

Abstract
Background
The hereditary Long QT Syndrome (LQTS) is a common cardiac disorder where ventricular repolarization is delayed, abnormally prolonging the QTc interval on ECG. LQTS is linked to various genetic loci including the KCNH2 (HERG) gene that encodes the α–subunit of the cardiac potassium channel that carries IKr. Here we report and characterize a novel pathologic missense mutation, G816V HERG, in a patient with sudden cardiac death.
Methods
Autopsy-derived tissue sample was used for DNA extraction and sequencing from an unexpected sudden death victim. The G816V HERG mutation was studied using heterologous expression in mammalian cell culture, whole cell patch clamp, confocal immunofluorescence, and immunochemical analyses.
Results
The mutant G816V HERG channel has reduced protein expression and shows a trafficking defective phenotype that is incapable of carrying current when expressed at physiological temperatures. The mutant channel showed reduced cell surface localization compared to wild-type HERG (WT HERG) but the mutant and wild-type subunits are capable of interacting. Expression studies at reduced temperatures enabled partial rescue of the trafficking defect with appearance of potassium currents albeit with reduced current density and altered voltage-dependent activation. Lastly, we examined a potential role for hypokalemia as a contributory factor to the patient’s lethal arrhythmia by possible low-potassium induced degradation of WT HERG and haplo-insufficiency of G816V HERG.
Conclusion
The G816V mutation in HERG causes a trafficking defect which acts in a partially dominant-negative manner. This intermediate severity defect agrees with the mild clinical presentation in other family members harboring the same mutation. Possible hypokalemia in the proband induced WT HERG degradation combined with haplo-insufficiency may have further compromised repolarization reserve and contributed to the lethal arrhythmia.
Keywords: G816V, HERG, KCHN2, IKr, LQT2, sudden death, hypokalemia, ventricular arrhythmia
INTRODUCTION
The hereditary Long QT Syndrome (LQTS) is a cardiac disorder where ventricular repolarization is delayed, abnormally lengthening the action potential, which is characteristically exhibited as a prolonged interval between the start of the Q wave and the end of T wave on body surface electrocardiograms (ECG). This electrophysiological situation predisposes patients to ventricular tachyarrhythmias that can lead to syncope or sudden cardiac death. In LQTS, electrical rhythm disturbances can degenerate into a dangerous form of polymorphic ventricular tachycardia also known as Torsades des Pointes.1 The clinical phenotype in hereditary LQTS is often variable, even in individuals from the same family with the same genotype.2 The reasons for this variability are not completely understood but have been ascribed to additional genetic modifiers, differing environmental triggers, and varying expression of mutant and wild type alleles. More than 300 different mutations in KCNH2 have been reported to cause LQTS and only a fraction of these have been phenotypically characterized for mechanism or degree of severity in channel defect.3 Thus, the specific deleterious nature of individual mutations (due to the site within the channel and the type of amino acid substitution, deletion or insertion) may play a major role in the ultimate clinical presentation.
The two main voltage-gated ion channels that contribute to ventricular repolarization are KCNH2 (HERG) that carries the IKr current and KCNQ1 (with its associated accessory subunit KCNE1) that carries the IKs current. The HERG gene (human ether-a-go-go related gene, locus LQT2 on chromosome 74) encodes the six-transmembrane spanning α-subunit of the rapidly-activating delayed rectifier potassium channel. HERG channels are (as all voltage-gated K channels) comprised of four α-subunits in a tertrameric complex. HERG channels may complex with accessory subunits encoded by KCNE1 and/or KCNE2.5, 6 The only part of HERG whose structure has been resolved to high resolution is the N-terminal portion that contains PAS (Per-Ant-Sim) domain.7 In the C-terminus there exists a domain with homology to cyclic nucleotide binding proteins and is capable of binding cAMP with moderate affinity.8 Both of these domains in HERG appear to be fairly intolerant of genetic variations and appear to be relative hot spots of LQTS mutations.9
Currently, there are 13 different proposed loci associated with hereditary LQTS.2 Mutations in the HERG gene are responsible for approximately 40% of all cases of the genotype-positive hereditary LQTS, whereas mutations in KCNQ1/KCNE1 account for nearly 50%. Mutations in KCNQ1 usually perturb the channel’s biophysical properties such as gating, voltage sensing or conduction. In contrast, mutations in HERG are most often deleterious to channel trafficking, processing and/or subunit-tetramerization.10 Symptoms due to arrhythmias in LQT2 patients are frequently triggered by startle stimulus.11 However, hypokalemia as a cause of lethal arrhythmias in LQT2 has also been reported.12 A frequent clinical scenario today is how to approach the family with suspected LQTS that harbors variants in the LQT genes that have not been phenotypically characterized. Furthermore, it is equally challenging when a genetic variant is discovered in a family and the members carrying the variant have little or no clinical evidence for LQTS.
In this study we report and functionally characterize the novel LQT2 mutation G816V HERG. We explore the case of a young adult female with a prolonged QT interval on ECG who succumbed to sudden cardiac death and was later found to have the G816V mutation. We discuss the intermediate clinical, biochemical and cell biological correlation of this LQT2 mutations and possible contributory environmental triggers for arrhythmia.
