

Non-technical summary
Activation of trigeminal neurons innervating the meninges and release of proinflammatory peptides (in particular calcitonin gene-related peptide (CGRP)) from their terminals are believed to play a key role in generating migraine pain. A monogenic subtype of migraine (familial hemiplegic migraine type-1 (FHM1)) is caused by gain-of-function mutations in a neuronal voltage-gated calcium channel (Cav2.1) involved in controlling neurotransmitter release from many synaptic terminals including those of trigeminal neurons at the meninges. Using a FHM1 transgenic mouse model, we show that the migraine mutation produces gain-of-function (i.e. it increases calcium influx) in a subpopulation of trigeminal neurons that do not innervate the meninges. In contrast, the calcium channels of trigeminal neurons innervating the meninges and releasing CGRP are not affected by the mutation. Congruently, the migraine mutation does not alter CGRP release at the meninges. Our findings suggest that the facilitation of CGRP actions at the meninges does not contribute to the generation of headache in FHM1.
Abstract
Abstract
Familial hemiplegic migraine type-1 (FHM1), a monogenic subtype of migraine with aura, is caused by gain-of-function mutations in CaV2.1 (P/Q-type) calcium channels. The consequences of FHM1 mutations on the trigeminovascular pathway that generates migraine headache remain largely unexplored. Here we studied the calcium currents and excitability properties of two subpopulations of small-diameter trigeminal ganglion (TG) neurons from adult wild-type (WT) and R192Q FHM1 knockin (KI) mice: capsaicin-sensitive neurons without T-type calcium currents (CS) and capsaicin-insensitive neurons characterized by the expression of T-type calcium currents (CI-T). Small TG neurons retrogradely labelled from the dura are mostly CS neurons, while CI-T neurons were not present in the labelled population. CS and CI-T neurons express CaV2.1 channels with different activation properties, and the CaV2.1 channels are differently affected by the FHM1 mutation in the two TG neuron subtypes. In CI-T neurons from FHM1 KI mice there was a larger P/Q-type current density following mild depolarizations, a larger action potential (AP)-evoked calcium current and a longer AP duration when compared to CI-T neurons from WT mice. In striking contrast, the P/Q-type current density, voltage dependence and kinetics were not altered by the FHM1 mutation in CS neurons. The excitability properties of mutant CS neurons were also unaltered. Congruently, the FHM1 mutation did not alter depolarization-evoked CGRP release from the dura mater, while CGRP release from the trigeminal ganglion was larger in KI compared to WT mice. Our findings suggest that the facilitation of peripheral mechanisms of CGRP action, such as dural vasodilatation and nociceptor sensitization at the meninges, does not contribute to the generation of headache in FHM1.
Introduction
Voltage-gated P/Q-type calcium (CaV2.1) channels play a prominent role in initiating neurotransmitter release at many mammalian synapses (Iwasaki et al. 2000; Pietrobon, 2005). Missense mutations in theCACNA1Agene that encodes the pore-forming subunit of CaV2.1 channels cause a rare autosomal dominant subtype of migraine with aura: familial hemiplegic migraine type 1 (FHM1) (Ophoff et al. 1996). Apart from the transient hemiparesis, the aura and headache symptoms of typical attacks of FHM1 are similar to those of the common forms of migraine with aura (Pietrobon & Striessnig, 2003). FHM1 mutations produce gain-of-function of human recombinant CaV2.1 channels, mainly due to channel activation at lower voltages and increased channel open probability (Tottene et al. 2002; Pietrobon, 2010). Accordingly, a larger P/Q-type calcium current density was measured in cortical pyramidal and other central neurons from knockin (KI) mice carrying FHM1 mutations (van den Maagdenberg et al. 2004, 2010; Tottene et al. 2009; Inchauspe et al. 2010). Induction and propagation of cortical spreading depression (CSD) were both facilitated in FHM1 KI mice (van den Maagdenberg et al. 2004, 2010; Eikerman-Haerter et al. 2009), as a consequence of increased glutamate release from cortical pyramidal cell synapses (Tottene et al. 2009). CSD is widely considered the phenomenon underlying migraine aura (Lauritzen, 1994; Ayata, 2009); animal studies indicate that CSD may also initiate the migraine headache mechanisms (Bolay et al. 2002; Zhang et al. 2010, 2011).
Migraine headache is thought to depend on the activation and sensitization of trigeminal sensory afferents innervating the meninges (Pietrobon & Striessnig, 2003; Olesen et al. 2009). The meningeal trigeminal afferents exhibit properties characteristic of nociceptors in other tissues including chemosensitvity and sensitization (Strassmann & Levy, 2006); most C-type and slow A-δ type rat dural afferents are activated by inflammatory agents applied to the dura (Strassmann et al. 1996) and most mechanosensitive C-type guinea pig dural afferents are activated by topical application of capsaicin (Bove & Moskowitz, 1997). Most rat dural afferents expressing the capsaicin receptor TRPV1 co-express the vasoactive neuropeptide calcitonin gene-related peptide (CGRP) (Shimizu et al. 2007) that plays a pivotal role in migraine (Villalon & Olesen, 2009; Ho et al. 2010). P/Q-type calcium channels contribute to control of CGRP release from capsaicin-sensitive perivascular meningeal sensory fibres and from trigeminal ganglion (TG) neurons (Hong et al. 1999; Akerman et al. 2003a; Xiao et al. 2008). P/Q-type calcium channels also contribute to the long-duration action potential (AP) typical of certain TG neurons (Grigaliunas et al. 2002), including capsaicin-sensitive neurons (Catacuzzeno et al. 2008). Thus, FHM1 mutations could have gain-of-function effects on several processes relevant in the pathophysiology of migraine headache (Pietrobon & Striessnig, 2003; Villalon & Olesen, 2009). However, the functional consequences of the FHM1 mutations on P/Q-type calcium channels and excitability of TG neurons are unknown.
Here, we studied the P/Q-type calcium current and the excitability properties of two defined subpopulations of small TG neurons in R192Q FHM1 KI mice. We show that the FHM1 mutation does not affect the P/Q-type calcium channels expressed in capsaicin-sensitive CS neurons, whereas it produces gain-of-function of the P/Q channels and prolongs the AP in capsaicin-insensitive CI-T neurons characterized by the presence of T-type calcium current. Recording from neurons retrogradely labelled from the dura indicate that a major fraction of small dural afferents are CS neurons and none are CI-T neurons. The FHM1 mutation does not affect depolarization-evoked CGRP release from the dura mater, but it increases CGRP release from the trigeminal ganglion.
Methods
Isolation of trigeminal ganglion neurons
Trigeminal ganglion neurons were isolated from 4- to 6-week-old C57BL/6J mice (Charles River Laboratories) or homozygous R192Q FHM1 knockin (KI) and wild-type (WT) littermates (van den Maagdenberg et al. 2004), as previously described (Catacuzzeno et al. 2008). KI and WT littermates were backcrossed for at least five generations with C57BL/6J mice; all mice used for the experiments had at least 97% C57BL/6J background (van den Maagdenberg et al. 2004). Confirmatory genotyping was performed for KI and WT littermates (van den Maagdenberg et al. 2004). A similar number of male and female mice were used. Unless otherwise stated, neurons were studied 12–24 h after plating in dishes coated with poly-d-lysine (Sigma). Only neurons without or with very short processes were used.
To identify dural afferents, the retrograde tracer DiI (Neurotrace, Molecular Probes) was applied to a section of the dura surrounding the superior sagittal sinus. Briefly, 4- to 6-week-old male WT C57BL/6J mice were anaesthetized with an i.p. injection of 3 ml kg−1 of a mixture of ketamine (30 mg ml−1) and xylazine (3 mg ml−1), and placed in the ‘flat-skull’ position in a stereotaxic frame, without the use of ears bars, with their head supported by a custom foam block. Depth of anaesthesia was assessed by monitoring breathing rate and determining reflex responses following pinching of the foot. A midline incision was made to expose the top of the skull. A dental drill was used to make a small hole (approx. 2 mm diameter) halfway between bregma and lambda to expose the dura and the superior sagittal sinus. A small DiI crystal or small amount of DiI in Vaseline (Neurotrace, Molecular Probes) was placed onto the dural surface and covered in saline-soaked gelfoam. The surface of the skull was then sealed with a thin layer of dental acrylic, the skin sutured and then dusted with antibiotic powder (Tricin). Mice received Carprofen (5 mg kg−1s.c.) while recovering from anaesthesia, and again 24 and 48 h following surgery. At least 7 days later, cells were isolated from trigeminal ganglia as described in Borgland et al. (2001), and used for up to 8 h after dissociation.
All animal experiments were performed in accordance with the guidelines of the respective national legislations and with the Animal Care and Use Committee guidelines of the Universities of Padova and Perugia; experiments involving dural tracing (on 14 animals) were approved and monitored by the Australian Joint Royal North Shore Hospital/University of Technology Animal Ethics Committee, a body constituted in accordance with NSW State Legislation and overseen by the Department of Primary Industry. All animal experiments conform to the principles of UK regulations.
