

Abstract
Cycloalkanones are easily converted into aryl-substituted cyclic alkenes by the addition of an aryl Grignard reagent followed by dehydration. These alkenes are good substrates for asymmetric epoxidation. We have found that the addition of allylic and benzylic Grignard reagents can occur preferentially at the benzylic position of the derived epoxides, to give the quaternary stereogenic center. This approach led to a short synthesis of the nanomolar serotonin re-uptake inhibitor (−)-mesembrine.
Introduction
Alicyclic rings containing quaternary stereogenic centers1 with attached aromatic rings are a common motif both among physiologically-active alkaloids, including mesembrine 1 and morphine 2, and in medicinal chemistry. We report what promises to be a general enantioselective method for the construction of such stereogenic centers. We have illustrated the power of this approach with a short synthesis of (−)-mesembrine 1.2–4

(−)-Mesembrine 1 was isolated as the major alkaloid component of Sceletium tortuosum, chewed for its stimulant properties by the indigenous population of the Kalahari. In a limited trial,5 the dried and powdered plant preparation, a mild stimulant, was shown to have marked anxiolytic properties. This crude preparation was also shown to be a powerful anti-addictive. The pure alkaloid 1 was recently shown6 to be a nanomolar inhibitor of serotonin re-uptake, with efficacy being observed at a once-a-day oral dose of 100 micrograms.
Results and Discussion
We recently observed (Scheme 1)7 that allylic and benzylic Grignard reagents would open epoxides such as 3 to give selectively the product 4. We envisioned that an epoxide such as 5, with an attached arene,8 might be similarly activated.
Scheme 1.

In the event (Table 1), we were not disappointed. Even a simple non-activating arene9–11 (entry 1) gave a substantial portion of the desired product. An activating p-methyl (entry 2) increased the fraction of the desired secondary alcohol. With the still more activating p-methoxy (entry 3), the cyclic quaternary stereogenic center was formed smoothly.
Table 1.
Opening of Arene-Substituted Cyclic Epoxides
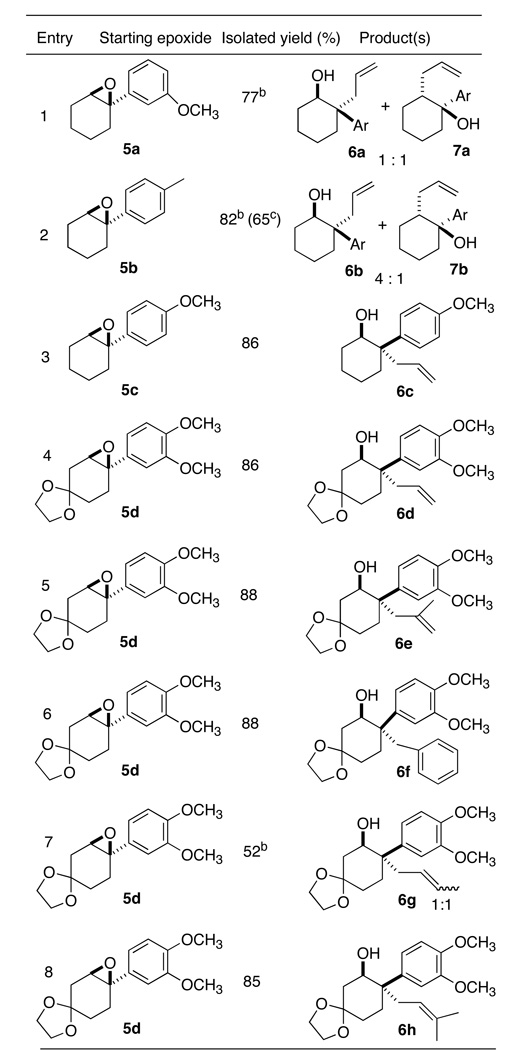 |
Ratio of two isomers was determined by 1H NMR.
Combined yields of two isomers.
Isolated yield of 6b
We also explored alternative nucleophiles with the epoxide 5d10 (Table 1). We found that allylic and benzylic Grignard reagents gave the desired ring-opening products. It was striking that although the alkyne-activated epoxide (Scheme 1) gave the branched product 4 with crotyl magnesium chloride, the arene-activated epoxide gave only the complementary linear product 6g. The less reactive and more basic alkyl, phenyl and vinyl Grignard reagents gave primarily undesired side products. This difference is attributable at least in part to the lack of reactivity of these nucleophiles toward the trisubstituted cyclic expoxide. Et3Al8d gave rearrangement to the cyclohexanone followed by hydride reduction, while Et2AlCN12 gave only rearrangement to the ketone.