METHODS
Genetic Analysis
Genomic DNA was extracted from dried blood on a ~3 × 3 mm piece of bloodstain card (Whatman, GE Healthcare Waukesha, WI) using a MagAttract DNA Mini M48 Kit (Qiagen, Valencia, CA) and Qiagen’s BioRobot M48 following manufacture’s instruction. All the coding regions and intron-exon boundaries of five long-QT genes, KCNQ1, KCNH2, SCN5A, KCNE1 and KCNE2, were amplified by the polymerase chain reaction. All sequence specific primers were designed by online programs ExonPrimer or Primer3 and purchased from Integrated DNA Technologies (Coralville, IA). For high throughput sequencing, the primers were tagged with universal primer sequences M13 on the 5′ end of forward primers and FKS on the 5′ end of reverse primers. The KCNH2-G816V mutation is located on exon 10; the primers used were as follows: KCNH2_10F+M13 5′-TGTAAAACGACGGCCAGTAAGGTGCCTGCTGCCTG-3′; KCNH2_10R+FKS 5′-TCGAGGTCGACGGTATCGATAATGTCACACAGCAAAGGGG-3′. For PCR two types of master mixtures were used: Master Mix A (1.5 mM MgCl2, 100 μM dNTPs and 0.625 U AmpliTaq Gold with 1X PCR Buffer II (Applied Biosystems, Foster City, CA)) for the majority of amplicons, and Master Mix B (3 mM MgCl2, 200 μM dATP, 200 μM dCTP, 200 μM dTTP, 100 μM dGTP, 100 μM 7-deaza-dGTP (USB, Cleveland, OH), 0.6 M betaine, 5% DMSO and 1.25 U AmpliTaq Gold with 1X PCR Buffer II), for amplicons with high GC content. Approximately 5 ng of genomic DNA were used for each amplicon in a 25 μl reaction volume. The reaction also included fluorescence labeled primers 6-FAM-M13 and NED-FKS (0.1 μM each for Master Mix A, and 0.04 and 0.08 μM respectively for Master Mix B) to visualize the PCR products for sizing on an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA). The PCR program was as follows: 95°C × 5 min, followed by 35 cycles of denaturing at 95°C × 40 s, annealing at 60°C × 60 s, and extension at 72°C × 60 s, followed by additional extension at 72°C × 5 min. Amplicons were treated with ExoSAP-IT (USB, Cleveland, OH) to remove remaining primers and dNTPs and, following sizing, subjected to bi-directional cycle sequencing using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) as per manufacturer’s instructions. Following the cycle sequencing reaction samples were precipitated with 250 mM sodium acetate and 10 mM EDTA in 70% ethanol, washed with 70% ethanol and resuspended in 10 μl Hi-Di formamide (Applied Biosystems, Foster City, CA). Sequencing was performed on an ABI Prism 3130xl Genetic Analyzer. Sequencing data analysis and editing were performed using Sequencher 4.2 (Gene Codes Corporation, Ann Arbor, MI). Each chromatograph must meet rigorous quality standards for correct base-calling. Sequence variants are numbered based on cDNA reference sequences in National Center for Biotechnology Information (NCBI)
Plasmids, Cell Culture, and Transfection
The G816V mutation was introduced into pCI-neo-HERG-myc using site-directed mutagenesis with the following primers: forward 5′-ATGCAAGGCCTGTCAAGTCGAA-3′ and reverse 5′-TTCGACTTGACAGGCCTTGCAT-3′. PCR conditions using Pfu Turbo polymerase (Stratagene) were as follows: 98°C for 3 min for 1 cycle, and 96°C for 1 min, 60°C for 1 min, 72°C for 18 min for 18 cycles. Mutated cDNAs were verified by automated bi-directional DNA sequencing.
For electrophysiology, Chinese Hamster Ovary cells (American Type Culture Collection) were maintained in Ham’s F12 media (Mediatech Inc.) supplemented with 10% fetal bovine serum (Hyclone) and 10,000 IU Penicillin/Streptomycin under 5% CO2 at 37°C or 30°C. Transient co-transfections were done using FuGene 6 (Roche) with GFP as an identifier of positively transfected cells. All recordings were done at room temperature (22°C) and at 48 or 72–96 hours post-transfection as indicated.
For biochemistry and immunofluorescence assays, HEK 293 (ATCC) cells were maintained similarly in supplemented RPMI1640 (Mediatech) or supplemented low potassium media as indicated. Cells were kept at 37°C or 30°C with 5% CO2. Stably transfected HEK-HERG cells were created via limiting dilution cloning as previously described.5 Stably transfected, polyclonal HEK-HERG G816V cells were created by FuGene 6 (Roche) transfection with 800 ug/mL G418 selection for 14 days. Stable expression of HERG G816V was verified by Western blot at 10 and 30 days and cells were maintained under non-selective RPMI media.
Transient co-transfections were done using FuGene 6 with GFP as an identifier of positively transfected cells. Both myc-tagged and 3x FLAG-tagged versions of WT HERG and myc-tagged HERG G816V were used in transient transfections as indicated. 3x FLAG- and myc-tagged HERG fusion proteins have been previously characterized for lack of functional differences.5, 13 Experiments were performed at 48, 72, or 96 hours post-transfection as noted.
Electrophysiology
CHO cells were used for electrophysiological experiments since they lack endogenous voltage-gated potassium currents. Transiently transfected CHO cells were grown on sterile glass coverslips and placed in an acrylic/polystyrene perfusion chamber (Warner Instruments) mounted in an inverted microscope outfitted with fluorescence optics and patch pipette micromanipulators. Transfected HEK cells were grown on gelatin coated coverslips and used as an additional control. The extracellular solution consisted of NaCl 150 mM, CaCl2 1.8 mM, KCl 4 mM, MgCl2 1 mM, glucose 5 mM, and HEPES buffer 10 mM (pH 7.4) at room temperature (20–22°C). Intracellular pipette solution contained KCl 126 mM, Mg-ATP 4 mM, MgSO4 2 mM, EGTA 5 mM, CaCl2 0.5 mM, and HEPES buffer 25 mM (pH 7.2) at room temperature (20–22°C). The whole-cell configuration of the patch clamp technique was used to measure potassium currents.14 A MultiClamp 700B patch-clamp amplifier was used and protocols were controlled via PC using pCLAMP10 acquisition and analysis software (Molecular Devices). Patch clamp pipettes were manufactured and tips were heat-polished to obtain a tip resistance of 2–3 megaOhm in the test solutions. The pipette offset potential in these solutions was corrected to zero just prior to seal formation. Whole cell capacitance (generally 10–25 pF) was compensated electronically through the amplifier. Whole cell series resistance was compensated to 85–90% using amplifier circuitry such that the voltage errors for currents of 2 nA were always less than 6 mV. A standard holding potential was −80 mV, and figure insets show specific voltage protocols. Data was filtered using an 8-pole Bessel filter at 1 kHz for subsequent analyses using CLAMPFIT (Molecular Devices).
Western Blots and Immunoprecipitation
Forty-eight hours after transfection with the desired expression plasmids, HEK 293 cells were lysed with ice-cold NDET buffer (1% NP-40, 0.4% Deoxycholic acid, 5 mM EDTA, 25 mM Tris, 150 mM NaCl, pH 7.5) with complete protease inhibitor cocktail (Roche) for 15 minutes. Cell lysates were centrifuged at 13,000 rpm at 4°C for 10 minutes, and the supernatants were mixed with SDS-PAGE loading buffer. Proteins were separated by 7.5% SDS-PAGE and transferred to nitrocellulose membranes (BioRad) by semi-dry blotting unit (FisherBiotech). The nitrocellulose membranes were blocked with Tris-buffered saline containing 0.5% Tween-20 (TBS-T) and 5% non-fat dry milk for 30 minutes at room temperature, and then incubated with primary antibody overnight at 4°C. Antibodies against HERG were rabbit polyclonal H175 and mouse monoclonal 9e10 anti-myc antibodies (Santa Cruz Biotech). Loading control antibodies used were pan-cadherin (Abcam), calnexin (Santa Cruz Biotech), Na-K-ATPase (Upstate) and tubulin (Sigma). The membrane was washed with TBS-T and then incubated in the corresponding infrared-fluorescence IRDye® 800 conjugated donkey anti-mouse and IRDye® 700DX conjugated donkey anti-rabbit secondary antibodies (1:10,000, Rockland Immunochemicals Inc.) for 30 minutes at room temperature in the dark followed by washing with TBS-T. The membranes were then scanned to visualize the signal at 680 nm or 780 nm by the Odyssey detection system (Li-Cor Biosciences) and densitometry of the protein bands was performed using Li-Cor software.
