

Abstract
High affinity and selective small molecule agonists of the S1P4 receptor (S1P4-R) may have significant therapeutic utility in diverse disease areas including autoimmune diseases, viral infections and thrombocytopenia. A high-throughput screening (HTS) of the Molecular Libraries-Small Molecule Repository library identified 3-(2-(2,4-dichlorophenoxy)ethoxy)-6-methyl-2-nitropyridine as a moderately potent and selective S1P4-R hit agonist. Design, synthesis and systematic structure-activity relationships study of the HTS-derived hit led to the development of novel potent S1P4-R agonists exquisitely selective over the remaining S1P1–3,5–Rs family members. Remarkably, the molecules herein reported provide novel pharmacological tools to decipher the biological function and assess the therapeutic utility of the S1P4–R.
Keywords: S1P4 receptor, selective small molecule S1P4–R agonists, autoimmune diseases, viral infections, thrombocytopenia
Sphingosine-1-phosphate (S1P) is a sphingolipid mediator formed by the phosphorylation of sphingosine (SPH) and involved in multiple physiological and pathological processes. Among them, lymphocyte trafficking, angiogenesis, tumorigenesis as well as vascular development and permeability have been extensively explored.1–4 The involvement of S1P in these processes results from its ability to modulate important cellular events such as cytoskeletal changes, chemotaxis, survival and proliferation.5–7 It has become clear that the role of S1P in immunological response is not restricted to the regulation of lymphocyte trafficking but extends to the control of immune cell function.8 The generation of S1P is mediated by two cytosolic sphingosine kinase isoforms (SPK1 and SPK2) and occurs preferentially in the plasma membrane.6,8 The site of action of S1P is not merely intracellular since it is exported out of the cell and binds to five G-protein-coupled receptors named S1P1–5-Rs, in a paracrine or autocrine manner.4,6 In addition to its effects on S1P1–5-Rs, S1P can also affect cell function by either binding or modifying putative intracellular targets or by affecting the relative levels of other lipid products, particularly SPH and ceramide whose biological effects oppose those of S1P.6,7 S1P1,2,3-Rs are ubiquitously expressed in almost all organs in mice and humans whereas S1P4-R and S1P5-R expression is restricted to specific organs and cell types. S1P4-R is predominatly expressed in lymphoid and hematopoietic cells.9 S1P4–R couples to Gαi, Gαo and Gα12/13 proteins leading to the stimulation of MAPK/ERK signaling pathways, as well as PLC and Rho-Cdc42 activation.10,11
Both, S1P1-R and S1P4-R are the most widely expressed S1P-Rs on lymphocytes and dendritic cells (DCs).12,13 In contrast to S1P1-R, the function of S1P4-R in fundamental immunological processes has been poorly characterized. However, the contribution of S1P4–R to the immune response is becoming increasingly evident. S1P induces migratory response of murine T-cell lines expressing both S1P1–R and S1P4–R mRNA. In D10.G4.1 and EL-4.IL-2 murine cells S1P-induced migration was significantly inhibited by treatment with (S)-FTY720-phosphate, a potent agonist at S1P1–R and S1P4–R. In murine CHO cells co-expressing S1P4–R and S1P1–R on the cell surface S1P-induced T-cell migration involved the activation of Rho family small GTPase, Cdc42 and Rac. These results have suggested that the association of S1P4–R and S1P1–R may play an important role in the migratory and recirculation response of T-cells toward S1P.14 Intratracheal delivery of synthetic sphingosine analogs with mixed activity over S1P1,3–5–Rs efficiently inhibited the T-cell response to influenza virus infection by impeding the accumulation of DCs in draining lymph nodes. The inhibitory effects were not observed upon specific chemical activation of S1P1–R, and persisted in S1P3–R null mice. Based on these findings and on the observation that S1P5-R expression in DCs is very low whereas S1P4–R is highly expressed, it has been hypothesized that the S1P4–R modulation in the lung may be effective at controlling the immunopathological response to viral infections.15,16.
S1P4-R signaling has been proposed to negatively affect T-cell proliferation and modify the cytokine secretion profile of T-lymphocytes.17 However, these observations were derived from S1P4-R overexpressing cell lines and need further confirmation. Recently, S1P4-R has been implicated in the regulation of DC function and TH17 T-cell differentiation in a murine model.18 Interestingly, S1P4-R-mediated S1P signaling also modifies the course of various immune diseases in a murine model. Hence, it has been hypothesized that S1P4-R may constitute an interesting target to influence the course of various autoimmune diseases.
