioxin (24) led to a completely inactive compound. The acetamide was also replaced with a more rigid oxindole group (25). The role of the phenyl group was further explored. Replacing the phenyl group with a more hydrophobic thiophene (26) preserved inhibitory activity for HDAC6. Replacing the aromatic ring with an aliphatic chain (27–30) was not tolerant to its inhibition activity although 30 retained some activity for HDACs 1, 2, 3, 5, and 6. The linker region between the carbonyl group and phenyl group was also examined by synthesizing compounds 31–33. Although most of the modifications described here lead to inactive analogs, several of the compounds retained inhibition of HDAC6, which is possibly due its unique enzymatic structure consisting of two catalytic domains.8
Table 2.
SAR on the phenyl moiety and respective IC50 values.
 |
HDAC IC50 (μM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 14 |
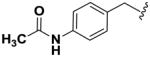
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 15 |
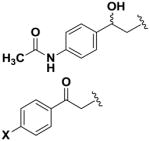
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 16 | X = H | >100 | >100 | >100 | >100 | >100 | 13.7 | >100 | >100 | >100 |
| 17 |
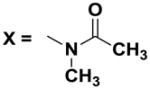
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 18 |
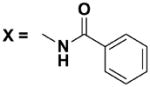
|
>100 | >100 | >100 | >100 | >100 | 10.8 | >100 | >100 | >100 |
| 19 | X = OCH3 | >100 | >100 | >100 | >100 | >100 | 60.9 | >100 | >100 | >100 |
| 20 | X = tBu | >100 | >100 | >100 | >100 | 94.3 | >100 | >100 | >100 | >100 |
| 21 | X = Cl | 82.3 | 67.0 | 86.4 | >100 | >100 | 12.9 | >100 | >100 | >100 |
| 22 | X = OH | >100 | >100 | >100 | >100 | >100 | 40.1 | >100 | >100 | >100 |
| 23 |
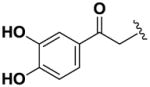
|
>100 | >100 | >100 | >100 | >100 | 16.7 | >100 | >100 | >100 |
| 24 |
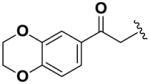
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 25 |
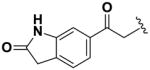
|
>100 | >100 | >100 | >100 | >100 | 32.6 | >100 | >100 | >100 |
| 26 |
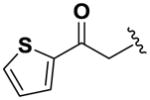
|
>100 | >100 | >100 | >100 | >100 | 24.8 | >100 | >100 | >100 |
| 27 |
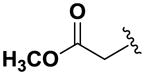
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 28 |
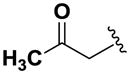
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 29 |
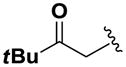
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 30 |
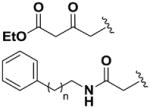
|
77.2 | 69.3 | 40.0 | >100 | 48.6 | 7.2 | >100 | >100 | >100 |
| 31 | n = 3 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 32 | n = 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 33 | n = 5 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
Values are means of >2 experiments
We examined the effects of selected compounds on both histone and α-tubulin lysine acetylation in cells.16 Tubulin acetylation is regulated by HDAC617 and many of the analogs inhibited its activity. HeLa cells were plated in 384-well plates with 2000 cells/well and treated with compounds at various concentrations for 16 h. Acetylation levels were measured using antibodies that recognize acetylated α-tubulin or acetylated lysine residues in post-translationally modified proteins. The fluorescence levels were measured for each antibody and normalized to the DMSO control. Treatment with BRD-2577 leads to an increase in both total lysine and tubulin acetylation (Fig. 3), consistent with our biochemical assay data demonstrating that it inhibits class I and class II HDACs. Other compounds that inhibited HDAC6 in the enzymatic assay were also evaluated for their activity in cells but did not show a significant increase in acetylation level at a concentration of 20 μM (data not shown). Our positive control compound, SAHA, increases both global lysine and α-tubulin acetylation levels, consistent with the ability to inhibit both class I and class II HDACs. MS-275, a control compound, increased global lysine acetylation, but caused little change in tubulin acetylation, which is consistent with its selective inhibition of HDAC1-3 versus HDAC6 in our biochemical assay.
Figure 3.

Immunofluorescence microscopy of cells treated with BRD-2577. (A) Acetylated lysine and tubulin levels plotted against increasing concentration of BRD-2577. (B) Representative images of Hoechst (cyan), acetylated lysine (red), acetylated tubulin (green) for DMSO, BRD-2577 (6 μM), SAHA (0.6 μM), and MS-275 (0.6 μM).