For immunoprecipitation, confluent 60 mm dishes were lysed as above. Centrifuged lysate was then pre-cleared with non-specific antibodies and protein A/G beads (Santa Cruz) for 1 hour at 4°C. The pre-cleared lysate was then immuno-precipitated overnight at 4°C. The antibodies used were anti-Flag-M2 (Sigma) and 9e10 anti-myc (Santa Cruz). The beads were washed three times in ice-cold lysis buffer and eluted in 4x Laemmli sample buffed for 30 minutes at room temperature. Samples were separated using 7.5% SDS-PAGE and blotted as above.
Immunofluorescence
HEK 293 cells were used for these experiments because their subcellular architecture is better suited for microscopic analysis with immunofluorescence, compared to CHO cells. HEK 293 cells were transiently transfected for 48 hours as described above. Twenty-four hours after transfection, cells were plated onto 35 mm glass bottom culture dishes (MatTek Corporation). Forty-eight hours after transfection, cells were fixed in 3.7% formaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature, followed by permeabilization with 0.2% triton-X100 in PBS at room temperature for 10 minutes and then blocking with 5% BSA in 0.2% triton-X100 in PBS at room temperature for 30 minutes. The cells were then incubated with rabbit anti-HERG (1:50, Santa Cruz) and mouse anti-Cadherin (1:100, Novus) or goat anti-Calnexin (1:50 Santa Cruz) antibodies in 5% BSA, 0.2% triton-X100 in PBS at room temperature for one hour. The cells were washed with 0.2% triton-X100 in PBS at room temperature for five minutes three times before incubation with Alexa Fluor 488 donkey anti-rabbit (Molecular Probes) and Alexa Fluor 568 donkey anti-mouse (Molecular Probes) or Alexa Fluor 568 donkey anti-goat secondary antibodies (1:1000) in 5% BSA, 0.2% triton-X100 in PBS at room temperature for 1 hour in the dark. After incubation, the cells were washed with 0.2% triton-X100 in PBS at room temperature three times, mounted in GelMount™ (Biømeda Corp.), and kept at 4°C in the dark until subjected to fluorescence microscopy examination. Images were collected with a Leica TCS SP2 AOBS confocal microscope (Mannheim, Germany) with 63X oil immersion optics. Laser lines at 488 nm and 561 nm for excitation of Alexa Fluor 488 and 568 dyes were provided by an Argon laser and a diode laser. Detection ranges were set to eliminate crosstalk between fluorophores. Fluorescent images were analyzed with the Image Correlation Analysis plug-in for ImageJ software (http://rsbweb.nih.gov/ij/)15, with Pearson’s correlation quotient (ranging from 0 to 1) obtained as a quantification of HERG colocalization.
Cell Surface Biotinylation
Cell surface protein expression was determined by labeling with the membrane-impermeant biotinylation reagent, NHS-SS-biotin (ThermoFisher). Cells were washed twice with PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS-Ca/Mg), and incubated on ice in NHS-SS-biotin (1.5 mg/mL) in 20 mM HEPES, pH 9.0, 2 mM CaCl2, and 150 mM NaCl for 45 minutes on ice. After labeling, the cells were rinsed briefly with PBS-Ca/Mg and incubated in 100 mM glycine in PBS-Ca/Mg for 15 minutes on ice to quench unreacted NHS-SS-biotin. Cells were lysed in 100 μl of lysis buffer (50 mM Tris-HCl (pH 7.5), 1% Triton X-100, 1% SDS, 150 mM NaCl, 5 mM EDTA, complete protease inhibitor cocktail (Roche)) with gentle shaking on ice for 15 min. The cell lysates were then diluted by the addition of 900 μl of lysis buffer without SDS and then homogenized by QiaShredder (Qiagen). Protein concentration of each sample was determined by Micro BCA Assay (ThermoFisher). Equal amounts of biotinylated proteins were precipitated from the supernatant solution with UltraLink Immobilized Streptavidin (ThermoFisher) and overnight incubation at 4°C with gentle agitation. The beads were washed three times with lysis buffer, twice with high-salt lysis buffer (lysis buffer with 500 mM NaCl and 0.1% Triton X-100), and once with 50 mM Tris-HCl (pH 7.5). The biotinylated surface proteins were eluted from the beads with SDS sample loading buffer at room temperature for 30 minutes. The remaining supernatant represented the unlabeled proteins that had not yet been trafficked to the cell surface. Proteins were then subjected to SDS-PAGE and immunoblotting. Cadherin was used as a positive control for cell-surface pull down and calnexin was used as the negative control for intracellular proteins.
Low-Potassium Assays
This media formulation was adapted from Guo et. al. 200916. The zero-potassium media was based on a MEM formulation using Earle’s Balanced Salt Solution excluding potassium salts. Exact formulation is as follows: 5.56 mM D-glucose, 117.24 mM NaCl, 26.19 mM NaHCO3, 1.36 mM CaCl2 (dihydrate), 0.644 mM MgSO4 (anhydrous) and 1.01 mM NaH2PO4 (anhydrous). The zero-potassium media was supplemented with penicillin/streptomycin (Mediatech), L-glutamine (Hyclone), BME vitamins (Sigma), and RPMI 1640 amino-acids (Sigma). The solution was sterile filtered, osmolarity was adjusted to 285±5 mOsm using D-mannitol and pH was adjusted to 7.5. The media was lastly supplemented with 10% dialyzed fetal bovine serum (Sigma) and the potassium concentration was later adjusted using sterile 1M KCl. Cells were treated with the low-potassium media for 24 hours at 37°C and lysed for Western as previously described.
Statistics
All data are expressed as the mean +/− SEM. The two-tailed Student’s T-test was used and a p-value of less than 0.05 was considered significant.