In vitro and in vivo experiments have indicated an additional potential therapeutic application of S1P4–R molecule modulators in the terminal differentiation of megakaryocytes. Namely, the application of S1P4–R antagonists might be exploited for inhibiting potentially detrimental reactive thrombocytosis, whereas S1P4–R agonists represent a potential therapeutic approach for stimulating platelet repopulation after thrombocytopenia.19
To date, despite the S1P4–R therapeutic potential, the in vivo function of the target receptor remains largely unknown due to the paucity of selective small molecules S1P4-R modulators.
Recently, our research group identified the 3-methyl-2-((2-methoxyethyl)imino)thiazolidin-4-one 1 as a novel and selective S1P4-R agonist (Figure 1).20 Herein we report on the discovery and structure-activity relationships (SAR) of novel potent and selective S1P4-R agonists based on the hit 3-(2-(2,4-dichlorophenoxy)ethoxy)-6-methyl-2-nitropyridine 2. A high-throughput screening (HTS) of the Molecular Libraries-Small Molecule Repository (MLSMR) library identified 2 as a novel, moderately potent and selective S1P4-R agonist. The structural integrity of the hit was corroborated by the resynthesis (Scheme 1) of the title compound that showed confirmed EC50’s of 162 nM at S1P4-R and no agonistic activity at S1P1–3,5-Rs at concentrations up to 25 μM (Figure 1).
Figure 1.

Novel and selective S1P4-R agonists
Scheme 1.

Synthesis of 8a–8s
Reagents and conditions (i) 3 (1 equiv.), 4 (4 equiv.), K2CO3 (2 equiv.), DMSO, 40°C, 4h, 65%; (ii) 5 (1 equiv.), 6 or 7 (2 equiv.), K2CO3 (2 equiv.), DMSO, 40°C, 4h, 50–98%.
Our SAR studies commenced varying the 2,4-dichlorophenyl coil of the hit as outlined in Scheme 1. Alkylation of 3-hydroxypyridine 3 with 1,2-dibromoethane 4 led to the key intermediate 5, which was subsequently reacted with various hydroxyaryl derivatives 7 to furnish compounds 8a–8s. The biological results of the entitle molecules are listed in Table 1.23
Table 1.
S1P4-R agonist activity of 8a–8s
| cpd | Ar | EC50 (nM)α | |
|---|---|---|---|
| 8a | I | 2-chlorophenyl | NA |
| 8b | II | 4-chlorophenyl | NA |
| 8c | III | 2,5-dichlorophenyl | NA |
| 8d | IV | 2,3-dichlorophenyl | NA |
| 8e | V | 3,4-dichlorophenyl | NA |
| 8f | VI | phenyl | NA |
| 8g | VII | 2-bromo-4-chlorophenyl | 232 |
| 8h | VIII | 4-bromo-2-chlorophenyl | 220 |
| 8i | IX | 2-chloro-4-methoxy phenyl | 3900 |
| 8j | X | 4-chloro-2-methoxyphenyl | 5400 |
| 8k | XI | 2,4-dimethylphenyl | 7200 |
| 8l | XII | 2-chloro-4-phenylphenyl | NA |
| 8m | XIII | 4-chloro-2-fluorophenyl | 3100 |
| 8n | XIV | 2-chloro-4-fluorophenyl | 2600 |
| 8o | XV | 2,4-difluorophenyl | 40% 50μM |
| 8p | XVI | 2-fluoro-4-nitrophenyl | 2800 |
| 8q | XVII | 5-chloro-3-nitropyridin-2-yl | 3500 |
| 8r | XVIII | 4-bromo-2-(trifluoromethyl)phenyl | 601 |
| 8s | XIX | 2,4,6-trichlorophenyl | 570 |
Data are reported as mean of n = 3 determinations. NA = not active at concentrations up to 25 μM.