The animals were killed by being rendered unconscious by 4% isofluorane in air and then killed by exsanguination, or by cervical dislocation.
CGRP release
The experiments of CGRP release from the hemisected skull preparation were performed as described in Ebersberger et al. (1999). Briefly, the skin and galea were cut along the midline and retracted from the skull, which was then divided into two equal halves along the sagittal suture. The cerebral hemispheres were removed without lesioning the dura mater encephali that remained attached to the skull. The skull cavities were washed for 30 min at 37°C with carbogen (95% O2, 5% CO2)-gassed synthetic interstitial fluid (SIF) containing (mm): 108 NaCl, 26 NaHCO3, 9.6 sodium gluconate, 7.6 sucrose, 5.5 glucose, 3.5 KCl, 1.7 NaH2PO4, 1.5 CaCl2 and 0.69 MgSO4, pH 7.4. The skull halves were then mounted in a water bath that formed a closed humid chamber with a baseline temperature of 37°C. The cut edges of the dura and the outer surrounding tissue were covered with Vaseline to prevent loss of the incubating solutions. During the experiment the skull cavity was filled with 100 μl of either SIF or a potassium-modified SIF (whose osmolarity was kept constant by substituting NaCl with an equivalent concentration of KCl). This volume of fluid was sufficient to cover completely the supratentorial dura mater. After 5 min incubation in any given solution, the solution was removed and mixed with 240 μm EIA buffer (provided with the enzyme immunoassay kit by SPIbio, France), and stored on ice until the determination of the CGRP content. We performed preliminary experiments to identify the potassium concentration capable of inducing a submaximal CGRP release, by incubating the dura in succession with SIF, 35 mm and 60 mm K+-modified SIF. Under the conditions used in our experiments, 35 mm K+ evoked a significant CGRP release (from 5.6 ± 0.6 pg of CGRP released under basal conditions to 10.0 ± 1.3 pg of CGRP released in 35 mm K+, n = 3, P< 0.05), which was, however, significantly smaller than the CGRP released by 60 mm K+ (28.2 ± 4.3 pg, n = 3, P< 0.05vs.the CGRP release in 35 mm K+). Based on these results, to study the effect of the FHM1 mutation on depolarization-evoked CGRP release from the peripheral terminals of dural trigeminovascular afferents, the hemisected skulls from WT and KI mice were filled in succession with SIF (control), 35 mm K+-modified SIF (35K), SIF, SIF (wash).
The experiments of CGRP release from intact isolated trigeminal ganglia were performed as in Eberhartd et al. (2008). Briefly, both TGs were dissected and the dura was carefully removed from the ganglia under stereomicroscopic control. The ganglia were then immersed in SIF for 30 min at 37°C before the experimental protocol was applied. During the experiments, the two TGs dissected from the same mouse were incubated in succession with 200 μl of SIF (control), 35 mm K+-modified SIF (35K), and SIF again (wash). After 10 min incubation each solution was removed and mixed with 240 μm EIA buffer, and stored on ice until the determination of the CGRP content.
For the assessment of CGRP content, the samples were processed with a commercial enzyme immunoassay (EIA) kit for CGRP (SPIbio, France). All solutions used throughout the experiments did not exceed the detection levels for CGRP content (2 pg ml−1). All samples were measured in duplicate and the two CGRP contents were averaged. CGRP content was always expressed as picograms of CGRP released by either one hemisected skull or two ganglia from the same mouse.
Electrophysiological recordings
Whole-cell patch-clamp recordings were made at room temperature (20–25°C) following standard techniques (Hamill et al. 1981) using an Axopatch-200B or Axopatch-1D patch-clamp amplifier and pCLAMP software (Axon Instruments) or an EPC10 amplifier and Patchmaster software (HEKA, Germany).
Recordings of the different calcium current components were performed as in Fletcher et al. (2001). Unless otherwise stated, the pipette solution contained (in mm): caesium methanesulfonate 100, Hepes 30, MgCl2 1, EGTA 10, CsCl 8, MgATP 4, NaGTP 0.5, cAMP 1 (pH 7.4 with CsOH); the seal and break into the cell were done in external Tyrode solution and the cell was then perfused with the recording calcium current solution containing (in mm): TEACl 147, CaCl2 2, Hepes 10, glucose 10, 5 μm nimodipine (pH 7.4 with TEAOH). Three minutes were allowed to equilibrate the pipette solution with the intracellular compartment; the cell was then repeatedly stimulated (every 30 s) with a 136 ms depolarizing pulse to –12 mV (after liquid junction potential correction) (Neher 1992), from a holding potential of –72 mV, to monitor the high-voltage activated (HVA) calcium currents. In the large majority of cells, the N- and P/Q-type calcium current components were dissected pharmacologically using sequential applications of ω-conotoxin-GVIA (GVIA, 1 μm) and ω-conotoxin-MVIIC (MVIIC, 3 μm) in the continuous presence of nimodipine (5 μm) as shown in Fig. 2A. Cytochrome c (0.1 mg ml−1) was included in all recording solutions to block non-specific peptide toxin binding. In a few cells (mainly CS neurons), 30 μm Cd2+ was used in place of MVIIC. Given the low expression of R-type calcium channels in these cells (Borgland et al. 2001; Fig. 2B), the Cd2+-sensitive current under these conditions essentially represents the P/Q-type component. Under each pharmacological condition tested, a series of 50 ms depolarizing pulses from –62 mV to +18 mV (in 10 mV steps) were applied to construct current–voltage (I–V) relationships, followed by an action potential (AP)-like voltage stimulation to record the AP-evoked calcium current (AP clamp using a typical AP recorded in current clamp from a TG neuron in response to a suprathreshold depolarizing stimulus). Depolarizing pulses of 50 ms from –52 to –32 mV, preceded by a 500 ms preconditioning pulse to –102 mV, were applied to verify the presence of low-voltage-activated (LVA) T-type calcium currents. In a number of experiments dedicated to the study of the properties of the LVA calcium currents, cells were held at –92 mV. Unless otherwise stated, I–Vrelationships were obtained only from cells with a voltage error of <5 mV. At the end of the experiment, the extracellular solution was changed back to Tyrode solution, and 1 μm capsaicin was applied to verify the presence of a capsaicin-activated current in the cell under study (at –72 or –92 mV). Recordings from preparations containing labelled dural afferents were made as outlined in Borgland et al. (2001). Solutions were similar to the above except that the internal solution contained CsCl instead of caesium methansulfonate, no cAMP, and 0.2 mm NaGTP, while the external solution contained either 1.25 or 2.5 mm CaCl2, 1 mm MgCl2 and no nimodipine. Ca2+ currents were elicited fromVh = –96 mV to test potentials between –66 mV and +54 mV.
Figure 2. P/Q-type calcium currents in CS and CI-T trigeminal ganglion neurons.

A, peak whole-cell Ca2+ currentvs. time recorded from a CS neuron at –12 mV (every 30 s from a holding potential of –72 mV) during a typical experiment carried out to dissect pharmacologically the N- and P/Q-type Ca2+ current components. Bars indicate the time periods during which nimodipine, ω-conotoxin GVIA (GVIA) and ω-conotoxin MVIIC (MVIIC) were applied. The labelI–Vindicates the time point at which a family of 50 ms pulses in the range –62 mV to +18 mV was applied. Inset on the left: representative current traces at timesa, bandc; on the right: N- and P/Q-type Ca2+ current traces obtained as the difference between traces at timesaandb, andbandc, respectively. Cell WT6D.B, percentage of nimodipine-insensitive Ca2+ current inhibited by GVIA (N-type), MVIIC (P/Q-type) and remaining in the presence of MVIIC (R-type) in CS and CI-T neurons.C, average normalized total nimodipine-insensitive Ca2+ current as a function of voltage in CS (n = 8) and CI-T (n = 13) neurons (obtained in cells with voltage error ≤10 mV). Dotted lines are fits of equation:I = Gmax(V–Erev){1+exp[(V1/2–V)/k]}−1 withV1/2 = –12.0 ± 0.4 mV (k = 5.6 ± 0.2 mV) for CS neurons andV1/2 = –16.7 ± 0.2 mV (k = 5.7 ± 0.1 mV) for CI-T neurons. Experimental points for CI-T neurons were fitted atV≥–22 mV.D, average normalized P/Q-type Ca2+ current as a function of voltage in CS (n = 12) and CI-T (n = 13) neurons (obtained in cells with voltage error ≤5 mV). Dotted lines are fits withV1/2 = –16.5 ± 0.3 mV (k = 5.4 ± 0.2 mV) for CS neurons andV1/2 = –19.8 ± 0.3 mV (k = 5.1 ± 0.2 mV) for CI-T neurons. Experimental points for CI-T neurons were fitted atV≥–32 mV. The hint of a shoulder at low voltages in theI–Vof CI-T neurons is a consequence of a small rundown of the R-type (and possibly residual slow LVA) calcium current and the fact that the P/Q current is obtained as difference current. Inset: pooled P/Q-type Ca2+ current traces at –12 mV obtained by pharmacological subtraction in CS (black) and CI-T (grey) neurons. Mean capacitance of CS and CI-T neurons: 11 ± 1 pF (n = 12) and 12 ± 1 pF (n = 13), respectively.