The beauty of this approach is that it allows the construction of a quaternary stereogenic center on an existing ring. It is particularly exciting that using the Shi epoxidation,13 one can convert a prochiral cyclic alkene to the enantiomerically-defined product. To establish the viability of this approach, and to confirm the sense of absolute stereocontrol of the process, we converted the monoprotected cyclohexanone 6 (Scheme 2) to (−)-mesembrine (1).
Scheme 2a.

a (a) THF, 0 °C to 20 °C, overnight; (b) benzene, PTSA (cat.), ethylene glycol (excess), reflux; (c) Shi's catalyst, DME-acetonitrile-H2O, 0 °C, 4 h; (d) allylmagnesium chloride, THF, 0 °C to 20 °C, overnight; (e) 10% aqueous HCl, THF, reflux, 1 h; (f) O3, MeOH, −78 °C; CeCl3·7H2O (1.0 equiv), NaBH4 (8.0 equiv), 0°C; (g) TsCl (1 equiv), Et3N, CH2Cl2, 20 °C, overnight; (h) 40% aqueous CH3NH2, THF, 65 °C, 1 h; (i) MnO2, CH2Cl2, 20 °C, 3 h.
The cyclohexanone 8 reacted with the aromatic Grignard reagent 9 to give the known10 alkene 10, after dehydration using PTSA in the presence of excess ethylene glycol. Shi expoxidation13 of 10 followed by ring-opening of the resulting crude epoxide by allylmagnesium chloride gave the enantiomerically enriched secondary alcohol 6d.10 The enantiomers of 6d were well resolved by chiral HPLC (Chiralcel OD, 15% isopropanol / hexane, 1 mL / min, retention time: 17.6 min for the minor enantiomer and 34.2 min for the major enantiomer). The alcohol 6d derived from Shi epoxidation had 96% ee.
Exposure of 6d to 10% aqueous HCl in THF gave the enone 11.4b Selective ozonolysis of the terminal double bond in 11 followed by treatment14 of the resulting ozonide in situ with NaBH4 in the presence of CeCl315 furnished the diol 113h as a 4:1 mixture of diastereomers. The primary hydroxyl group in 11 was selectively converted into the corresponding tosylate 12, which on heating with 40% aqueous methylamine followed by oxidation using activated MnO23h gave (−)- mesembrine (1), ([α]22D = −55.4, lit. = −62.8,3a −63.33d, −53.0, 3e −59.3,3h), identical (1H, 13C NMR) with authentic material.
Conclusion
We believe that the approach outlined here will be a useful strategy for the enantioselective assembly of cyclic quaternary stereogenic centers having attached aromatic rings, complementary to such recently-developed methods as enantioselective allylation16 and enantioselective arylation.17
Experimental
Epoxide (5c)
A solution of 1-(4-methoxyphenyl)-cyclohexene (0.752g, 3.68 mmol) in 2 mL of acetone was added to a solution of dimethyldioxirane4 (50 mL, ~ 0.07 M) in acetone at room temperature. The yellow color of dimethyldioxirane disappeared immediately. After evaporation of acetone, the residue was treated immediately with 10 mL of 2.0 M allylmagnesium chloride as below.
Typical procedure for the ring opening of epoxides with Grignard reagents
To a solution of epoxide (2 mmol) in anhydrous THF (15 mL) was added dropwise a solution of Grignard reagent (10 mmol, 5 equiv) over two minutes at 0 °C. The reaction mixture was warmed slowly to room temperature, then stirred overnight. The reaction mixture was partitioned between saturated aqueous NH4Cl and ether. The combined organic extract was dried (Na2SO4) and concentrated, and the residue was chromatographed.