Removal of the chlorine from positions 2 and/or 4 of the aromatic ring (8a, 8b, 8f) led to complete loss of activity. Dichloro-regioisomers 8c, 8d, 8e and 2-chloro-4-phenyl analog 8l were also inactive. Drastic loss of potency was observed when the chlorines were substituted with fluorine at one or both positions (8m, 8n, 8o) as well as for the 2- or 4-methoxy derivatives (8j, 8i) and for the 2,4-dimethyl analog (8k). Interestingly, the 4-nitro derivative 8p showed week but similar activity compared to the 4-chloro analog 8m. The pyridinyl derivative 8q resulted in a 22-fold decrease in potency compared to the hit. Conversely, the potency remained similar by replacing the chlorine at position 2 or 4 with bromine (8g, 8h). Interestingly, the 4-bromo-2-trifluoromethyl analog 8r was only 3-fold less potent than the corresponding 2-chloro analog 8h. The symmetrically substituted 2,4,6-trichloro-phenyl analog 8s was also 3-fold less potent than the hit suggesting a steric clash of the trisubstituted phenyl ring within the binding pocket.
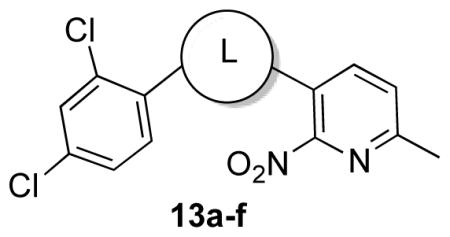
Next, we synthesized compounds 13a–13g (Schemes 2–6) in order to explore the ethylenedioxy spacer while maintaining the 2,4-dichlorophenyl coil and the 6-methyl-2-nitropyridine polar head.
Scheme 2.

Synthesis of 13a, 13b, 13c
Reagents and conditions: (i) 6 (1 equiv.), 1,3-dibromopropane (4 equiv.), K2CO3 (2 equiv.), DMSO, 40°C, 4h, 65%; (ii) 10 (1 equiv.), PBr3 (0.33 equiv.), CH2CI2, 0°C to rt, 3h, 85%; (iii) 9 or 11 or 12 (1 equiv.), 3 (1 equiv.), K2CO3 (2 equiv.), 40°C, 4h, 75–90%.
Scheme 6.

Synthesis of 13g
Reagents and conditions: (i) 19 (1 equiv.), 14 (1.5 equiv.), PPA, 130°C, 2h, 45%.
Intermediate 9 was synthesized by alkylating 2,4-dichlorophenol 6 with 1,3-dibromopropane under basic conditions. Alcohol 10 was reacted with phosphorus tribromide (PBr3) at room temperature to furnish the corresponding bromide 11. Compounds 13a, 13b and 13c were obtained by the alkylation of 3 with 9, 11 and 12 respectively under basic conditions (Scheme 2).
Commercially available carboxylic acid 14 was converted into the corresponding acyl chloride using thionyl chloride (SOCl2) followed by condensation with 3 to furnish the desired product 13d (Scheme 3).
Scheme 3.

Synthesis of 13d
Reagents and conditions: (i) 14 (1 equiv.), SOCI2, reflux, 2h; (ii) 3 (1.2 equiv.), DIPEA (1.2 equiv.), CH2CI2, 0°C to rt, overnight, 80% (over two steps).
The cyclopentene oxide 15 was reacted with 6 followed by mesylation of the alcohol intermediate to furnish the mesylated derivative 16. The final cyclic compound 13e was obtained by condensation of 3 with 16 (Scheme 4).
Scheme 4.

Synthesis of 13e
Reagents and conditions: (i) 6 (1 equiv.), 15 (2.1 equiv.), K2CO3 (4 equiv.), EtOH, 80°C, overnight; (ii) MeSO2CI (2 equiv.), pyridine, 0°C, 6h, 70% (over 2 steps); (iii) (a) 3 (1 equiv.), KOH, H2O/EtOH, 80°C, 30 min; (b) 16 (1 equiv.), DMF, 120°C, overnight, 60%.
The aza-analog 13f was obtained via aromatic nucleophilic substitution of commercially available triflate 17 with amine 18 (Scheme 5).
Scheme 5.

Synthesis of 13f
Reagents and conditions: (i) 17 (1 equiv.), 18 (1 equiv.), DMF, 100°C, 12h, 65%.
The oxazolo[4,5-b]pyridine 13g was synthesized by condensing hydroxypyridine 19 with carboxylic acid 14 using polyphosphoric acid (PPA) (Scheme 6).
The biological results of 13a–13f are listed in Table 2.
Table 2.