Currents were sampled at 5–20 kHz and low-pass filtered at 1–2 kHz. Compensation (typically 70–80%) for series resistance was used. Cell capacitance was obtained by integrating the capacitative current elicited by 5 mV voltage steps or by zeroing the amplifier capacitance compensation circuit.
Current-clamp experiments were performed using an intracellular solution containing (in mm): KCl 135, CaCl2 1, MgCl2 2, EGTA 10, Hepes 10, NaATP 4 and NaGTP 1 (pH 7.2 with KOH) and an extracellular solution containing (in mm): NaCl 145, KCl 5, CaCl2 2, MgCl2 1, Hepes 10 and glucose 10 (pH 7.4 with NaOH). Voltage traces were sampled at 25 kHz and low-pass filtered at 3 kHz. Cell capacitance, and membrane and access resistance were measured in voltage-clamp configuration at the beginning of the experiment (after about 2 min from achieving the whole-cell configuration) using the automatic compensation circuitry of the EPC-10 amplifier, at a holding potential of –60 mV. After recording the cell resting potential for 1 min, short (1 ms) depolarizing current pulses, to evoke single action potentials, and then long (500 ms) depolarizing current pulses, to evoke multiple firing, were applied. Long (500 ms) hyperpolarizing current pulses were applied to verify the presence of a rebound depolarization (RDP). Finally, the responsiveness to bath application of 1 μm capsaicin at –60 mV of applied potential was verified after switching back to voltage-clamp mode.
RDPs due to the opening of low-voltage-activated (LVA or T-type) calcium channels and RDPs due to theIh current could be easily distinguished on the basis of their time course and the presence of a voltage sag during the hyperpolarizing pulse: slow RDPs with sigmoidal time course without voltage sag were considered as indicative of T-type calcium channels, whereas fast RDPs with a voltage sag during the hyperpolarizing pulse (observed in only a few cells) were considered as indicative ofIh. In a dedicated set of experiments we verified that the slow RDP was selectively present in neurons expressing LVA calcium currents; first, we established the presence or absence of a slow RDP as described above, then we switched to voltage clamp to measure LVA calcium currents after perfusion with a solution in which Na+ and Ca2+ ions were replaced by TEA+ and Ba2+ ions, respectively.
All drugs were stored as stock solutions at –20°C: 1 mm capsaicin (Sigma) in DMSO, 250 μmω-conotoxin GVIA (Bachem, Budendorf, Switzerland), 250 μmω-conotoxin MVIIC (Bachem), 100 mm NiCl2 (Sigma), 10 mm CdCl2 (Sigma) and 2 m TEACl (Sigma) in distilled water. Carprofen, tricin, ketamine and xylazine were from Cenvet, Huntingwood, Australia.
Data analysis
TheI–Vrelationships were fitted with the equation:
wherek = RT/zF.
Results are expressed as mean ± standard error of the mean (SEM). Statistical differences between means were analysed using attest which does not assume equal variances. In multiple comparisons, thettest was performed when the one-way ANOVA analysis detected a significant difference (P< 0.05). *, ** and *** in figures indicateP< 0.05, < 0.01 and < 0.001 in thettest, respectively.
Results
We measured calcium currents and capsaicin responses in isolated adult mouse TG neurons of small size (capacitance, C≤ 20 pF). According to our previous findings, this population of small-diameter cells comprise most of the TG neurons with tonic or slowly adapting firing and almost all the capsaicin-sensitive TG neurons (Catacuzzeno et al. 2008). Capsaicin elicited an inward current in 58% (41 out of 71) of small (C≤ 20 pF) TG neurons (Fig. 1A). Low-voltage-activated (LVA or T-type) calcium channels were not expressed in the capsaicin-sensitive (CS) neurons, as shown by the absence of a measurable Ca2+ current at –52 mV (after liquid junction potential correction) (Fig. 1A) following depolarization from a holding potential, Vh, of –92 mV (as in Fig. 1) or after a 500 ms prepulse to –102 mV fromVh = –72 mV (Figs 2–4). Capsaicin-insensitive neurons were subdivided into two classes depending on the presence (CI-T neurons) or absence (CI neurons) of a LVA calcium current; CI-T neurons accounted for 27% (19 out of 71) and CI neurons for 15% (11 out of 71) of the small TG neuron population (Fig. 1A). Given the small number of CI neurons, the present study was restricted to the two most abundant groups, CS and CI-T neurons.
Figure 1. Sensitivity to capsaicin and presence of LVA calcium currents distinguish three subgroups of small trigeminal ganglion neurons.

A, small-diameter TG neurons (C≤ 20 pF) were subclassified into three subgroups, based on the sensitivity to capsaicin and the presence of a LVA (T-type) calcium current: capsaicin-sensitive, T-type negative (CS) neurons, capsaicin-insensitive, T-type positive (CI-T) neurons, and capsaicin-insensitive, T-type negative (CI) neurons.Aalso shows a chart histogram with the frequency of occurrence of the three neuronal subgroups.B, top: LVA calcium current at –52 mV in a representative CI-T neuron in control (tracea) and in the presence of 20 μm Ni2+ (traceb), 50 μm Ni2+ (tracec) and 100 μm Ni2+ (traced). The corresponding normalized current tracesa, bandd, together with the normalized difference trace (a – brepresenting the LVA current inhibited by 20 μm Ni2+) are shown on the bottom. The time course of inactivation of the latter current (tracea – b) was best fitted by a single exponential with time constant of 12 ms, whereas that of the total LVA current (tracea) required two exponentials with time constants of 14 ms (82%) and 79 ms (18%). The residual current in the presence of 100 μm Ni2+ (traced) could be well fitted with the same components by increasing the fraction of the slow component from 18 to 70%. 95% of the peak current (T95) was reached after 8.1, 8.9 and 13 ms for the Ni2+-sensitive (tracea – b), total LVA (tracea) and residual (traced) currents, respectively. Cell T16D.C, Ca2+ current as a function of voltage and corresponding Ca2+ current traces at –82, –72, –62, –52 and –42 mV of a CI-T neuron with a largely prevailing fast LVA current component. The time course of inactivation during 136 ms pulses at –52 mV was best fitted by two exponentials with time constants of 13 ms (88%) and 94 ms (12%). T95 = 6 ms. Cell T16I.D, Ca2+ current as a function of voltage and corresponding Ca2+ current traces of a CI-T neuron with a largely prevailing slow LVA current component. The time course of inactivation at –52 mV was best fitted by a single exponential with time constant of 68 ms and a constant component of 43 pA, that possibly represents a small R-type Ca2+ current; the best fit required such a small constant value in most neurons with prevailing slow LVA and in some neurons with prevailing fast LVA current. T95 = 22 ms. Right inset: LVA Ca2+ current at –52 mV in control (tracea) and in the presence of 100 μm Cd2+ (traceb). The time course of inactivation of the residual current in the presence of 100 μm Cd2+ was best fitted by a single exponential with time constant of 103 ms. Cell T10A.
Figure 4. Selective gain of function of P/Q-type calcium current in CI-T trigeminal ganglion neurons from R192Q KI mice.

A, average P/Q-type Ca2+ current density as a function of voltage in CI-T neurons from WT and R192Q KI mice obtained by using the pharmacological protocol illustrated in Fig. 2A. Average normalizedI–Vcurves (obtained in cells with voltage error ≤5 mV: WT, n = 13; KI, n = 8) were multiplied by the average maximal current densities from all cells (WT, n = 15; KI, n = 12). Dotted lines are fits (as in Fig. 2) withV1/2 = –19.8 ± 0.3 mV (k = 5.1 ± 0.2 mV) for WT neurons andV1/2 = –24 ± 0.3 mV (k = 4.5 ± 0.2 mV) for KI neurons. Insets: pooled P/Q-type Ca2+ current traces at –32, –22 and –12 mV obtained by pharmacological subtraction in WT (black) and KI (red) CI-T neurons. Mean capacitance of WT and KI CI-T neurons: 12±1 pF (n = 15) and 11 ± 1 pF (n = 12), respectively.B, average action potential-evoked P/Q-type Ca2+ current densities in CI-T neurons from WT and R192Q KI mice, obtained as in Fig. 3B. The average normalized P/Q currents (obtained in cells with voltage error ≤5 mV: WT, n = 10, KI, n = 8) were multiplied by the average maximal current densities from all cells (WT, n = 15; KI, n = 12).