RESULTS
Case Presentation
The proband was a 27-year-old white female with a history of obesity. Previous weight-loss strategies included multiple liquid diets and purging. Eventually a laparoscopic gastric banding procedure was performed, and the peri-operative and immediate post-operative periods were uneventful. She reported to her family that she had frequent vomiting and had difficulty keeping liquids or solids down for any length of time over the following weeks. Two months after the operation she was discovered deceased in her home. Resuscitation attempts failed. Prior to the gastric surgery, two ECGs were done that showed prolonged QT intervals. The first QTc was 483 msec (Figure 1A–B) and a second was taken after beginning a liquid diet and showed a QTc of 516 msec. She had no prior history of syncope, palpitations, lightheadedness or seizures. The post-mortem examination was negative for any anatomical pathology or evidence of infection, and routine toxicology screening revealed no substances that could contribute as a cause of death.
Figure 1. Family Pedigree and Location of KCNH2-G816V Mutation.

A) The proband is indicated by the arrow. Each individual is assigned as male (squares) or female (circles), carrying the KCNH2-G816V mutation (filled black) or those not carrying the mutation (white). Numbers below indicate QTc intervals from one or more ECGs. B) Leads V4 and II of a 12-lead ECG obtained at rest from the proband showing a prolonged QT interval. C) Schematic representation of the HERG protein topology indicating the position of residue 816 within the C-terminal cyclic-nucleotide binding domain (CNBD).
Members of the patient’s immediate family were subsequently examined and found to have no history of arrhythmia, syncope, seizures or sudden death. The proband’s mother had borderline QTc prolongation over multiple ECGs at rest, 450–472 msec (Figure 1A). The brother of the proband also has a resting QTc of 434–440 msec. The results of a treadmill stress-test while on beta-blockers showed some degree of QTc prolongation during the recovery phase, from 440 msec at rest to 470 msec at 2 minutes and 50 seconds of recovery. Neither the father nor the two sisters showed any QTc prolongation (Figure 1A). A post-mortem DNA sample from the proband was submitted for commercial sequencing (Familion) of LQT loci. Subsequent exon-specific genetic analyses of family members was performed.
Genetic Analysis
The coding regions and intron-exon boundaries of five Long-QT genes, KCNQ1, KCNH2, SCN5A, KCNE1 and KCNE2, were bi-directionally sequenced. From this set of candidate genes, the KCNH2 2447 G-to-T mutation was found within exon 10. This mutation resulted in the amino acid change of glycine to valine at position 816 of the KCNH2 protein. This site is predicted to reside in the proximal C-terminus within the cyclic-nucleotide binding domain (CNBD) (Figure 1C). Family screening for the KCNH2 G2447T mutation was positive in the mother and brother.
Channel Protein Expression
HERG appears as two distinct bands at 135 kD and 155 kD on SDS-PAGE. These two bands represent glycosylation variants where the 135 kD band is the ER glycosylated immature form and the 155 kD band is the Golgi processed mature form. Channel glycosylation is not necessary to achieve functional potassium current but is considered a surrogate marker for mature and correctly trafficked channel protein.17 Our results showed that at 48 hours post-transfection, wild-type HERG (WT HERG) displayed two bands at 135 kD and 155 kD and G816V HERG displayed only the 135 kD band. The mutant G816V HERG showed lower protein expression in HEK 293 cells given the same amount of DNA for the transient transfection (Figure 2A–B). At 72 hours the quantity of both 135 kD and 155 kD HERG increased for WT HERG, whereas for G816V HERG, only the 135 kD band increased while the 155 kD band was still absent. In all three samples at both time points the majority of HERG protein expression was comprised of the immature 135 kD band (Figure 2C). Quantification of total HERG expression for G816V showed a 40% baseline expression level compared to 100% WT HERG at both time points (Figure 2B). When we mixed and co-expressed WT and mutant HERG in a 50/50 ratio there was an intermediate amount of HERG expressed (75–80%), which is less than the amount of 100% WT HERG (with the same plasmid DNA concentration) at both time points.
Figure 2. Baseline Protein Expression of G816V HERG compared to WT HERG in HEK 293 cells.

A) Western blot shows whole cell lysate of HEK 293 cells transfected with either WT HERG or G816V HERG plasmid DNA alone or in a 50/50 mix at 48 and 72 hours post-transfection. Antibodies used were against the Myc-tag for both types of HERG and against Na+/K+ ATPase as a loading control. The molecular weight marker appears at left and HERG appears as a doublet of two bands at 135 kDa and 155 kDa (arrows) separated on 7.5% SDS-PAGE. B) Histogram showing summary data of total HERG protein expression levels normalized to Na+/K+ ATPase. Graph shows densitometry quantification of total HERG where protein expression of total G816V HERG compared to total WT HERG is reduced both at 48 and 72 hours, n=5 with ** p-value < 0.01 and ***p-value < 0.001. C) Histogram showing the distribution of 135 kD and 155 kD HERG bands for wild-type, 50–50 mix and G816V with totals normalized to 1.
Electrophysiological Characterization
To determine the functional effect of G816V on HERG currents we used whole-cell patch clamp techniques. WT HERG and G816V HERG were expressed in various ratios using transient transfection in CHO cells. Using a standard activation protocol, we found that G816V HERG did not produce a current when expressed on its own (Figure 3A). We confirmed the same result in transiently transfected HEK cells. HEK cells transfected with G816V HERG did not produce any measurable current at 48 hours whereas cells transfected with WT HERG had a mean current density of 30.98 ± 6.45 pA/pF with n=8 for G816V and n=6 for WT HERG (data not shown). We then examined whether heterozygous expression of mutant G816V would result in dominant-negative current-suppression over co-expressed WT HERG. We co-transfected WT and mutant HERG in 100/0, 75/25, 50/50, 25/75, and 0/100 WT/G816V ratios. Figure 3B plots our results against the ideal curve of a fully dominant-negative mutant (dashed line) and the ideal curve of a completely non-dominant-negative mutant (dotted line). Our results showed that the current density decreased linearly with the amount of WT HERG in the mixed samples (Figure 3B). Therefore, G816V HERG caused partially dominant negative current suppression when mixed in various proportions with WT HERG. Comparison of the 50/50 WT-to-G816V mix with the current produced by transfection of 50% WT HERG and 50% empty vector revealed equivalent current densities (Figure 3C). Further electrophysiological characterization was carried out only on the 50/50 transfection ratio. The normalized current voltage relationships were similar for WT HERG and 50/50 mixed samples (Figure 3D). The voltage-dependent activation curves for WT HERG and 50/50 mixed samples were similar in shape and did not have a significant shift in the V1/2 values (Figure 3E).