Alcohol (6b)
Clear oil (58% yield), TLC Rf = 0.46 (PE: acetone, 3:1), Rf = 0.43 for the minor isomer 5f. IR (neat, cm−1): 3456 (s), 2935 (s), 2863 (s), 1638 (w), 1514 (m), 1454 (m), 1239 (m); 1H NMR (400 MHz, CDCl3) δ 7.15–7.30 (m, 4H), 5.23 (m, 1H), 4.92 (d, J= 30.4 Hz, 1H), 4.89 (d, J=23.2 Hz, 1H), 4.02 (t, J=3.2 Hz, 1H), 2.34–2.64 (m, 2H), 2.34 (s, 3H), 1.45–2.10 (m, 9H); 13C NMR (CDCl3, 100 MHz) δ u: 141.9, 135.9, 117.1, 45.2, 41.3, 28.3, 27.0, 21.2, 20.1; d: 134.7, 129.5, 127.0, 74.1, 21.1; HRMS calcd for C16H21 (M+H-H2O) 213.1643, found 213.1644.
Alkene (10)
To a Grignard reagent prepared from Mg (1.82 g, 75.0 mmol) and 4-bromoveratrole (13.0 g, 60.0 mmol) in 40 mL anhydrous THF at 0 °C was added 1,4-dioxaspiro[4,5]decan-8-one 8 (7.80 g, 50.0 mmol) in 20 mL anhydrous THF over 5 min under nitrogen. After stirring at 0 °C for 30 min, then ambient temperature for 2 h, the reaction mixture was partitioned between ether and saturated aqueous NH4Cl. The combined organic extract was dried (Na2SO4) and concentrated. To the residue was added benzene (200 mL), ethylene glycol (30 mL) and p-toluenesulfonic acid (10 mg). A Dean-Stark water separation apparatus was set up, and the solution was heated to reflux until about 0.9 mL of water had been collected. The reaction mixture was partitioned between ether and saturated aqueous NaHCO3. The organic extract was dried (Na2SO4) and evaporated, and the residue was chromatographed to give 8.86 g of alkene 10 (64% yield from 8) as white solid: mp = 76–78 °C (lit.10 = 77.5–78 °C). 1H NMR (400 MHz, CDCl3) δ 6.80–6.95 (m, 3H), 5.91 (m, 1H), 4.01 (s, 4H), 3.88 (s, 3H), 3.87 (s, 3H), 2.64 (m, 2H), 2.46 (br, 2H), 1.90 (t, J= 6.4 Hz, 2H).
Alcohol (6d)
To a 500 mL three –necked flask was added alkene 10 (2.31 g, 8.37 mmol), 85 mL of a 2:1 mixture of dimethoxymethane and acetonitrile, 50 mL of potassium carbonate-acetic acid buffer solution, tetrabutylammonium hydrogen sulfate (63 mg) and Shi’s chiral ketone (0.756 g, 2.93 mmol). The flask was connected to two dropping funnels, one charged with a solution of oxone (7.72 g, 12.6 mmol) in 30 mL of aqueous 4×10−4 Na2EDTA and the other charged with 30 mL of 1.47 M aqueous KOH. The two solutions were added dropwise at the same rate over 65 min to the cold reaction mixture which was stirred vigorously at 0 °C. After 3 h, ether (100 mL) was added. The biphasic mixture was partitioned between ether and, sequentially, water and brine. The organic extract was dried (Na2SO4) and concentrated. The residue was dissolved in 50 mL of anhydrous THF. To this solution allylmagnesium chloride (21 mL, 2.0 M) was added dropwise over 5 min at 0 °C under N2. The reaction solution was warmed slowly from 0 °C to room temperature, then maintained at ambient temperature overnight. The reaction mixture was partitioned between saturated aqueous NH4Cl and ether. The combined organic extract was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed to give 2.04 g of alcohol 6d (73% yield from 10) as a colorless oil. TLC Rf = 0.56 (ether), [α]22D = +37.3 (c 1.6, THF); IR (neat, cm−1): 3514 (s), 2954 (s), 1637 (w), 1587 (w), 1518 (s), 1464 (m), 1256 (s), 1150 (m), 1028 (m); 1H NMR (400 MHz, CDCl3) δ 6.83–6.95 (m, 3H), 5.28 (m, 1H), 4.96 (d, J= 23.6 Hz, 1H), 4.93 (d, J= 17.2 Hz, 1H), 4.16 (t, J= 3.6 Hz, 1H), 3.97 (m, 4H), 3.88 (s, 3H), 3.86 (s, 3H), 3.08 (d, J=8.0 Hz, 1H), 1.65–2.50 (m, 8H); 13C NMR (CDCl3, 100 MHz) δ u: 148.6, 147.1, 137.2, 117.3, 108.7, 64.6, 64.1, 44.6, 41.6, 36.7, 30.5, 25.2; d: 134.3, 119.1, 110.8, 110.3, 74.3, 55.8, 55.7; HRMS calcd for C19H26O5 (M+) 334.1780, found 334.1774.