S1P4-R agonist activity of 13a–13f
 13a–f | ||
|---|---|---|
| cpd | L | EC50 (nM)α |
| 13a |
|
NA |
| 13b |
|
920 |
| 13c |
|
NA |
| 13d |
|
NA |
| 13e |
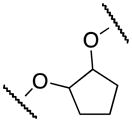
|
NA |
| 13f |
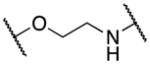
|
NA |
Data are reported as mean of n = 3 determinations. NA = not active at concentrations up to 25 μM.
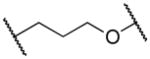
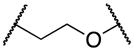
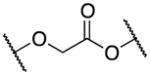
Elongating the alkyl chain (13a) as well as introducing a carbonyl group (13d) resulted in complete loss of activity. Furthermore, the constrained 5-membered ring analog 13e was inactive. Interestingly, the methylene analogs 13c and 13b were respectively inactive and 5–6 fold less active than the hit suggesting that the replaced oxygen might be involved in a hydrogen bond interaction. Surprisingly, the aza-analog 13f was inactive suggesting that the hydrogen bond acceptor capability in this portion of the molecule is an essential binding requirement. Merging the spacer and the head led to oxazolo[4,5-b]pyridine 13g, which was found inactive in concentrations up to 25 μM. Cumulatively, these data suggest that the ethylenedioxy spacer is an essential structural feature for the binding activity and is highly restricted in terms of length, bulkiness and conformation.
Successively, we focused on the study of the polar head while retaining the 2,4-dichlorophenyl coil and the ethylenedioxy chain. The synthesis of the target molecules 22a–22aa is represented in Scheme 7. The alkyation of 6 with 4 yielded the key bromide intermediate 20. A diverse set of hydroxyaryl analogs 21I–21XVIII was reacted with 20 to furnish the desired products 22a–22e, 22h–22s, 22v. Hydrolysis of methylester 22e under standard conditions yielded the carboxylic acid 22g, which was converted into 22f via standard amide coupling conditions. Methyl pyridine analog 22p was reacted with meta-chloroperbenzoic acid (mCPBA) to form the corresponding N-oxide which was converted into the alcohol derivative 22t using the Boekelheiden reaction.21 Oxidation of 22t using manganese dioxide (MnO2) under standard conditions gave aldehyde 22u. Oxidation of alcohol 22v using previously described conditions furnished the aldehyde 22w. Analogs 22y and 22z were then synthesized employing the Wittig reaction between aldehyde 22w and the corresponding phosphonium ylide. The 6-methoxymethyl analog 22x was synthesized by metylation of the alcohol 22v with methyl iodide. The fluoro derivative 22aa was obtained from alcohol 22v via displacement of the mesylate using tetrabutylammonium fluoride (TBAF).
Scheme 7.

Synthesis of 22a–22aa
Reagents and conditions: (i) 6 (1 equiv.), 4 (4 equiv.), K2CO3 (2 equiv.), DMSO, 40°C, 6h, 75%; (ii) 20 (1 equiv.), 21I–21XVIII (1.1 equiv.), K2CO3 (1.5 equiv.), DMSO, 40°C, 6h, 70–95%; (iii) 22v (1 equiv.), MnO2 (5 equiv.), CHCI3, rt, 3h, 67%; (iv) (a) CH3PPh3+Br− (2 equiv.), BuLi (2 equiv.), THF, 0°C, 30min; (b) 22w (1 equiv.), 0°C to rt, 2h, 60%; (v) (a) (CH3)2CHPPh3+Br− (2 equiv.), BuLi (2 equiv.), THF, 0°C, 30 min; (b) 22w (1 equiv.), 0°C to rt, 2h, 40%; (vi) 22v (1 equiv.), NaH (1.5 equiv.), Mel (5 equiv.), DMF, 0°C to rt, overnight, 72%; (vii) (a) 22v (1 equiv.), MsCI (1.1 equiv.), Et3N (3 equiv.), 0°C to rt, overnight; (b) TBAF (1M THF) (1.5 equiv.), acetonitrile, 50°C, 12h, 60% (over two steps); (viii) 22e (1 equiv.), LiOH (1.1 equiv.), MeOH/THF/H2O (3:1:1), rt, 2h, 70%; (ix) 22g (1 equiv.), NHMe2. HCI (1.2 equiv.), DIPEA (1.2 equiv.), EDCI (1.3 equiv.), HOBt (1.3 equiv.), CH2CI2, rt, 3h, 88%; (x) (a) 22p (1 equiv.), MCPBA (12 equiv.), CH2CI2, rt, 6h; (b) (CF3CO)2O (1 equiv.), DMF, 0°C to rt, 24h; (c) Na2CO3, rt, 1h, 39% (over three steps); (xi) 22t (1 equiv.), MnO2 (5 equiv.), CHCI3, rt, 3h, 65%.