The investigation of the effect of low concentrations of Ni2+ on the calcium current recorded at –52 mV in CI-T neurons revealed the presence of two LVA calcium current components with different Ni2+ sensitivity and different kinetics of activation and inactivation (Fig. 1B). Ni2+ at 20 μm selectively inhibited a LVA calcium current component characterized by fast kinetics of activation and inactivation (Fig. 1B); the average time constant of inactivation of this Ni2+-sensitive fast LVA current component was 14 ± 1 ms (n = 9), and the time to reach 95% of peak current (T95) was 6.2 ± 0.4 ms. The residual LVA current in the presence of 20 μm Ni2+ was characterized by slower kinetics of activation and inactivation and was less sensitive to Ni2+ block, as shown by the relatively small further inhibition by 50 and 100 μm Ni2+ (Fig. 1B). On average, 20, 50 and 100 μm Ni2+ inhibited 65 ± 2% (n = 12), 66 ± 4% (n = 9) and 74 ± 2% (n = 4), respectively, of the calcium current at –52 mV. Distinct kinetics of activation, inactivation and deactivation of the LVA calcium current were observed in different CI-T neurons, reflecting different relative fractions of the fast and slow LVA calcium current components. Figure 1CandD shows theI–Vrelationships and the LVA calcium current traces at different voltages recorded from two CI-T neurons with largely prevailing fast and slow LVA calcium current components, respectively. On average, the time constant of inactivation of the calcium current at –52 mV in neurons with prevailing fast LVA was 15 ± 1 ms (n = 9); in many neurons a small slower component with average time constant of 110 ± 7 ms (14 ± 1%, n = 7) was present. In neurons with prevailing slow LVA the time constant of inactivation was on average 81 ± 11 ms (n = 10). T95 was 6.6 ± 0.8 ms (n = 9) and 22 ± 2 ms (n = 10) in neurons with prevailing fast and slow LVA, respectively. Although we did not carry out detailed kinetic analysis of deactivation, neurons with prevailing fast LVA and slow LVA also differed in the kinetics of tail currents, which were slower in neurons with slow LVA. Typically, the first measurable LVA calcium current was observed at –72 mV in CI-T neurons with prevailing fast LVA (Fig. 1C) and at –62 mV in CI-T neurons with prevailing slow LVA (Fig. 1D). Holding the cell at –72 mV led to complete inactivation of the LVA current in CI-T neurons with prevailing fast LVA, whereas a small residual LVA current could still be observed in CI-T neurons with prevailing slow LVA (not shown). The right inset in Fig. 1D shows that a high concentration of Cd2+ (100 μm, a concentration that completely blocks the HVA calcium current, not shown) blocked a relatively small fraction of the slow LVA calcium current component.
The contributions of N- and P/Q-type calcium channels to the high-voltage activated calcium current of CS and CI-T neurons were dissected by recording the Ca2+ current at –12 mV fromVh = –72 mV during successive applications of ω-conotoxin-GVIA (GVIA, 1 μm) and ω-conotoxin MVIIC (MVIIC, 3 μm) in the continuous presence of the L-type channel blocker nimodipine (5 μm) (Fig. 2A) (Tottene et al. 2009). The N- and P/Q-type calcium current components were obtained as the difference current before and after GVIA (a–b) and MVIIC (b–c), respectively, as shown in Fig. 2A. CS and CI-T neurons express different relative fractions of N- and P/Q-type calcium channels: in CS neurons, N-type channels accounted for a relatively larger fraction of the nimodipine-insensitive calcium current than P/Q-type channels while in CI-T neurons the proportions were reversed (Fig. 2B). The larger fraction of P/Q-type channels in CI-T neurons is mainly due to a lower density of N-type channels rather than a larger density of P/Q-type channels, since the P/Q-type current density was similar in CI-T and CS neurons (38 ± 4 pA pF−1vs.30 ± 4 pA pF−l) whereas the N-type current density at –12 mV was significantly smaller in CI-T neurons (25 ± 4 pA pF−1 in CI-Tvs. 54 ± 11 pA pF−1 in CS;P< 0.05). CS neurons did not show a significant residual (R-type) current in the presence of MVIIC, whereas a small residual current was observed in CI-T neurons (Fig. 2B). This R-type calcium current, together with the small slow LVA calcium current that is not inactivated atVh = –72 mV, accounts for the small shoulder that characterizes the averageI–Vrelationship of CI-T neurons at low voltages (Fig. 2C). CS and CI-T neurons showed differentI–Vcurves of both the total nimodipine-insensitive (Fig. 2C) and the P/Q-type calcium current obtained as current difference before and after MVIIC (Fig. 2D). Fitting of the normalizedI–Vrelationships of the P/Q-type calcium current in individual CS and CI-T neurons gave significantly different mean half-activation voltages: –16.6 ± 0.8 mV (n = 12) and –19.6 ± 0.7 mV (n = 13) in CS and CI-T neurons, respectively (P< 0.05; with similar mean slopes of 5.2 ± 0.2 and 4.9 ± 0.2 mV). This result suggests that CS and CI-T neurons express different types of P/Q calcium channels.
We investigated the functional consequences of FHM1 mutations on the P/Q-type calcium channels of CS and CI-T neurons by measuring the P/Q-type calcium current density as a function of voltage in neurons from FHM1 KI mice carrying the R192Q missense mutation. In striking contrast with previous findings in central neurons of R192Q KI mice (van den Maagdenberg et al. 2004; Tottene et al. 2009; Inchauspe et al. 2010), the mean half-activation voltage of the P/Q-type calcium current as well as the P/Q-type calcium current density were similar in CS neurons from R192Q KI mice and WT mice (Fig. 3A). The mean half-activation voltage obtained by fitting the normalizedI–Vrelationships of the P/Q-type calcium current in individual CS neurons from R192Q KI mice was –17.9 ± 0.9 mV (n = 14; mean slope: 5.4 ± 0.2 mV), a value not significantly different from that obtained for CS neurons from WT mice (–16.6 ± 0.8 mV; mean slope 5.2 ± 0.2 mV). Likewise, the kinetics of activation and inactivation of the P/Q-type current recorded in CS neurons at different voltages (see pooled traces in inset of Fig. 3A) as well as the P/Q-type calcium currents elicited by action potential waveforms (previously recorded from a WT CS neuron: Fig. 3B) were similar in R192Q KI and WT mice.
Figure 3. The FHM1 mutation does not significantly affect the P/Q-type calcium current in CS trigeminal ganglion neurons from R192Q KI mice.

A, average P/Q-type Ca2+ current density as a function of voltage in CS neurons from WT and R192Q KI mice obtained by using the pharmacological protocol illustrated in Fig. 2A. Average normalizedI–Vcurves (obtained in cells with voltage error ≤5 mV: WT, n = 12, KI, n = 14) were multiplied by the average maximal current densities from all cells (n = 15 for both WT and KI). Continuous lines are fits (as in Fig. 2) withV1/2 = –16.5 ± 0.3 mV (k = 5.4 ± 0.2 mV) for WT neurons andV1/2 = –18.0 ± 0.3 mV (k = 5.7 ± 0.2 mV) for KI neurons. Insets: pooled P/Q-type Ca2+ current traces at –32, –22 and –12 mV obtained by pharmacological subtraction in WT (black) and KI (red) CS neurons. Mean capacitance of WT and KI CS neurons: 11 ± 1 pF (n = 15) and 10 ± 1 pF (n = 15), respectively.B, top: action potential waveform elicited by suprathreshold stimulation in a WT CS neuron; bottom: average P/Q-type Ca2+ current densities in response to the action potential used as voltage stimulus in CS neurons from WT and R192Q KI mice. Action potential-evoked P/Q-type current was obtained as difference current before and after MVIIC (see Fig. 2A); the average normalized P/Q currents (obtained in cells with voltage error ≤5 mV: WT, n = 6, KI, n = 7) were multiplied by the average maximal current densities from all cells (n = 9 for both WT and KI).
By contrast, in CI-T neurons from R192Q KI mice the activation of the P/Q-type calcium current was shifted to lower voltages and the P/Q-type calcium current density was significantly larger at relatively mild depolarizations compared to WT mice (Fig. 4A). Fitting of the normalizedI–Vrelationships of the P/Q-type calcium current in individual KI and WT CI-T neurons gave mean half-activation voltages of –24 ± 0.5 mV (n = 8) and –19.6 ± 0.7 mV (n = 13), respectively (P< 0.05; with similar mean slopes of 4.4 ± 0.2 and 4.9 ± 0.2 mV:P> 0.05). AtV> 0 mV, P/Q current densities were similar in KI and WT CI-T neurons, indicating similar densities of functional CaV2.1 channels (van den Maagdenberg et al. 2004; Tottene et al. 2009). Also similar were the N- and R-type calcium current densities (at 0 mV, N-type: 25 ± 4 pA pF−1, n = 15, vs. 29 ± 6 pA pF−1, n = 11; R-type: 3 ± 1 pA pF−1, n = 15, vs. 5 ± 2 pA pF−1, n = 9, in WTvs. KI neurons), thus confirming the absence of compensatory changes, as previously shown for central neurons of R192Q KI mice (van den Maagdenberg et al. 2004; Tottene et al. 2009). Interestingly, the different activation curves of mutant and WT P/Q-type channels resulted in different P/Q-type calcium currents elicited by action potential waveforms in CI-T neurons; in particular, the mean AP-evoked P/Q-type calcium current density was larger in KI neurons during the shoulder that characterizes the slow repolarisation of the AP (Fig. 4B).