Enone (11)
To a solution of alcohol 6d (0.90 g, 2.7 mmol) in 10 mL of THF was added 10% aqueous HCl (4 mL). After heating to reflux for 1 h, the reaction mixture was partitioned between saturated aqueous Na2CO3 and CH2Cl2. The combined CH2Cl2 extract was dried (Na2SO4) and concentrated in vacuo. The residue was chromatographed to give 0.67 g of enone 11 (92% yield) as a colorless oil, TLC Rf = 0.69 (PE:EtOAc=1:1), [α]22D = −107.6 (c 0.8, THF); IR (neat, cm−1): 2935 (m), 1682 (s), 1639 (w), 1603 (w), 1518 (s), 1464 (m), 1255 (s), 1148 (m), 1027 (m); 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J=10.0 Hz, 1H), 6.86 (s, 3H), 6.17 (d, J=10.0 Hz, 1H), 5.56 (m, 1H), 5.11 (d, J= 15.2 Hz, 1H), 5.08 (d, J= 8.4 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 2.15–2.73 (m, 6H); 13C NMR (CDCl3, 90 MHz) δ u: 199.6, 149.1, 148.0, 135.4, 119.0, 46.3, 43.6, 36.1, 34.5; d: 155.3, 133.6, 129.5, 119.4, 111.1, 110.2, 56.1, 55.9; HRMS calcd for C17H21O3 (M+H) 273.1491, found 273.1490.
Diol (12)
To a solution of enone 11 (0.663 g, 2.43 mmol) in HPLC grade methanol (20 mL) was added an indicator amount of Sudan III. After purging with O2 at −78 °C for 2 min, O3 was bubbled through the solution until the red color of Sudan III disappeared. The reaction was purged with N2 for 5 min, then CeCl3·7H2O8 (1.00 g, 2.68 mmol) was added. The reaction mixture was cooled with an ice-water bath, then NaBH4 (554 mg, 14.6 mmol) was added in three portions over 5 min. After an additional 30 min, NaBH4 (92 mg, 2.43 mmol) was added followed by stirring for another 30 min. The latter procedure was repeated once more. The reaction mixture was partitioned between water and CH2Cl2. The combined organic extract was dried (Na2SO4) and concentrated, and the residue was chromatographed to give 0.474 g of diol 11 (4:1 mixture of two diasteromers, 73% yield) as a colorless oil. TLC Rf = 0.18 (wet ether), [α]22D = −23.5 (c 0.55, THF); IR (neat, cm−1): 3390 (b), 2941 (s), 1715 (m), 1520 (s), 1260 (s), 1026 (s); 1H NMR (400 MHz, major isomer, CDCl3) δ 6.78–6.82 (m, 3H), 5.86–5.95 (m, 2H), 4.21 (m, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.47–3.58 (m, 2H), 1.62–2.07 (m, 7H); 13C NMR (CDCl3, major isomer, 90 MHz) δ u: 148.8, 147.4, 139.1, 59.7, 45.0, 41.8, 35.0, 28.9; d: 134.9, 131.7, 119.4, 110.9, 110.5, 67.2, 56.1, 56.0; 1H NMR (400 MHz, minor isomer, CDCl3) δ 6.80–6.82 (m, 3H), 5.60–6.06 (m, 2H), 4.13 (m, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.55–3.65 (m, 2H), 1.62–2.10 (m, 7H); 13C NMR (CDCl3, minor isomer, 90 MHz) δ u: 148.9, 147.5, 139.0, 59.7, 44.7, 41.9, 32.6, 28.5; d: 136.5, 130.0, 119.0, 111.1, 110.3, 64.6, 56.1, 56.0; HRMS calcd for C16H22O4Na (M+Na) 301.1416, found 301.1412.