The biological results of 22a–22aa are listed in Table 3.
Table 3.
S1P4-R agonist activity of 22a–22aa
| cpd | Ar2 | EC50 (nM)α | |
|---|---|---|---|
| 22a | I |
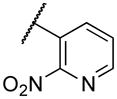
|
2980 |
| 22b | II |
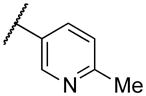
|
NA |
| 22c | III |
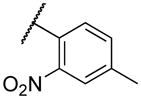
|
1300 (40%)β |
| 22d | IV |
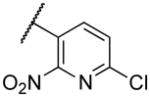
|
304 |
| 22e | V |
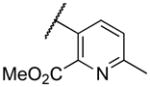
|
4800 |
| 22f |
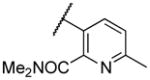
|
NA | |
| 22g |
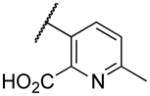
|
NA | |
| 22h | VI |
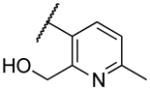
|
NA |
| 22i | VII |
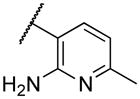
|
NA |
| 22j | VIII |
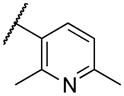
|
2600 (65%)β |
| 22k | IX |
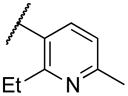
|
7300 |
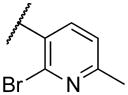
| 221 | X |
|
52 |
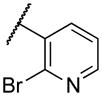
| 22m | XI |
|
1300 |
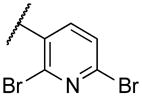
| 22n | XII |
|
46 |
| 22o | XIII |
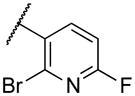
|
208 |
| 22p | XIV |
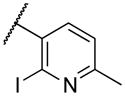
|
54 |
| 22q | XV |
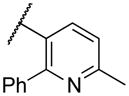
|
NA |
| 22r | XVI |
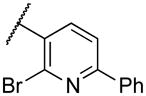
|
6000 (70%)β |
| 22s | XVII |
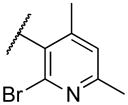
|
NA |
| 22t |
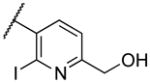
|
287 | |
| 22u |
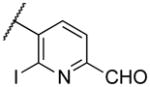
|
2100 | |
| 22v | XVIII |
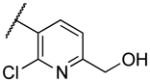
|
1500 |
| 22w |
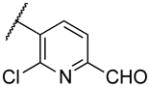
|
1800 | |
| 22x |
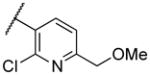
|
NA | |
| 22y |
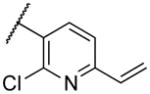
|
658 | |
| 22z |
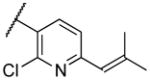
|
NA | |
| 22aa |
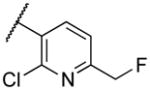
|
45 | |
Data are reported as mean of n = 3 determinations.
Percentage of response at given concentration. NA = not active at concentrations up to 25 μM.