Next, we investigated the functional consequences of the gain-of-function of mutant P/Q-type calcium channels on the excitability of CI-T neurons by measuring the action potentials elicited by short (1 ms) and long (500 ms) depolarizing current pulses using current-clamp recordings under near-physiological conditions. CI-T neurons were identified on the basis of their insensitivity to capsaicin and their ability to evoke a slow transient rebound depolarization (RDP) immediately after a long (500 ms) hyperpolarizing current pulse (Fig. 5A). In a dedicated set of experiments, we verified that the slow RDP was selectively present in neurons expressing LVA calcium currents (Fig. 5A and see Methods). A significant LVA calcium current at –50 mV, elicited after a 500 ms prepulse to –100 mV, was measured in all neurons with slow RDP (average density 25 ± 3 pA pF−1, n = 9), whereas none of the neurons without RDP (n = 15) showed a significant LVA calcium current. The slow RDP was inhibited by 100 μm Ni2+ to a similar extent (80 ± 10%, n = 3) as the LVA calcium current. All neurons with slow RDP on which capsaicin was tested turned out to be capsaicin insensitive. The capsaicin-insensitive neurons with slow RDP accounted for 34% (22 out of 65) of the total small WT TG neuron population. CS neurons were identified on the basis of their sensitivity to capsaicin and the lack of the slow RDP and they accounted for 54% of the small TG neurons, while CI neurons were identified on the basis of their insensitivity to capsaicin and the lack of the slow RDP and they accounted for 12% of the small TG neurons (Fig. 5B). The close match between the fractions of CI-T, CS and CI neurons, inferred from the presence of RDP and the response to capsaicin, and those shown in Fig. 1, obtained by direct recording of the T-type currents, is a strong indication of the reliability of the protocol adopted for the identification of the neuron subtypes in the current-clamp experiments. Given that all neurons with slow RDP in which capsaicin was tested were capsaicin insensitive, we included also a number of neurons with slow RDP in which capsaicin was not tested in the following comparison of the excitability properties of CI-T and CS neurons.
Figure 5. Excitability properties of CI-T and CS trigeminal ganglion neurons.
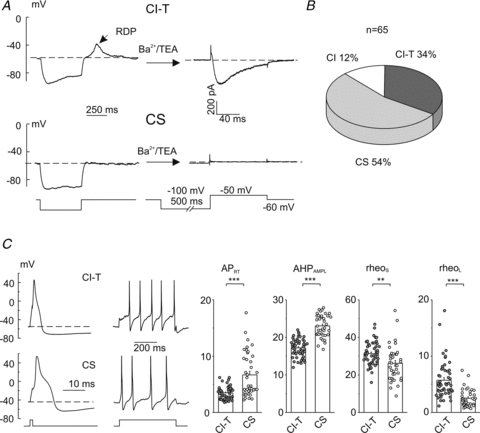
A, the presence of a slow rebound depolarization (RDP) under physiological ionic conditions correlates with the presence of a LVA Ca2+ current. The presence of a RDP was verified at the beginning of the experiment by applying, under current-clamp mode, a 500 ms hyperpolarizing current able to bring the membrane potential close to –80 mV (representative traces for a CI-T and CS neuron are shown on the left); then the cell was perfused with a solution in which Na+ and Ca2+ ions were replaced by TEA+ and Ba2+ ions and Ca2+ currents were measured in voltage clamp in response to depolarizing pulses preceded by 500 ms prepulses to –100 mV from a holding potential of –60 mV (right traces: Ca2+ current recorded at –50 mV). Cells TR170506E5 and TR120506E6.B, chart histogram with the frequency of occurrence of capsaicin-insensitive neurons with slow RDP (CI-T), capsaicin-sensitive neurons without slow RDP (CS) and capsaicin-insensitive neurons without slow RDP (CI). The sensitivity to capsaicin was verified at the end of the experiment, under voltage clamp at a holding potential of –60 mV, by extracellular application of 1 μm capsaicin.C, left: typical voltage responses evoked by applying short (1 ms) and long (500 ms) depolarizing current pulses at 1.5–2× rheoL in WT CI-T and CS neurons. Cells TR190506E4 and TR230207E1. Right: mean AP repolarizing time (APRT), afterhyperpolarization amplitude (AHPAMPL), rheoS and rheoL for CI-T (n = 51) and CS (n = 38) neurons. All these parameters were found significantly different (P< 0.05) between CI-T and CS neurons. Mean capacitance of CI-T and CS neurons: 11 ± 0.3 pF (n = 51) and 10 ± 0.5 pF (n = 38), respectively.
Long-duration action potentials (APs) with typical shoulder were elicited by 1 ms depolarizing pulses in both CI-T and CS neurons from WT mice (Fig. 5C). However, the AP of CI-T neurons was of shorter duration and was followed by an afterhyperpolarization (AHP) of lower amplitude than that in CS neurons; as a measure of the AP duration we considered the AP repolarizing time, APRT, defined as the time needed to repolarize from the peak of the AP down to the resting membrane potential. Higher current injections were necessary to evoke APs in CI-T than in CS neurons (by both short and long current pulses: rheoS and rheoL, respectively; Fig. 5C). Multiple tonic or slowly adapting firing was elicited by long depolarizing pulses in both CS and CI-T neurons, with mean frequency (at 1.5–2× rheoL) of 6.2 ± 0.7 Hz (n = 38) and 8.7 ± 0.7 Hz (n = 49), respectively (P< 0.05). Notably, in most CI-T neurons (41 out of 51: 80%) the multiple firing was preceded by a typical delay (of mean duration 110 ± 6 ms at 1.5–2× rheoL, n = 32) which was never observed in CS neurons (n = 38) (Fig. 5C). The properties of CI-T neurons with typical delay are similar to those of the delayed multiple firing (DMF) TG neuronal subpopulation that expresses a conspicuous A-type voltage-gated K+ current, which is responsible for the typical delay that precedes the multiple AP firing (Catacuzzeno et al. 2008).
The gain-of-function of the P/Q-type calcium channel in CI-T neurons from R192Q KI mice leads to significant changes in the duration and shape of the action potential. CI-T neurons from R192Q KI mice displayed a prolonged action potential compared to WT CI-T neurons (APRT: 5.2 ± 0.4 ms, n = 22, vs.4.1 ± 0.3 ms, n = 21, for KI and WT CI-T neurons, respectively;P< 0.05). This is illustrated in Fig. 6A, showing representative APs and the cumulative distributions of the AP repolarizing time in WT and KI CI-T neurons. The analysis of the first derivative of the voltage waveform shows that the more prolonged APRT in KI neurons is caused by a more delayed second rate of repolarization, as indicated by the smaller absolute value of the second minimum rate of repolarization rr2 (Fig. 6B; 24 ± 1 V s−1, n = 22, vs. 31 ± 2 V s−1, n = 21, for KI and WT CI-T neurons, respectively;P< 0.01). KI and WT CI-T neurons did not show significant differences in the minimal current injections necessary to elicit APs (rheoS: 34 ± 2 pA pF−1, n = 22, vs.35 ± 1 pA pF−1, n = 21; rheoL: 5.3 ± 0.7 pA pF−1, n = 22, vs.5.7 ± 0.5 pA pF−1, n = 21 for KI and WT CI-T neurons, respectively) and in firing frequency (9.9 ± 0.7 Hz, n = 20, vs.9.0 ± 1 Hz at 1.5× rheoL, n = 21 for KI and WT CI-T neurons, respectively).
Figure 6. Action potentials are prolonged in CI-T trigeminal ganglion neurons from R192Q KI mice.

A, left: representative superimposed APs upon 1 ms depolarizing pulses for a WT (black) and a KI (red) CI-T neuron. Cells TR160508E6 and TR151008E5. Right: cumulative distributions of the AP repolarizing time (APRT) in WT (n = 21) and KI (n = 22) CI-T neurons.B, left: superimposed first derivatives of the APs shown inA. Right: cumulative distributions of the second minimum rate of repolarization (rr2) in WT (n = 21) and KI (n = 22) CI-T neurons. Mean capacitance of both WT and KI CI-T neurons was 12 ± 0.6 pF.
As expected from the lack of effects of the R192Q mutation on the functional properties of P/Q-type currents in the CS subgroup, WT and R192Q KI CS neurons did not show significant differences in the action potential shape parameters or in any other excitability property (data not shown).