Figure 3. Effect of the G816V Mutation on IKr and IKs Currents.

A) Whole cell current traces of HERG channels with voltage protocol below and brackets indicate the portion of the current tracing that is shown. WT HERG and G816V HERG channels were transiently expressed in CHO cells either alone, in a 50% WT/50% G816V mix, or 50% WT/50% empty vector mix. B) Current density at 48 hrs shown as a function of the fraction of WT subunits. Measurements for current density were taken as peak amplitude at the −40 mV tail current. Numbers in parentheses indicate number of cells assayed. The dashed line indicates the expected relationship for a fully dominant-negative mutant that does not carry current in tetrameric assembly. The dotted line indicates the expected relationship for a non dominant-negative mutant in tetrameric assembly. C) Histogram comparing current density of 50% WT/50% empty vector mix and 50% WT/50% G816V HERG. The difference was not significant. D) Normalized current-voltage relationship shown for WT HERG and 50/50 mixed sample. The measurements for the current-voltage curve were taken at the end of the 3 second depolarization pulse. E) Normalized voltage-dependent activation curves. Curves were fitted using a Boltzmann function. V1/2 for WT HERG = −17.96±2.1 mV and WT/G816V HERG = −23.67±2.68 mV. n = 8–11, the difference was not significant. F) Whole cell current traces at the −120 tail current for IKs alone, IKs with WT HERG and IKs with G816V HERG. Voltage protocol is shown below and brackets indicate portion of the current trace that is shown. G) Histogram shows summary data for tau of deactivation and numbers in parentheses indicated number of cells assayed. Tail currents were fit with a single-exponential function and the difference in tau for deactivation between IKs alone and IKs with G816V HERG was significant * p-value < 0.05.
We also found that neither WT HERG nor G816V HERG affected the current density of IKs in CHO cells (data not shown). However, the presence of WT HERG decreased the deactivation time constant for IKs and the presence of G816V HERG decreased the IKs time constant to a greater degree (Figure 3F–G). The deactivation time constant was 301.13±27.89 msec for IKs alone, 226.08±30.85 msec for IKs with WT HERG and 155.20±21.4 msec for IKs with G816V HERG. The decrease in deactivation time constant for G816V HERG with IKs compared to IKs alone is significant with p-value < 0.05, n=6 and n=7.
Sub-cellular Channel Localization
Since mutations in the cyclic-nucleotide binding domain (CNBD) of HERG frequently lead to channel trafficking defects, we examined if G816V HERG had impaired trafficking to the plasma membrane. We used NHS-SS-Biotin to label only proteins at the surface of the cell membrane, and showed quantitatively that G816V HERG surface expression is reduced by 38% (Figure 4A–B). The quantification was normalized to HERG expression, total protein expression, and amount of biotinylated protein.
Figure 4. Cell Surface Expression of G816V HERG compared to WT HERG.

A) Immunoblot of cell surface proteins which were isolated by biotin labeling and pull-down from transiently transfected HEK cells. Samples were separated by 7.5% linear SDS-PAGE. Calnexin was the negative control for surface labeling and cadherin was the positive control for surface labeling in the lower panels. Western blot was performed with anti-HERG, anti-calnexin, and anti-cadherin antibodies. B) Summary data for part A. Densitometry analysis of the surface expression of HERG proteins was quantified as the amount of surface HERG divided by the total cellular HERG and also normalized for streptavidin pull-down of biotinylated protein (normalization calculated using cadherin and calnexin controls). WT HERG is normalized to 1.0. n=4, * p<0.05. C) Confocal immunofluorescence micrographs of HEK 293 cells transfected with either WT HERG or G816V HERG and counter-stained with anti-cadherin antibody to indicate the cell membrane. Pearson correlation of HERG and cadherin colocalization is 0.571±0.025 for WT HERG and 0.332±0.029 for G816V, p<0.001. n=15, scale bar=10 μm, DIC = differential interference contrast. D) Confocal immunofluorescence micrographs of HEK 293 cells transfected with either HERG-WT or HERG-G816V and counter-stained with anti-calnexin antibody to indicate the ER, an intracellular compartment. Pearson correlation of HERG and calnexin colocalization is 0.831±0.026 for WT HERG and 0.916±0.006 for G816V, p<0.01. n=13, scale bar=10 μm.
Confocal immunofluorescence showed an enhanced intracellular staining pattern for G816V HERG which corresponded more with calnexin, an ER marker, as compared to cadherin, a plasma membrane marker (Figure 4C–D). Pearson correlation of HERG and cadherin colocalization was 0.571±0.025 for WT HERG and 0.332±0.029 for G816V HERG, p<0.001 for 15 cells assayed. Pearson correlation of HERG and calnexin colocalization was 0.831±0.026 for WT HERG and 0.916±0.006 for G816V, p<0.01 for 13 cells assayed. This is consistent with greater retention of G816V HERG in the ER as compared to trafficking-competent WT HERG.
Interaction of Wild-type and Mutant HERG Subunits
Since the initial protein biochemistry, surface labeling, and electrophysiology experiments suggested a partial trafficking defective phenotype, we sought to further determine whether the wild-type and mutant HERG subunits were capable of interacting. We performed assays using differentially tagged WT HERG (3X-FLAG tag) and G816V HERG (Myc tag). Figure 5 shows that the wild-type and mutant subunits were able to co-immunoprecipitate together from whole cell lysates. The first panel (Figure 5A) shows transient transfections of HEK 293 with a range of combinations from 100% WT HERG to 100% G816V HERG. The whole cell lysates were immunoprecipitated (IP) using anti-Myc antibody for G816V HERG and for subsequent detection on the Western blot anti-FLAG antibody was used to detect WT HERG. In the IP lanes, the blot shows that G816V HERG preferentially pulled-down the immature 135 kD form of WT HERG. The lower panel shows the reverse experiment where whole cell lysates were immunoprecipitated using anti-FLAG antibody and subsequent detection with anti-Myc antibody. This showed that WT subunits can pull-down mutant subunits.
Figure 5. Interaction of WT HERG and G816V HERG subunits.

A) Co-immunoprecipitation and subsequent Western blot from transiently transfected HEK 293 cells shows 3x FLAG-tagged WT HERG detected from a pull-down of myc-tagged G816V HERG in 3 different mixed combinations with 100% WT HERG and 100% G816V HERG as controls, n=3. Lower panel shows the reverse experiment where myc-tagged G816V HERG was detected in a pull-down of 3x FLAG-tagged WT HERG from the same combinations, n=3. B) Immuno-blots of cell surface biotinylation of 3x FLAG-tagged WT HERG and myc-tagged G816V HERG alone or in a 50/50 mix. Proteins were separated by 7.5% SDS-PAGE. Top panel shows a blot with anti-FLAG antibody with cadherin and calnexin as controls. Lower panel shows a blot with anti-Myc antibody with cadherin and calnexin as controls, n=3.