Deletion of the 6-methyl from the hit led to 18-fold loss of activity (22a), while removal of the 2-nitro led to the inactive compound 22b. Interestingly, the phenyl analog 22c showed a 8-fold loss in potency and a reduced efficacy of 40% suggesting that the basic pyridinyl nitrogen may be involved in a hydrogen bond interaction. Replacing the 6-methyl with chlorine (22d) led to only 2-fold decrement in potency. Aware that most nitro-containing compounds can cause methaemoglobinemia and are potentially mutagenic, our synthetic efforts focused on the replacement of the nitro group by a different molecular feature.22 However, the installation of classical nitro-bioisosteres such as methyl ester (22e), amide (22f) and carboxylic acid (22g) led to a great or complete loss of activity. Next, analogs containing hydrogen-bond donor groups were synthesized. Unfortunately, hydroxymethyl (22h) and amino (22i) analogs were found inactive. These data suggest that the nitro group may not be involved in a hydrogen bond interaction but rather may be directing the ether spacer into the active conformation. In order to investigate this hypothesis, the methyl (22j), ethyl (22k) and phenyl (22q) analogs were synthesized. These analogs were found less active than the hit showing a decrement in potency correlated with the size of the substituent. Finally, we explored the possibility of installing halogen atoms to substitute the nitro group. Remarkably, the 2-bromo 22l (CYM50199) and the 2,6-dibromo 22n (CYM50179) analogs were found to be 3-fold more potent than the hit. Restricting the rotation of the spacer by attaching a methyl group at position 4 of 22l led to the inactive compound 22s. Removal of the bromine from position 6 (22m) led to significant loss of potency as previously observed with the nitro analog 22a. The 2-bromo-6-fluoro analog 22o was only 4-fold less potent than 22l, but 6-fold more potent than 22m, suggesting that substituents at position 6 modulate the potency. The 2-iodo-6-methyl analog 22p (CYM50138) was equipotent to the 2-bromo analog 22l. Based on the acquired information, the SAR at position 6 was further investigated. Substituting the methyl group with a phenyl ring (22r) led to a substantial loss of potency. Interestingly, hydroxymethyl 22t was only 4-fold less potent whereas aldehyde 22u was 38-fold less active than 22p, suggesting that a hydrogen-bond donor rather than an acceptor may be better tolerated in this region of the molecule. Interestingly, in the presence of chlorine at position 2 comparable activities were found for both the alcohol 22v and aldehyde 22w derivatives, which were respectively 5-fold less and slightly more active than the 2-iodine counterparts (22t, 22u). In line with the hypothesis that a hydrogen-bond acceptor is not well accepted in this region, the methyl ether 22x was inactive. Successively, we explored the influence of alkenyl substituents in this region. The ethylene analog 22y showed modest potency, while its dimethylated analog 22z was inactive suggesting that bulky substituents are detrimental for the potency. Taking into account that halogens directly attached at position 6 of the pyridine ring (22d, 22n, 22o) were well tolerated probably due to the formation of a dipole, we prepared the fluoromethyl derivative 22aa. Remarkably, 22aa (CYM50260) was found 3.5-fold more potent than the hit compound and was equipotent to the dibromine analog 22n. These data suggest that substitutents at positions 2 and 6 are essential to improve the potency. The position 2 tolerated nitro and halogen groups while small lipophilic or dipole-inducing groups were the most suitable substituents at position 6.
A set of the most active compounds was selected for selectivity assays against S1P1–3,5-R subtypes (Table 4). Remarkably, all tested compounds displayed exquisite selectivity against the other S1P-R family members.
Table 4.
S1P1–3,5-Rs selectivity counter screen
| cpd | EC50 nMα | ||||
|---|---|---|---|---|---|
| S1P4-R | S1P1-R | S1P2-R | S1P3-R | S1P5-R | |
| 2 | 162 | NA | NA | NA | NA |
| 8g | 232 | NA | NA | NA | NA |
| 8h | 220 | NA | NA | NA | NA |
| 8r | 601 | NA | NA | NA | NA |
| 8s | 570 | NA | NA | NA | NA |
| 22d | 304 | NA | NA | NA | NA |
| 22l | 52 | NA | NA | NA | NA |
| 22n | 46 | NA | NA | NA | NA |
| 22p | 54 | NA | NA | NA | NA |
| 22t | 287 | NA | NA | NA | NA |
| 22aa | 45 | NA | NA | NA | NA |
Data are reported as mean of n = 3 determinations. NA = not active at concentrations up to 25 μM.
In summary, we have reported the discovery, design and synthesis of novel small molecule S1P4-R agonists based on a 3-(2-(phenoxy)ethoxy)-6-alkyl-2-nitropyridine chemotype distinct from previously reported S1P4-R modulators. Systematic SAR analysis of the original MLSMR hit 2, a selective but moderately potent S1P4-R agonist, led to the development of novel potent and exquisitely selective S1P4-R agonists 22l, 22n, 22p, 22aa (CYM50199, CYM50179, CYM50138, CYM50260). Noteworthy, the studies herein reported provide novel pharmacological tools to decipher the biological function and assess the therapeutic utility of the S1P4–R. Further studies of our research program will be communicated in due curse.