To explore the possible implications of our findings for migraine pain, we investigated whether CI-T neurons innervate the dura by measuring the calcium currents in small TG neurons retrogradely labelled from the dura of WT mice. Calcium currents were also recorded from a similar number of small unlabelled TG neurons from the same preparation. CI-T neurons were identified on the basis of the presence of an LVA calcium current with the typical kinetics shown in Fig. 1 atV≤–36 mV (fromVh = –96 mV). None of the labelled dural TG neurons withC≤ 20 pF that we recorded (n = 24) showed an LVA calcium current, whereas this current was observed in 13 out of 35 (37%) of the unlabelled cells of similar size (Fig. 7). Capsaicin elicited an inward current in almost all labelled TG neurons tested (8 out of 9), but only in 10 out of 19 (53%) of unlabelled cells (Fig. 7). None of the capsaicin-sensitive neurons showed an LVA calcium current and all the unlabelled cells with LVA calcium current were capsaicin insensitive. The fractions of CS and CI-T neurons in the control population of unlabelled small TG neurons were similar to those shown in Figs 1 and 5. In contrast, according to our definition of CS and CI-T neurons, a major fraction of small dural afferents are CS neurons and none of them are CI-T neurons.
Figure 7. Small trigeminal ganglion neurons retrogradely labelled from the dura do not include CI-T neurons; most are CS neurons.
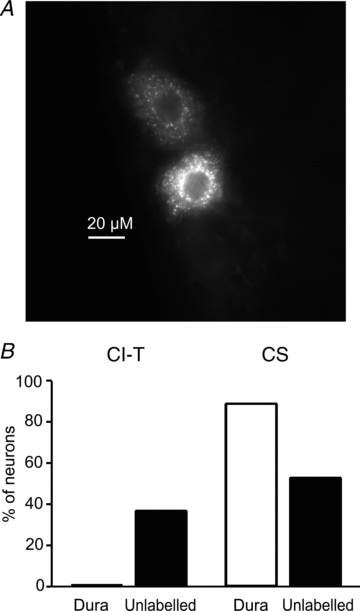
TG neurons were dissociated from wild-type mice in which the fluorescent tracer DiI was previously applied to the dura to label dural afferents. Whole-cell Ca2+ current was recorded from both labelled and unlabelled neurons withC≤ 20 pF. Ca2+ currents were elicited fromVh = –96 mV to test potentials between –66 mV and +54 mV. CI-T neurons were identified on the basis of the presence of LVA Ca2+ current and CS neurons on the basis of capsaicin sensitivity and absence of LVA Ca2+ current.A, image of TG neurons retrogradely labelled from dura from a 16 μm thick slice of a longitudinally sectioned TG. The image was taken using a 40× oil immersion lens.B, fractions of CI-T and CS neurons among small labelled dural and unlabelled TG neurons. The presence of LVA Ca2+ current was tested in 24 labelled dural afferents and 35 unlabelled TG neurons withC≤ 20 pF; 13 of the unlabelled cells were from animals that had surgery but in which DiI was placed in the bone just above the dura. Capsaicin was tested in a fraction of labelled dural (n = 9) and unlabelled (n = 19) TG neurons.
Several lines of evidence indicate that most small capsaicin-sensitive (CS) dural afferents are peptidergic neurons expressing CGRP (see Discussion). If the FHM1 mutation does not affect the P/Q calcium channels at the peripheral terminals of CS neurons, one would predict unaltered CGRP release from CS dural afferents of R192Q KI mice. To test this prediction, we measured K+-evoked CGRP release from dura mater in fluid-filled hemisected skulls from WT and R192Q KI mice. Neither the basal CGRP release nor the CGRP release evoked by 35 mm KCl (a submaximal depolarizing stimulus) were significantly different in KI compared to WT mice (Fig. 8A). In contrast, CGRP release evoked by the same depolarizing stimulus from TG neuron cell bodies in intact isolated trigeminal ganglia was enhanced in KI compared to WT mice (Fig. 8B).
Figure 8. The FHM1 mutation does not significantly affect depolarization-evoked CGRP release from the dura, but it increases depolarization-evoked CGRP release from the trigeminal ganglion.
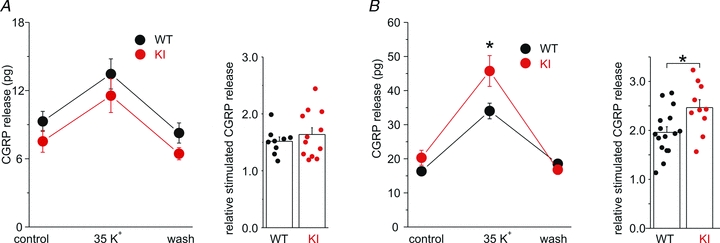
A, left: CGRP released from the dura in hemisected skulls from WT (n = 9) and R192Q KI mice (n = 12) in control solution, after stimulation with 35 mm K+ and after wash. Right: relative stimulation of CGRP release, obtained by dividing the CGRP released in 35 mm K+ by the average of the CGRP released in control and after wash.B, left: CGRP released from intact isolated trigeminal ganglia from WT (n = 16) and R192Q KI mice (n = 10) in control solution, after stimulation with 35 mm K+ and after wash. Right: relative stimulation of CGRP release, obtained as inA.
Discussion
We studied the calcium currents and excitability of two populations of small (C≤ 20 pF) trigeminal ganglion neurons from adult WT and R192Q FHM1 KI mice: capsaicin-sensitive neurons without T-type calcium currents (CS) and capsaicin-insensitive neurons characterized by the expression of T-type calcium currents (CI-T). Both CS and CI-T neurons showed APs of long duration with typical shoulder and tonic or slowly adapting AP firing. However, in CI-T neurons APs were on average of shorter duration and the multiple firing was preceded by a typical delay, which was never observed in CS neurons. CS and CI-T neurons also differed in the relative fraction of P/Q-, N- and R-type calcium channels and in the voltage dependence of activation of the P/Q-type calcium current component. While the molecular mechanisms underlying the different activation properties of P/Q-type calcium channels in CS and CI-T neurons remain unknown, possible mechanisms include the expression of different splicing variants of the α1 subunit and/or the expression of different auxiliary subunits and/or other modulatory proteins.
One of the key finding of this study is the demonstration that the CaV2.1 channels expressed in CS and CI-T neurons are differently affected by the R192Q FHM1 mutation. The activation of the P/Q-type calcium current was shifted to lower voltages and the P/Q-type calcium current density was larger in a wide voltage range in CI-T neurons from R192Q KI compared to WT mice. Although quantitatively smaller, the gain-of-function effect of the R192Q mutation on the CaV2.1 channels of CI-T neurons is qualitatively similar to that previously reported for CaV2.1 channels in cerebellar, cortical and brainstem neurons (van den Maagdenberg et al. 2004; Tottene et al. 2009; Inchauspe et al. 2010). Interestingly, the shift to lower voltages of channel activation produced by the FHM1 mutation in CI-T neurons resulted in a larger action potential-evoked P/Q-type calcium current during the slow repolarization phase of the AP (Fig. 4), and also in an AP of longer duration due to a delayed slow repolarization and more pronounced AP shoulder (Fig. 6). In striking contrast, neither the P/Q-type calcium current density nor its voltage dependence were altered by the FHM1 mutation in CS neurons from R192Q KI mice.
While differential effects of FHM1 mutations on recombinant CaV2.1 channels containing either different auxiliary β subunits (Mullner et al. 2004) or different α1 splice variants (Adams et al. 2009) were previously reported, this is the first demonstration of a differential effect on native P/Q-type calcium channels expressed in different neurons and the first evidence that the gating properties of specific neuronal CaV2.1 channels may be unaffected by FHM1 mutations. This finding has an important general implication for familial migraine mechanisms, in that neuron subtype-specific alterations of CaV2.1 channels by FHM1 mutations may help to explain why a mutation in a calcium channel that is widely expressed in the nervous system (Westenbroek et al. 1995) produces the specific neuronal dysfunctions leading to migraine. Tottene et al. (2009) have recently shown enhanced glutamate release at cortical pyramidal cell synapses but unaltered GABA release at cortical fast-spiking interneuron synapses in R192Q KI mice, and suggested that the synapse-specific effect of FHM1 mutations could lead to disruption of the cortical excitation–inhibition balance and to the neuronal hyperactivity that might be the basis for episodic vulnerability to CSD ignition. According to unpublished evidence, a differential effect of the R192Q mutation on P/Q-type calcium channels of cortical pyramidal cells and fast-spiking inhibitory interneurons and a different duration of the action potential in the two types of cortical neurons (Inchauspe et al. 2010) both contribute to explain the synapse-specific effect of the mutation (D. Vecchia, A. Tottene and D. Pietrobon, unpublished observations).
In this study, we explored the possible implications for headache mechanisms of the differential effect of FHM1 mutations on CS and CI-T neurons by performing experiments in TG neurons retrogradely labelled from the dura. The finding that small adult WT mouse trigeminal ganglion neurons retrogradely labelled from the dura (C≤ 20 pF) did not include neurons with T-type calcium current suggests that small CI-T neurons are not dural afferents. Based on their sensitivity to capsaicin and the absence of LVA calcium current, a major fraction of small TG neurons retrogradely labelled from the dura can be classified as CS neurons. This is consistent within vivorecordings showing that topical application of capsaicin to the dura activates the majority of C-type mechanosensitive guinea pig dural fibres (Bove & Moskowitz, 1997), and that most C-type dural afferents show multiple slowly adapting firing in response to a mechanical stimulus (Levy & Strassmann, 2002).