While the co-immunoprecipitation suggests that WT and mutant subunits are capable of interacting, it is important to examine if they interact at the plasma membrane. Figure 5B shows cell surface biotinylation using 100% Flag-WT HERG, 50/50 mixed, and 100% Myc-G816V HERG. 100% WT HERG and 100% G816V HERG were pulled down from the surface as shown in Figure 4A. In the 50/50 mixed sample however, FLAG-tagged WT HERG could be pulled-down from the surface whereas Myc-tagged G816V HERG was not. This suggests that co-expressed WT subunits did not enhance surface expression of interacting mutant subunits but that WT-mutant heterotetramers are more likely to be mis-trafficked. Together, these data suggest that those interacting heterotetramers that do form, do so early (in the ER) and are retained for early recycling but that there may also be reduced interaction between WT and G616V subunits, accounting for the partially dominant phenotype.
Protein Expression and Electrophysiology at Reduced Temperature
Reduced temperature is known to help restore trafficking of some defective mutant proteins by enhancing correct protein folding.13, 18 To examine if mutant protein misfolding is the mechanism underlying G816V defect we performed biochemical and electrophysiological studies on cells grown at 30°C after transient transfection. Reduced temperature led to a small but consistent increase in more mature forms of G816V mutant protein at 72 and 96 hours after transfection (noted as an enhanced signal above 135 kD, Figure 6A). As demonstrated in previous studies the WT HERG also shows enhanced maturation at reduced temperatures.13, 18
Figure 6. Protein Expression and Electrophysiology of G816V HERG at Reduced Temperature.

A) Western blots of whole cell lysate from transiently transfected HEK 293 cells with WT HERG, 50/50 mixed, or G816V HERG at 48, 72 and 96 hours post-transfection. Anti-HERG antibody and anti-Na+/K+ ATPase antibodies were used. Top panels show the experiment done at 37°C incubation and middle panels show the same experiment done at 30 degrees incubation. Bottom graph shows a histogram of densitometry quantification of percent of 155 kD over total HERG for the two temperatures at the 96 hour time point, n=3. B) Whole cell current traces of HERG channels patched at 30°C with voltage protocol shown below. WT HERG and G816V HERG channels were transiently expressed in CHO cells either alone, or in a 50% WT/50% G816V mix. C) Current density at 72–96 hours and 30°C shown as a function of the amount of WT subunits. Numbers in parentheses indicate number of cells assayed. D) Normalized current-voltage relationship shown for WT HERG and G816V HERG at 30°C. E) Normalized voltage-dependent activation curves. Curves were fitted using a Boltzmann function. V1/2 for WT HERG = −8.43±3.8 mV and G816V HERG = −22.64±2.8 mV. n = 8–11. The difference was statistically significant, p-value < 0.05.
We also tested if lowering temperature would rescue functionality. Incubation at 30°C for 72–96 hours produced a small amplitude current for G816V HERG that was never seen when cells were grown at 37°C (Figure 6B). The current amplitude for WT HERG was also larger at 30°C compared to 37°C. Mutant tetramers are capable of carrying current at less than 10% of the current density of WT HERG. In the 50/50 mix there was 60% of current density as compared to WT HERG as opposed to 50% when cells were grown at 37°C (Figure 6C). The current voltage relationship for G816V HERG compared to WT HERG was similar in shape however; G816V HERG showed altered voltage-dependent activation (Figure 6D–E). The voltage dependence of activation for G816V HERG had a hyperpolarizing shift in the V1/2 −22.64±2.8 mV as compared to WT HERG V1/2 −8.43±3.8 mV.
Potassium Effect on HERG Trafficking
As first reported by Guo and colleagues, low-extracellular potassium causes degradation of the 155 kD form of HERG through an endocytic mechanism involving mono-ubiquitination.16, 19, 20 This group showed that internalization and degradation of the 155 kD band of HERG occurs in low-potassium media in the range of 0–1.0 mM K+ and as early as 4 hours. Given the potential hypokalemia that the proband may have experienced ante mortem we sought to determine whether low potassium had preferential affects on WT or mutant HERG subunits. Our experiments were done with HEK 293 cells stably expressing WT HERG and stably expressing G816V HERG. The left panel of figure 7 shows whole cell lysate and Western blot of stable HEK-WT HERG cells treated with low potassium media. The right panel shows a Western blot of stable HEK-G816V cells treated with the same low-potassium media. We show that zero-potassium induced degradation of the WT HERG 155 kD band and increased the amount of the 135 kD band at 24 hours, which is consistent with the previous work. We also show that neither low nor high potassium concentration dramatically affected the amount of G816V HERG protein (Figure 7A). In the context of our proband, this shows that hypokalemia could have triggered an arrhythmia by effectively reducing IKr through inducing degradation of mature WT HERG leaving G816V HERG subunits relatively unperturbed.
Figure 7. Low Potassium Induced Degradation of HERG.
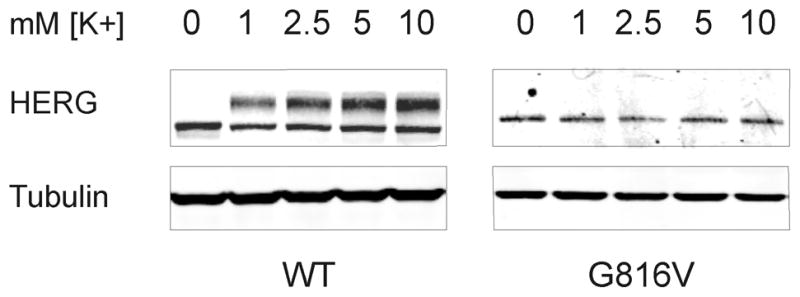
Western blots showing whole cell lysate of HEK 293 cells stably expressing WT HERG (left) or G816V HERG (right) treated with low-potassium media for 24 hours. The potassium concentration is indicated at the top and the concentration of potassium in regular media is 5.3 mM. Tubulin was used as the loading control, n=4.