Tosylate (13)
To a solution of diol 12 (major isomer, 133 mg, 0.478 mmol) in 5 mL of anhydrous CH2Cl2 was added triethylamine (145 mg, 1.44 mmol) and tosyl chloride (91.2 mg, 0.478 mmol). After stirring under N2 at ambient temperature for 16 h, the reaction mixture was partitioned between saturated aqueous Na2CO3 and CH2Cl2. The combined organic extract was dried (Na2SO4) and concentrated. The crude product was chromatographed to give 0.186 g of tosylate 13 (90% yield) as a clear oil. TLC Rf = 0.34 (ether), [α]22D = −11.4 (c = 0.35, THF); IR (neat, cm−1): 3390 (b), 2938 (s), 1598 (s), 1464 (m), 1359 (s), 1252 (s), 1176 (s); 1H NMR (400 MHz, CDCl3) δ7.69 (d, J=8.0 Hz, 2H), 7.30 (d, J=8.0 Hz, 2H), 6.75 (br, 3H), 5.90 (dd, J=10.0 Hz, 1.2 Hz, 1H), 5.78 (d, J=10.0 Hz, 1H), 4.19 (br, 1H), 3.81–3.98 (m, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 2.44 (s, 3H), 1.67–2.17 (m, 7H); 13C NMR (CDCl3, 100 MHz) δ u: 149.0, 147.7, 144.9, 137.8, 133.2, 67.8, 41.8, 40.9, 35.0, 28.8; d: 133.7, 132.5, 129.9, 127.9, 119.4, 111.1, 110.3, 67.0, 56.1, 56.0, 21.8; HRMS calcd for C23H28O6S (M+) 432.1607, found 432.1605.
(−)-Mesembrine (1)
To a sealed thick-walled reaction flask was added tosylate 13 (144 mg, 0.33 mmol), methylamine (2.0 mL, 40% in water), and THF (4.0 mL). The reaction mixture was heated at 65 °C for 1 h, then concentrated in vacuo, and the residual oil was dissolved in CH2Cl2 (15 mL). To this solution activated MnO2 (220 mg, 2.53 mmol) was added, and the mixture was stirred at rt for 3 h. After filtration through a thin layer of Celite, the filtrate was concentrated and the residue was chromatographed to give 58.8 mg of (−)-mesembrine (61% yield) as a colorless oil. TLC Rf = 0.21(wet ether), [α]22D = −55.4 (c = 0.54, MeOH); IR (neat, cm−1): 2944 (m), 1716 (s), 1520 (s), 1454 (m), 1253 (s), 1148 (m), 1027 (m); 1H NMR (400 MHz, CDCl3) δ 6.93 (dd, J= 8.4, 2.4 Hz, 1H), 6.90 (d, J=2.0 Hz, 1H), 6.84 (d, J=8.4 Hz, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.13–3.17 (m, 1H), 2.96 (t, J=3.6 Hz, 1H), 2.61 (m, 2H), 2.35–2.46 (m, 2H), 2.33 (s, 3H), 2.04–2.26 (m, 5H); 13C NMR (CDCl3, 90 MHz) δ u: 211.5, 149.3, 147.8, 140.4, 55.1, 47.8, 40.8, 39.1, 36.4, 35.5; d: 118.2, 111.3, 110.3, 70.6, 56.2, 56.1, 40.3; HRMS calcd for C17H23NO3 (M+) 289.1678, found 289.1671.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH (GM60287) for financial support of this study, and DSM for a gift of Shi ketone.
Footnotes
SUPPORTING INFORMATION. General experimental details, characterization of the products from Table 1, and spectra for all new compounds and for 1. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.For reviews on the enantioselective construction of quaternary stereogenic centers, see: Martin SF. Tetrahedron. 1980;36:419. Fuji K. Chem. Rev. 1993;93:2037. Corey EJ, Guzman-Perez A. Angew. Chem. Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. Christoffers J, Mann A. Angew. Chem. Int. Ed. 2001;40:4591. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v. Dennisova I, Barriault L. Tetrahedron. 2003;59:10105. Douglas CJ, Overman LE. Proc. Nat. Acad. Sci. 2004;101:5363. doi: 10.1073/pnas.0307113101.
- 2.For the isolation and structural determination of (−)-mesembrine, see Popelak A, Haack E, Lettenbauer G, Spingler H. Naturwissenschaften. 1960;47:156. Smith E, Hosansky N, Shamma M, Moss JB. Chem. & Ind. 1961:402.