In summary, we have identified a novel, pan-selective HDAC inhibitor from an SMM-based binding screen and demonstrated inhibitory activity against HDACs using an in vitro biochemical assay and cellular acetylation assays. The scaffold does not resemble previously established pharmacophore models for HDAC inhibitors. This novel HDACi structure may be explored further using structure-guided approaches to elucidate binding mechanism and to enhance the potency and selectivity through various structural modifications, leading towards broad biological and therapeutic applications.
Supplementary Material
Acknowledgments
This research was supported in part by a grant from the National Institute of General Medical Sciences (NIGMS 38627 to S.L.S.) and in part with Federal funds from the National Cancer Institute’s Initiative for Chemical Genetics (ICG), under Contract No. N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. S.L.S is an investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cole PA. Nat Chem Biol. 2008;4:590. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Biochem J. 2003;370:737. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taunton J, Hassig CA, Schreiber SL. Science. 1996;272:408. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 4.Witt O, Deubzer HE, Milde T, Oehme I. Cancer Lett. 2009;277:8. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, Bufler P, Fantuzzi G, Goncharov I, Kim SH, Pomerantz BJ, Reznikov LL, Siegmund B, Dinarello CA, Mascagni P. Proc Natl Acad Sci USA. 2002;99:2995. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. J Neurosci. 2007;27:3571. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]; (c) Suzuki T. Chem Pharm Bull (Tokyo) 2009;57:897. doi: 10.1248/cpb.57.897. [DOI] [PubMed] [Google Scholar]
- 7.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. J Hematol Oncol. 2010;3:5. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marks PA, Xu WS. J Cell Biochem. 2009;107:600. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. J Am Chem Soc. 2008;130:11219. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dokmanovic M, Clarke C, Marks PA. Mol Cancer Res. 2007;5:981. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]; (c) Miller TA, Witter DJ, Belvedere S. J Med Chem. 2003;46:5097. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]; (d) Singh EK, Ravula S, Pan CM, Pan PS, Vasko RC, Lapera SA, Weerasinghe SVW, Pflum MKH, McAlpine SR. Bioorg Med Chem Lett. 2008;18:2549. doi: 10.1016/j.bmcl.2008.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sternson SM, Wong JC, Grozinger CM, Schreiber SL. Org Lett. 2001;3:4239. doi: 10.1021/ol016915f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bieliauskas AV, Pflum MKH. Chem Soc Rev. 2008;37:1402. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. J Am Chem Soc. 2010;132:10842. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. J Med Chem. 2008;51:3437. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]; (d) Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. J Med Chem. 2008;51:4370. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G. J Med Chem. 2005;48:3344. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]; (f) Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. Embo J. 2002;21:6820. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Paris M, Porcelloni M, Binaschi M, Fattori D. J Med Chem. 2008;51:1505. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]; (h) Suzuki T, Matsuura A, Kouketsu A, Hisakawa S, Nakagawa H, Miyata N. Bioorg Med Chem. 2005;13:4332. doi: 10.1016/j.bmc.2005.04.002. [DOI] [PubMed] [Google Scholar]; (i) Tang W, Luo T, Greenberg EF, Bradner JE, Schreiber SL. Bioorg Med Chem Lett. 2011;21:2601. doi: 10.1016/j.bmcl.2011.01.134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wang DF, Helquist P, Wiech NL, Wiest O. J Med Chem. 2005;48:6936. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- 12.Vegas AJ, Fuller JH, Koehler AN. Chem Soc Rev. 2008;37:1385. doi: 10.1039/b703568n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Donald ADG, Clark VL, Patel S, Day FA, Rowlands MG, Wibata J, Stimson L, Hardcastle A, Eccles SA, McNamara D, Needham LA, Raynaud FI, Aherne W, Moffat DF. Bioorg Med Chem Lett. 2010;20:6657. doi: 10.1016/j.bmcl.2010.09.016. [DOI] [PubMed] [Google Scholar]; (b) Moffat D, Patel S, Day F, Belfield A, Donald A, Rowlands M, Wibawa J, Brotherton D, Stimson L, Clark V, Owen J, Bawden L, Box G, Bone E, Mortenson P, Hardcastle A, van Meurs S, Eccles S, Raynaud F, Aherne W. J Med Chem. 2010;53:8663. doi: 10.1021/jm101177s. [DOI] [PubMed] [Google Scholar]
- 14.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Nat Chem Biol. 2010;6:238. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wegener D, Wirsching F, Riester D, Schwienhorst A. Chem Biol. 2003;10:61. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 16.(a) Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Proc Natl Acad Sci USA. 2003;100:4389. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wong JC, Hong R, Schreiber SL. J Am Chem Soc. 2003;125:5586. doi: 10.1021/ja0341440. [DOI] [PubMed] [Google Scholar]
- 17.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. Nature. 2002;417:455. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.