DISCUSSION
In the present study, we report and characterize the novel and clinically significant HERG mutation G816V. Our clinical data show a proband with an asymptomatic phenotype until the first symptom which resulted in sudden cardiac death in the setting of suspected hypokalemia. Our biochemical and biophysical data show that mutant G816V HERG channel was unable to carry current by itself at physiological temperatures, exerted a partial dominant negative effect on wild-type HERG currents, and displayed a combination of biophysical and trafficking defects. Furthermore, we provide evidence that mutant and wild-type subunits were capable of interacting early in biosynthesis and that interaction reduced trafficking to the surface together. The fundamental defect is likely to be that of mutant subunit misfolding with ensuing partial reduction of interaction with WT subunits, ER-retention and degradation, alternative trafficking pathways, and non-functional channels.
The categories of LQT mutations have been classified by mechanism as follows: class (1) defective channel synthesis, (2) disrupted protein-trafficking, (3) altered gating, (4) changes in permeation.21, 22 A trafficking defect is the most common cellular phenotype for LQT2 mutants in the HERG CNBD and C-terminal regions.10 Analysis of a panel of five reported LQT2 mutants in the CNBD showed that it is a conserved domain that is important for cell-surface localization.23 Several groups have also shown importance of more distal C-terminal residues in ER retention and tetramer formation.24–26 Taken together, these results suggest a key role for various sections of the C-terminus of HERG to dictate the ability of monomers to assemble and traffic to the surface properly.
Based on the combination of our results, we consider the G816V HERG effect on WT HERG currents as partially dominant negative. Furthermore, we propose that certain combinations of WT-G816V heterotetramers may allow current (those with more WT subunits) and some may not (those with more mutant subunits). We speculate that this may depend on the degree to which the HERG mutant monomer is misfolded because when extended time and lower incubation temperature allowed for more productive folding, the mutant tetramers were capable of carrying some current. Recent work in the field of protein folding suggests that there are multiple pathways and multiple intermediate states on the way to achieving the native folded state27, 28. This is contrary with the earlier notions that there is one sequential pathway with distinct intermediate forms. The HERG monomers may exist in multiple misfolded states which combine to form different populations of heterotetramers. If the presence of a mutant subunit decreases interaction with WT subunits, then this could account for the modest reduction in total protein expression but the even greater reduction in functional expression for the 50/50 mixed case.
Mechanisms for the forward trafficking of HERG remain an area of active investigation. Recently, the group of Delisle and colleagues showed that HERG undergoes COPII-dependent ER export and endosomal trafficking mediated by small GTPases which determine its plasma membrane expression.29 That a portion of immature HERG G816V homotetramers was detected on the surface despite lack of function suggests that misfolded channels may traverse additional atypical pathways arriving at the plasma membrane. It has also been shown that trafficking defective LQT2 mutants are subsequently degraded by the ER-associated degradation pathway (ERAD) and the ubiquitin proteasome pathway.13, 30 Our data shows a modest reduction in protein expression in the 50/50 mixed sample and this could result from G816V and WT HERG heterotetramers which are capable of assembly but may get shunted to the proteasome for degradation instead of being trafficked to the surface. Also, if a significant portion of the G816V monomers are misfolded and then degraded early on, it may explain why the G816V protein expression by itself is reduced to 40% of WT.
The mechanisms of retrograde trafficking suggest that HERG takes an unconventional route that is not clathrin mediated. Recent studies have also examined the role of extracellular potassium in the endocytosis and degradation of HERG. The work of Guo and colleagues has provided a biochemical basis and mechanistic approach to study the behavior of HERG in low-potassium conditions. The 155 kDa form of HERG undergoes endocytic internalization and proteasomal degradation through a mechanism involving caveolin.20, 31 Further work was done by Massaeli and colleagues who studied the behavior of pore-lining mutations in HERG under zero-potassium conditions but did not examine C-terminal mutants.19 Expressed alone, G816V HERG was not affected by zero-potassium conditions that induce internalization and degradation of wild-type HERG.
Importantly, this same group used in vivo studies to show that rabbits on a low-potassium diet had prolonged QTc intervals and that IKr was reduced in isolated ventricular myocytes from hypokalemic rabbits.16 Thus, when taken together, hypokalemia may represent an additional trigger in a patient whose IKr is partially compromised by a G816V mutation. This is consistent with the previously noted observation that hypokalemia has been associated with Torsades des Pointes and other lethal arrhythmias in LQT patients.32 The idea of hypokalemia as a trigger for lethal arrhythmias in LQT2 patients has been proposed before by Berthet and colleagues who studied and sequenced four families where the proband had documented hypokalemia and lethal arrhythmias.12 Two of the mutations found in their study were HERG S818L and V822M which are also within the CNBD and very close to G816V. The S818L mutation was later electrophysiologically characterized in Xenopus oocytes and found to be similarly incapable of carrying current by itself, exerting a partial dominant negative effect on WT HERG channels, and causing a hyperpolarizing shift to the voltage-dependent activation.33
In our case, hypokalemia may have been the triggering factor for sudden cardiac death. She had an asymptomatic clinical phenotype (as do those family members carrying the same mutation) that corresponded to the incomplete dominant nature of the G816V channel defect. She likely relied on her repolarization reserve where the sum total of multiple simultaneous potassium currents results in ventricular repolarization.34 If one allele is compromised by a loss of function mutation, the other normal allele and complementary channels can compensate. Since the QTc duration prolonged after instituting a liquid diet, and the fatal event occurred in the setting of protracted vomiting these facts suggest that a hypokalemic state that may have provided the final arrhythmic trigger. In this situation, hypokalemia likely exacerbated an already reduced repolarization reserve which may have lead to the arrhythmia. Recently, there have been a few studies showing recovery of prolonged QTc intervals in LQT2 patients with combination therapy including potassium supplementation and aldosterone-inhibition 35–37. This provides a generally well-tolerated, potential therapeutic intervention which may be considered in patients with mild clinical phenotypes.
CONCLUSION
The G816V missense mutation in HERG causes a trafficking defect which acts in a partially dominant-negative manner and therefore reduced current density. In the proband, suspected hypokalemia induced WT HERG degradation combined with haplo-insufficiency to compromise the repolarization reserve which may have contributed to a lethal arrhythmia.
The accompanying article describes the S277L mutation in KCNQ1 which caused both a biophysical and trafficking defect, thus reducing IKs. The proband similarly had a mild clinical phenotype. Sudden cardiac death, however, was precipitated by pharmacological impairment of IKr. A common etiology for arrhythmia in both of these patients would be a primary reduction in repolarization reserve due to an inherited mutation combined with a secondary exogenous triggering factor. These reports highlight the utility of mechanistic investigation into newly discovered genetic variants in suspected hereditary arrhythmia syndromes where the clinical picture is uncertain.