- 3.For leading references to previous enantioselective syntheses of (−)-mesembrine, see Takano S, Imamura Y, Ogasawara K. Tetrahedron Lett. 1981;22:4479. Takano S, Samizu K, Ogasawara K. Chem. Lett. 1990:1239. Fukumoto K, Tanabe T, Nemoto H. J. Org. Chem. 1995;60:6785. Mori M, Kuroda S, Zhang C, Sato Y. J. Org. Chem. 1997;62:3263. doi: 10.1021/jo9701187. Denmark SE, Marcin LR. J. Org. Chem. 1997;62:1675. Langlois Y, Dalko PI, Brun V. Tetrahedron Lett. 1998;39:8979. Ogasawara K, Yamada O. Tetrahedron Lett. 1998;39:7747. Taber DF, Neubert TD. J. Org. Chem. 2001;66:143. doi: 10.1021/jo001237g. For leading references to previous enantioselective syntheses of (+)-mesembrine see Meyers AI, Hanreich R, Wanner KT. J. Am. Chem. Soc. 1985;107:7776. Kosugi H, Miura Y, Kanna H, Uda H. Tetrahedron Asymmetry. 1993;4:1409.
- 4.For leading references to the synthesis of racemic mesembrine, see Chavan SP, Khobragade DA, Pathak AB, Kalkote UR. Tetrahedron Lett. 2004;45:5263. Kulkarni MG, Rasne RM, Davawala SI, Doke AK. Tetrahedron Lett. 2002;43:2297. Rigby JH, Dong W. Organic Lett. 2000;2:1673. doi: 10.1021/ol005706c.
- 5.For the cultural background and some limited activity trials of Sceletium tortuosum, see Smith MT, Crouch N, Gericke N, Hirst M. J. Ethnopharm. 1996;50:119. doi: 10.1016/0378-8741(95)01342-3.
- 6.For the in vitro and in vivo activity of (−)-mesembrine, see Gericke NP, VanWyk B-E. PCT Int. Appl. 1997 WO 9746234 CAN 128:80030.
- 7.Taber DF, He Y, Xu M. J. Am. Chem. Soc. 2004;126:13900. doi: 10.1021/ja045849k. [DOI] [PubMed] [Google Scholar]
- 8.For reports of the opening of arene-activated trisubstituted epoxides at the more substituted carbon to establish a quaternary center, see: Gerteisen TJ, Kleinfelter DC. J. Org. Chem. 1971;36:3255. Krause N, Seebach D. Chem. Ber. 1988;121:1315. Kauffmann T, Neiteler C, Robbe S. Chem. Ber. 1992;125:2409. Jansen J, Knopp M, Amberg W, Bernard H, Koser S, Müller S, Münster I, Pfeiffer T, Riechers H. Org. Process Research & Development. 2001;5:16.
- 9.For the preparation of the alkenes corresponding to 5a and 5c, see Balsamo A, Crotti P, Macchia B, Macchia F. Tetrahedron. 1973;29:2183. (b) For the preparation of the alkenes corresponding to 5b and 5c, see Doan L, Bradley K, Gerdes S, Whalen DL. J. Org. Chem. 1999;64:6227.
- 10.For the preparation of 5d, see Hoshino O, Sawaki S, Shimamura N, Onodera A, Umezawa B. Chem. Pharm. Bull. 1987;35:2734.
- 11.We found that the sensitive epoxides could be prepared efficiently from the alkenes with dimethyl dioxirane: Murray RW, Singh M. Org. Syn. 1997;74:91.
- 12.Newbold RC, Shih TL, Mrozik H, Fisher MH. Tetrahedron Lett. 1993;34:3825. [Google Scholar]
- 13.Tu Y, Wang Z-X, Shi Y. J. Am. Chem. Soc. 1996;118:9806. [Google Scholar]
- 14.Sousa JA, Bluhm AC. J. Org. Chem. 1960;25:108. [Google Scholar]
- 15.Gemal AL, Luche J-L. J. Am. Chem. Soc. 1981;103:5454. [Google Scholar]
- 16.(a) Trost BM, Pissot-Soldermann C, Chen I, Schroeder GM. J. Am. Chem. Soc. 2004;126:4480. doi: 10.1021/ja0497025. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]
- 17.(a) Aahman J, Wolfe JP, Troutman MV, Palucki M, Buchwald SL. J. Am. Chem. Soc. 1998;120:1918. [Google Scholar]; (b) Hamada T, Chieffi A, Ahman J, Buchwald SL. J. Am. Chem. Soc. 2002;124:1261. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.