Most small capsaicin-sensitive (CS) dural afferents are peptidergic neurons expressing CGRP. In fact, immunoreactivities for CGRP, substance P (SP) and the TRPV1 receptor in the dura are located predominantly in small sensory fibres and most TRPV1 receptor-immunoreactive fibres are also CGRP immunoreactive (Messlinger et al. 1993; Shimizu et al. 2007); likewise, most TG neurons retrogradely labelled from the rat dura expressing TRPV1 receptors also expressed CGRP (Shimizu et al. 2007). CGRP and SP immunoreactivities in the dura and around pial vessels were almost completely eliminated after intravenous capsaicin in guinea pig (Jansen et al. 1990). Several lines of evidence indicate that CGRP plays a pivotal role in migraine, but its mechanisms of action during a migraine attack remain elusive (Villalon & Olesen, 2009; Ho et al. 2010). The localization of CGRP receptors in the trigeminovascular system points to multiple possible mechanisms at both peripheral and central sites (Lennerz et al. 2008; Villalon & Olesen, 2009; Ho et al. 2010). CGRP-induced dural vasodilatation and CGRP-induced dural mast cell degranulation leading to release of proinflammatory mediators and development of neurogenic inflammation, with consequent initiation and maintenance of peripheral sensitization at the trigeminal terminals around meningeal blood vessels, are among the peripheral mechanisms of CGRP action that have been involved in generation of migraine pain (Levy, 2009; Villalon & Olesen, 2009).In vivo, topical application of capsaicin to the rat dura causes vasodilatation of dural blood vessels primarily mediated by CGRP released from the capsaicin-sensitive sensory fibres (Dux et al. 2003; see also Akerman et al. 2003b, 2004). Local mast cell degranulation in the rat dura produces prolonged excitation chiefly in mechanosensitive C units, most of which express CGRP (Levy et al. 2007), and some mast cell mediators increase CGRP release from capsaicin-sensitive dural afferents (Dux et al. 2009). Thus, CGRP release from small capsaicin-sensitive perivascular dural fibres probably plays a key role in the peripheral mechanisms of CGRP action that have been implicated in migraine pain.
P/Q-type calcium channels contribute to control of CGRP release from capsaicin-sensitive perivascular meningeal sensory fibres (Hong et al. 1999; Akerman et al. 2003a). Our finding that K+-evoked CGRP release from dura mater was unaltered in R192Q KI mice is consistent with and supports the conclusion that the FHM1 mutation does not affect the P/Q-type Ca2+ channels at the peripheral terminals of CGRP-expressing dural afferents, including the CS dural afferents. A specific implication of this finding for headache mechanisms is that the facilitation of peripheral mechanisms of CGRP action, such as dural vasodilatation and peripheral sensitization at the meninges, does not contribute to the generation of headache in FHM1. The recent evidence that a single CSD, induced by focal stimulation of rat visual cortex, can lead to a long-lasting increase in ongoing activity of dural afferents and of trigeminovascular neurons in the trigeminal nucleus caudalis supports the view that CSD can activate the trigeminovascular pathway underlying the headache phase (Zhang et al. 2010, 2011; see also Bolay et al. 2002). In this view, the fact that the FHM1 mutations facilitate CSD ignition (van den Maagdenberg et al. 2004, 2010; Eikerman-Haerter et al. 2009) may explain not only the aura phase but also the initiation of headache in FHM1.
There isin vivoandin vitroevidence for non-synaptic intraganglionic release of CGRP, substance P and ATP from TG neurons cell bodies in response to depolarizing stimuli (Neubert et al. 2000; Matsuka et al. 2001; Ulrich-Lai et al. 2001), and it has been suggested that intraganglionic excitatory coupling mediated by somatic release may contribute to peripheral sensitization during inflammation (Neubert et al. 2000). Both the larger action potential-evoked P/Q-type Ca2+ current (Fig. 4) and the longer duration of the action potential (Fig. 6) in CI-T neurons of R192Q KI compared to WT mice should lead to increased somatic intraganglionic release from mutant CI-T neurons. Recently, it has been shown that prolonged application of CGRP to TG neurons in culture leads to alterations in gene expression and/or membrane targeting of specific receptors in neurons, and to increased release of NO, cytochines (e.g. IL-1β) and other inflammatory mediators from satellite glial cells; these inflammatory mediators can sensitize TG neurons (leading to further CGRP release) and act back on glial cells, further activating them (Fabbretti et al. 2006; Zhang et al. 2007; Li et al. 2008; Capuano et al. 2009). This evidence for CGRP-dependent intraganglionic crosstalk led to the suggestion that prolonged elevations of CGRP within the ganglion may promote and maintain a neuron–glia inflammatory cycle that may contribute to persistent peripheral trigeminal sensitization (and hence, to allodynia and hyperalgesia in migraine) (Vilallon & Olesen, 2009; Ho et al. 2010).
In agreement with, and complementing recent data obtained in cultured TG neurons (Ceruti et al. 2011), we have shown that K+-evoked CGRP release from intact trigeminal ganglia is enhanced in R192Q KI compared to WT mice. Thus, in the ganglion there are CGRP-expressing neurons in which the FHM1 mutation produces gain-of-function of P/Q channels leading to enhanced somatic release. Whether these neurons are CI-T neurons and, if they are, the relevance for headache mechanisms of enhanced intraganglionic CGRP release from neurons that do not innervate the dura, remain to be established by future studies. In this context, it is interesting that NO has been reported to stimulate CGRP release from TG neurons in culture via a mechanism involving T-type calcium channels, with a sensitivity to Ni2+ and Cd2+ block similar to that of the slow LVA calcium current of CI-T neurons (Bellamy et al. 2006). A possible working hypothesis, that might be explored in future studies, is that the TG (CI-T?) neurons accounting for enhanced intraganglionic CGRP release in FHM1 KI mice are involved in neuron–glia crosstalk within the ganglion, and that the enhanced somatic release from these neurons facilitates peripheral sensitization at the ganglion level. Interestingly, some evidence suggesting facilitation of CGRP-mediated neuron to glia crosstalk following exposure to proinflammatory stimuli in TG neurons in culture from R192Q mice has been recently reported (Ceruti et al. 2011).
Conclusions
FHM1 mutations have neuron subtype-specific effects on CaV2.1 channel function. In particular, the analysis of the P/Q-type calcium current in two subtypes of small trigeminal ganglion neurons from R192Q KI mice shows that the FHM1 mutation produces gain-of-function of the CaV2.1 channels in CI-T neurons, but does not affect the CaV2.1 channels of capsaicin-sensitive CS neurons. Neurons retrogradely labelled from the dura do not include CI-T neurons; a major fraction can be classified as CS neurons. The FHM1 mutation does not affect depolarization-evoked CGRP release from the dura mater, but it increases CGRP release from the trigeminal ganglion. Our findings implicate that the facilitation of peripheral mechanisms of CGRP action, such as dural vasodilatation and nociceptor sensitization at the meninges, does not contribute to the generation of headache in FHM1.
Acknowledgments
This work was supported by the Telethon Italy grant GGP06234 (to D.P.), the Italian Ministry of University and Research PRIN2007 (to D.P.), the EU'EUROHEAD’ grant LSHM-CT-2004-504837 (to D.P.) and NH&MRC Project Grant 512159 (to M.C.) and grants from the Brain Foundation and Lincoln Centre (to M.C. and M.B.G.-D.). We thank Dania Vecchia for help with some of the figures.
Glossary
Abbreviations
- AHP
afterhyperpolarization
- AP
action potential
- CI-T
capsaicin-insensitive with T-type calcium current
- CS
capsaicin-sensitive
- CSD
cortical spreading depression
- FHM1
familial hemiplegic migraine type 1
- GVIA
ω-conotoxin-GVIA
- KI
knockin
- LVA
low-voltage activated
- MVIIC
ω-conotoxin-MVIIC
- RDP
rebound depolarization
- rheo
rheobase
- TG
trigeminal ganglion
- WT
wild-type
Author contributions
Conception and design of the experiments: D.P., F.F., M.C., A.v.d.M. Collection, analysis and interpretation of data: B.F., D.P., L.C., L.S., F.F., M.C., M.B.G.-D. Drafting the article: D.P. Revising the article critically: B.F., F.F., M.C., A.v.d.M., L.C., L.S., M.B.G.-D. All authors approved the final version of the manuscript. The experiments were performed at the University of Padova, the University of Perugia and the University of Sydney.