Acknowledgments
Funding: NHLBI (F30HL096279 to YK and HL077929, to TVM) and American Heart Association (to TVM).
Financial Support: This work was supported in part by grants from the NIH/NHLBI (HL093440 and 1RC1HL100756) and American Heart Association.
Footnotes
Conflicts of Interest: None
Author Contributions:
Concept/design: TM, YK, Data collection: YK, RZ, CW, YT, TM. Data analysis/interpretation: TM, YK, RZ, YT. Drafting article: YK, TM. Critical revision of article TM, CW, YT.
Contributor Information
Yamini Krishnan, Department of Molecular Pharmacology, Albert Einstein College of Medicine.
Renjian Zheng, Department of Medicine, Division of Cardiology, Albert Einstein College of Medicine.
Christine Walsh, Department of Pediatrics, Division of Cardiology, Montefiore Medical Center.
YingYing Tang, Department of Forensic Biology, Office of Chief Medical Examiner.
Thomas V. McDonald, Departments of Medicine and Molecular Pharmacology, Albert Einstein College of Medicine.
References
- 1.Anantharam A, Markowitz SM, Abbott GW. Pharmacogenetic considerations in diseases of cardiac ion channels. J Pharmacol Exp Ther. 2003;307:831–838. doi: 10.1124/jpet.103.054569. [DOI] [PubMed] [Google Scholar]
- 2.Saenen JB, Vrints CJ. Molecular aspects of the congenital and acquired Long QT Syndrome: Clinical implications. J Mol Cell Cardiol. 2008 doi: 10.1016/j.yjmcc.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009;30:1486–1511. doi: 10.1002/humu.21106. [DOI] [PubMed] [Google Scholar]
- 4.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- 6.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 7.Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- 8.Cui J, Melman Y, Palma E, Fishman GI, McDonald TV. Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr Biol. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- 9.Jackson HA, Accili EA. Evolutionary analyses of KCNQ1 and HERG voltage-gated potassium channel sequences reveal location-specific susceptibility and augmented chemical severities of arrhythmogenic mutations. BMC Evol Biol. 2008;8:188. doi: 10.1186/1471-2148-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, et al. Most LQT2 mutations reduce Kv11. 1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 12.Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, et al. C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation. 1999;99:1464–1470. doi: 10.1161/01.cir.99.11.1464. [DOI] [PubMed] [Google Scholar]
- 13.Kagan A, Yu Z, Fishman GI, McDonald TV. The dominant negative LQT2 mutation A561V reduces wild-type HERG expression. J Biol Chem. 2000;275:11241–11248. doi: 10.1074/jbc.275.15.11241. [DOI] [PubMed] [Google Scholar]
- 14.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 15.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 16.Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, Zhang S. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–2757. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong Q, Anderson CL, January CT, Zhou Z. Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am J Physiol Heart Circ Physiol. 2002;283:H77–84. doi: 10.1152/ajpheart.00008.2002. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–31126. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- 19.Massaeli H, Guo J, Xu J, Zhang S. Extracellular K+ is a prerequisite for the function and plasma membrane stability of HERG channels. Circ Res. 2010;106:1072–1082. doi: 10.1161/CIRCRESAHA.109.215970. [DOI] [PubMed] [Google Scholar]
- 20.Sun T, Guo J, Shallow H, Yang T, Xu J, Li W, Hanson C, et al. The Role of Monoubiquitination in Endocytic Degradation of Human Ether-a-go-go-related Gene (hERG) Channels under Low K+ Conditions. J Biol Chem. 2011;286:6751–6759. doi: 10.1074/jbc.M110.198408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrin MJ, Subbiah RN, Vandenberg JI, Hill AP. Human ether-a-go-go related gene (hERG) K+ channels: Function and dysfunction. Progress in Biophysics and Molecular Biology. 2008;98:137–148. doi: 10.1016/j.pbiomolbio.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- 23.Akhavan A, Atanasiu R, Noguchi T, Han W, Holder N, Shrier A. Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J Cell Sci. 2005;118:2803–2812. doi: 10.1242/jcs.02423. [DOI] [PubMed] [Google Scholar]
- 24.Akhavan A, Atanasiu R, Shrier A. Identification of a COOH-terminal segment involved in maturation and stability of human ether-a-go-go-related gene potassium channels. J Biol Chem. 2003;278:40105–40112. doi: 10.1074/jbc.M307837200. [DOI] [PubMed] [Google Scholar]
- 25.Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to anendoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 27.Lindberg MO, Oliveberg M. Malleability of protein folding pathways: a simple reason for complex behaviour. Curr Opin Struct Biol. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Delisle BP, Underkofler HA, Moungey BM, Slind JK, Kilby JA, Best JM, Foell JD, et al. Small GTPase determinants for the Golgi processing and plasmalemmal expression of human ether-a-go-go related (hERG) K+ channels. J Biol Chem. 2009;284:2844–2853. doi: 10.1074/jbc.M807289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem. 2005;280:19419–19425. doi: 10.1074/jbc.M502327200. [DOI] [PubMed] [Google Scholar]
- 31.Massaeli H, Sun T, Li X, Shallow H, Wu J, Xu J, Li W, et al. Involvement of caveolin in low K+-induced endocytic degradation of cell-surface human ether-a-go-go-related gene (hERG) channels. J Biol Chem. 2010;285:27259–27264. doi: 10.1074/jbc.M110.124909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J. 1986;111:1088–1093. doi: 10.1016/0002-8703(86)90010-4. [DOI] [PubMed] [Google Scholar]
- 33.Nakajima T, Kurabayashi M, Ohyama Y, Kaneko Y, Furukawa T, Itoh T, Taniguchi Y, et al. Characterization of S818L mutation in HERG C-terminus in LQT2. Modification of activation-deactivation gating properties. FEBS Lett. 2000;481:197–203. doi: 10.1016/s0014-5793(00)01988-8. [DOI] [PubMed] [Google Scholar]
- 34.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 35.Etheridge SP, Compton SJ, Tristani-Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42:1777–1782. doi: 10.1016/j.jacc.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Compton SJ, Lux RL, Ramsey MR, Strelich KR, Sanguinetti MC, Green LS, Keating MT, et al. Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium. Circulation. 1996;94:1018–1022. doi: 10.1161/01.cir.94.5.1018. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu W, Aiba T, Antzelevitch C. Specific therapy based on the genotype and cellular mechanism in inherited cardiac arrhythmias. Long QT syndrome and Brugada syndrome. Curr Pharm Des. 2005;11:1561–1572. doi: 10.2174/1381612053764823. [DOI] [PMC free article] [PubMed] [Google Scholar]