Acknowledgments
This work was supported by the National Institute of Health Molecular Library Probe Production Center grant U54 MH084512 (Edward Roberts, Hugh Rosen) and AI074564 (Michael Oldstone, Hugh Rosen). We thank Mark Southern for data management with Pub Chem, Pierre Baillargeon and Lina DeLuca (Lead Identification Division, Scripps Florida) for compound management.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Sanchez T, Hla T. J Cell Biochem. 2004;92:913–922. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 2.Schwab SR, Cyster JG. Nature Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 3.Kono M, Allende ML, Proia RL. Biochim Biophys Acta. 2008;1781:435–441. doi: 10.1016/j.bbalip.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsolais D, Rosen H. Nat Rev Drug Discov. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fyrst H, Saba JD. Nat Chem Biol. 2010;6:489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiegel S, Milstien S. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 7.Hannun YA, Obeid LM. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 8.Rivera J, Proia RL, Olivera A. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gräler MH, Bernhardt G, Lipp M. Genomics. 1998;53:164–169. doi: 10.1006/geno.1998.5491. [DOI] [PubMed] [Google Scholar]
- 10.Toman RE, Spiegel S. Neurochem Res. 2002;27:619. doi: 10.1023/a:1020219915922. [DOI] [PubMed] [Google Scholar]
- 11.Kohno T, Matsuyuki H, Inagaki Y, Igarashi Y. Genes Cells. 2003;8:685. doi: 10.1046/j.1365-2443.2003.00667.x. [DOI] [PubMed] [Google Scholar]
- 12.Goetzl EJ, Rosen H. J Clin Invest. 2004;114:1531–1537. doi: 10.1172/JCI23704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T, Willart MA, Hijdra D, Hoogsteden HC, Lambrecht BN. J Clin Invest. 2006;116:2935–2944. doi: 10.1172/JCI28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuyuki H, Maeda Y, Yano K, Sugahara K, Chiba K, Kohno T, Igarashi Y. Cell Mol Immunol. 2006;3:429. [PubMed] [Google Scholar]
- 15.Maeda Y, Matsuyuki H, Shimano K, Kataoka H, Sugahara K, Kenji Chiba K. J Immunol. 2007;178:3437. doi: 10.4049/jimmunol.178.6.3437. [DOI] [PubMed] [Google Scholar]
- 16.Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H. Mol Pharmacol. 2008;74:896. doi: 10.1124/mol.108.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Graeler MH, Goetzl EJ. FASEB J. 2005;19:1731–1733. doi: 10.1096/fj.05-3730fje. [DOI] [PubMed] [Google Scholar]
- 18.Schulze T, Golfier S, Tabeling C, Räbel K, Gräler MH, Witzenrath M, Lipp M. FASEB J. 2011 doi: 10.1096/fj.10–179028. [DOI] [PubMed] [Google Scholar]
- 19.Golfier S, Kondo S, Schulze T, Takeuchi T, Vassileva G, Achtman AH, Gräler MH, Abbondanzo SJ, Wiekowski M, Kremmer E, Endo Y, Lira SA, Bacon KB, Lipp M. FASEB J. 2010;24:4701. doi: 10.1096/fj.09-141473. [DOI] [PubMed] [Google Scholar]
- 20.Urbano M, Guerrero M, Velaparthi S, Crisp M, Chase P, Hodder P, Schaeffer M, Brown S, Rosen H, Roberts E. Bioorg Med Chem Lett. 2011 doi: 10.1016/j.bmcl.2011.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontenas C, Bejan E, Aït Haddou H, Balavoine GGA. Synthetic Communications. 1995;25:629–633. [Google Scholar]
- 22.Smith GF. Prog Med Chem. 2011;50:1. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 23.The biological assays were performed using Tango S1P4-BLA U2OS cells containing the human Endothelial Differentiation Gene 6 (EDG6; S1P4–R) linked to a GAL4-VP16 transcription factor via a TEV protease site. The cells also express a beta-arrestin/TEV protease fusion protein and a beta-lactamase (BLA) reporter gene under the control of a UAS response element. Stimulation of the S1P4–R by agonist causes migration of the fusion protein to the GPCR, and through proteolysis liberates GAL4-VP16 from the receptor. The liberated VP16-GAL4 migrates to the nucleus, where it induces transcription of the BLA gene. BLA expression is monitored by measuring fluorescence resonance energy transfer (FRET) of a cleavable, fluorogenic, cell-permeable BLA substrate. As designed, test compounds that act as S1P4–R agonists will activate S1P4–R and increase well FRET. Compounds were tested in triplicate at a final nominal concentration of 25 micromolar