References
- Adams PJ, Garcia E, David LS, Mulatz KJ, Spacey SD, Snutch TP. CaV2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels (Austin) 2009;3:110–121. doi: 10.4161/chan.3.2.7932. [DOI] [PubMed] [Google Scholar]
- Akerman S, Kaube H, Goadsby PJ. Anandamide acts as a vasodilator of dural blood vesselsin vivoby activating TRPV1 receptors. Br J Pharmacol. 2004;142:1354–1360. doi: 10.1038/sj.bjp.0705896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Goadsby PJ. Voltage-dependent calcium channels are involved in neurogenic dural vasodilatation via a presynaptic transmitter release mechanism. Br J Pharmacol. 2003a;140:558–566. doi: 10.1038/sj.bjp.0705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Goadsby PJ. Vanilloid type 1 receptors (VR1) on trigeminal sensory nerve fibres play a minor role in neurogenic dural vasodilatation, and are involved in capsaicin-induced dural dilation. Br J Pharmacol. 2003b;140:718–724. doi: 10.1038/sj.bjp.0705486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C. Spreading depression: from serendipity to targeted therapy in migraine prophylaxis. Cephalalgia. 2009;29:1095–1114. doi: 10.1111/j.1468-2982.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006;23:2057–2066. doi: 10.1111/j.1460-9568.2006.04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Christie MJ. Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. J Physiol. 2001;536:35–47. doi: 10.1111/j.1469-7793.2001.t01-1-00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove GM, Moskowitz MA. Primary afferent neurons innervating guinea pig dura. J Neurophysiol. 1997;77:299–308. doi: 10.1152/jn.1997.77.1.299. [DOI] [PubMed] [Google Scholar]
- Capuano A, De Corato A, Lisi L, Tringali G, Navarra P, Dello Russo C. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain. 2009;5:43. doi: 10.1186/1744-8069-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catacuzzeno L, Fioretti B, Pietrobon D, Franciolini F. The differential expression of low-threshold K+ currents generates distinct firing patterns in different subtypes of adult mouse trigeminal ganglion neurones. J Physiol. 2008;586:5101–5118. doi: 10.1113/jphysiol.2008.159384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Villa G, Fumagalli M, Colombo L, Magni G, Zanardelli M, et al. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 knock-in mice: implications for basic mechanisms of migraine pain. J Neurosci. 2011;31:3638–3649. doi: 10.1523/JNEUROSCI.6440-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dux M, Rosta J, Sántha P, Jancsó G. Involvement of capsaicin-sensitive afferent nerves in the proteinase-activated receptor 2-mediated vasodilatation in the rat dura mater. Neuroscience. 2009;161:887–894. doi: 10.1016/j.neuroscience.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Dux M, Sántha P, Jancsó G. Capsaicin-sensitive neurogenic sensory vasodilatation in the dura mater of the rat. J Physiol. 2003;552:859–867. doi: 10.1113/jphysiol.2003.050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Hoffmann T, Sauer SK, Messlinger K, Reeh PW, Fischer MJM. Calcitonin gene-related peptide release from intact isolated dorsal root and trigeminal ganglia. Neuropeptides. 2008;42:311–317. doi: 10.1016/j.npep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Averbeck B, Messlinger K, Reeh PW. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience. 1999;89:901–907. doi: 10.1016/s0306-4522(98)00366-2. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119:99–109. doi: 10.1172/JCI36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti E, D'Arco M, Fabbro A, Simonetti M, Nistri A, Giniatullin R. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci. 2006;26:6163–6171. doi: 10.1523/JNEUROSCI.0647-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–1290. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- Grigaliunas A, Bradley RM, MacCallum DK, Mistretta CM. Distinctive neurophysiological properties of embryonic trigeminal and geniculate neurons in culture. J Neurophysiol. 2002;88:2058–2074. doi: 10.1152/jn.2002.88.4.2058. [DOI] [PubMed] [Google Scholar]
- Hamill O, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6:573–582. doi: 10.1038/nrneurol.2010.127. [DOI] [PubMed] [Google Scholar]
- Hong KW, Kim CD, Rhim BY, Lee WS. Effect ofw-conotoxin GVIA and w-agatoxin IVA on the capsaicin-sensitive calcitonin gene-related peptide release and autoregulatory vasodilation in rat pial arteries. J Cereb Blood Flow Metab. 1999;19:53–60. doi: 10.1097/00004647-199901000-00006. [DOI] [PubMed] [Google Scholar]
- Inchauspe CG, Urbano FJ, Di Guilmi MN, Forsythe ID, Ferrari MD, van den Maagdenberg AM, Uchitel OD. Gain of function in FHM-1 CaV2.1 knock-in mice is related to the shape of the action potential. J Neurophysiol. 2010;104:291–299. doi: 10.1152/jn.00034.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen I, Alafaci C, Uddman R, Edvinsson L. Evidence that calcitonin gene-related peptide contributes to the capsaicin-induced relaxation of guinea pig cerebral arteries. Regul Pept. 1990;31:167–78. doi: 10.1016/0167-0115(90)90003-f. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Lennerz JK, Ruhle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF, Messlinger K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol. 2008;507:1277–1299. doi: 10.1002/cne.21607. [DOI] [PubMed] [Google Scholar]
- Levy D. Migraine pain, meningeal inflammation, and mast cells. Curr Pain Headache Rep. 2009;13:237–240. doi: 10.1007/s11916-009-0040-y. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007;130:166–176. doi: 10.1016/j.pain.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Strassman AM. Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol. 2002;88:3021–3031. doi: 10.1152/jn.00029.2002. [DOI] [PubMed] [Google Scholar]
- Li J, Vause CV, Durham PL. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res. 2008;1196:22–32. doi: 10.1016/j.brainres.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuka Y, Neubert JK, Maidment NT, Spigelman I. Concurrent release of ATP and substance P within trigeminal ganglia in vivo. Brain Res. 2001;915:248–255. doi: 10.1016/s0006-8993(01)02888-8. [DOI] [PubMed] [Google Scholar]
- Messlinger K, Hanesch U, Baumgärtel M, Trost B, Schmidt RF. Innervation of the dura mater encephali of cat and rat: ultrastructure and calcitonin gene-related peptide-like and substance P-like immunoreactivity. Anat Embryol (Berl) 1993;188:219–237. doi: 10.1007/BF00188214. [DOI] [PubMed] [Google Scholar]
- Müllner C, Broos LA, van den Maagdenberg AM, Striessnig J. Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter Cav2.1 Ca2+ channel gating: evidence forb-subunit isoform-specific effects. J Biol Chem. 2004;279:51844–51850. doi: 10.1074/jbc.M408756200. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Neubert JK, Maidment NT, Matsuka Y, Adelson DW, Kruger L, Spigelman I. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal gangliain vivo. Brain Res. 2000;871:181–191. doi: 10.1016/s0006-8993(00)02440-9. [DOI] [PubMed] [Google Scholar]
- Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8:679–690. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr Opin Neurobiol. 2005;15:257–265. doi: 10.1016/j.conb.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Insights into migraine mechanisms and CaV2.1 calcium channel function from mouse models of familial hemiplegic migraine. J Physiol. 2010;588:1871–1878. doi: 10.1113/jphysiol.2010.188003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386–398. doi: 10.1038/nrn1102. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Toriumi H, Sato H, Shibata M, Nagata E, Gotoh K, Suzuki N. Distribution and origin of TRPV1 receptor-containing nerve fibers in the dura mater of rat. Brain Res. 2007;1173:84–91. doi: 10.1016/j.brainres.2007.07.068. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Levy D. Response properties of dural nociceptors in relation to headache. J Neurophysiol. 2006;95:1298–1306. doi: 10.1152/jn.01293.2005. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Cav2.1 knockin migraine mice. Neuron. 2009;61:762–773. doi: 10.1016/j.neuron.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A. 2002;99:13284–13289. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Flores CM, Harding-Rose CA, Goodis HE, Hargreaves KM. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: evidence for intraganglionic neurotransmission. Pain. 2001;91:219–226. doi: 10.1016/S0304-3959(00)00439-5. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/s0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Cav2.1 S218L mice. Ann Neurol. 2010;67:85–98. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- Villalón CM, Olesen J. The role of CGRP in the pathophysiology of migraine and efficacy of CGRP receptor antagonists as acute antimigraine drugs. Pharmacol Ther. 2009;124:309–323. doi: 10.1016/j.pharmthera.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TVB, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of thea1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Richter JA, Hurley JH. Release of glutamate and CGRP from trigeminal ganglion neurons: Role of calcium channels and 5-HT1 receptor signaling. Mol Pain. 2008;4:12. doi: 10.1186/1744-8069-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol. 2011;69:855–865. doi: 10.1002/ana.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci. 2010;30:8807–8814. doi: 10.1523/JNEUROSCI.0511-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Winborn CS, Marquez de Prado B, Russo AF. Sensitization of calcitonin gene-related peptide receptors by receptor activity-modifying protein-1 in the trigeminal ganglion. J Neurosci. 2007;27:2693–2703. doi: 10.1523/JNEUROSCI.4542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]