

Background: The Monod-Wyman-Changeux (MWC) mechanism is the preeminent conformational selection model for allosteric proteins.
Results: The novel allosteric ligand, BQCA, behaves according to a two-state MWC mechanism at the M1 muscarinic GPCR.
Conclusion: Chemical biological properties of GPCR allosteric ligands can be rationalized by the MWC model.
Significance: Application of our experimental framework to allosteric GPCR modulators can assist ligand classification and drug discovery.
Keywords: Allosteric Regulation, Chemical Biology, Drug Discovery, G Protein-coupled Receptors (GPCR), Pharmacology
Abstract
The Monod-Wyman-Changeux (MWC) model was initially proposed to describe the allosteric properties of regulatory enzymes and subsequently extended to receptors. Yet despite GPCRs representing the largest family of receptors and drug targets, no study has systematically evaluated the MWC mechanism as it applies to GPCR allosteric ligands. We reveal how the recently described allosteric modulator, benzyl quinolone carboxylic acid (BQCA), behaves according to a strict, two-state MWC mechanism at the M1 muscarinic acetylcholine receptor (mAChR). Despite having a low affinity for the M1 mAChR, BQCA demonstrated state dependence, exhibiting high positive cooperativity with orthosteric agonists in a manner that correlated with efficacy but negative cooperativity with inverse agonists. The activity of BQCA was significantly increased at a constitutively active M1 mAChR but abolished at an inactive mutant. Interestingly, BQCA possessed intrinsic signaling efficacy, ranging from near-quiescence to full agonism depending on the coupling efficiency of the chosen intracellular pathway. This latter cellular property also determined the difference in magnitude of positive cooperativity between BQCA and the orthosteric agonist, carbachol, across pathways. The lack of additional, pathway-biased, allosteric modulation by BQCA was confirmed in genetically engineered yeast strains expressing different chimeras between the endogenous yeast Gpa1 protein and human Gα subunits. These findings define a chemical biological framework that can be applied to the study and classification of allosteric modulators across different GPCR families.
Introduction
The concept of allosteric proteins was formalized nearly 50 years ago in the seminal MWC6 model, which proposed a conformational selection mechanism to account for ligand actions on regulatory enzymes (1). Since that time, the notion was extended to encompass behaviors across a broader range of protein families (2). The key statements of the MWC model are that allosteric proteins are oligomeric, they possess an axis of symmetry, they can exist in an equilibrium between (at least) two distinct states in the absence of ligand, and they possess multiple ligand recognition sites, binding to which stabilizes a subset of conformational states at the expense of others. These properties have substantial implications. For example, a large body of literature, supported by multiple high resolution crystal structures, now exists to indicate that numerous enzymes, ion-channels, and DNA-binding proteins possess an obligate oligomeric structure, suggesting that this is key for many proteins involved in signal transduction (2). In terms of chemical biology and pharmacology, the conformational selection mechanism that underpins the MWC, also referred to as a “two-state model” (3, 4), predicts that it should be possible to discover not only orthosteric, but allosteric, drugs that preferentially favor either active or inactive receptor states and can selectively modulate the properties of co-bound ligands in a manner that correlates with the efficacy (positive, negative, or neutral) of such ligands. Moreover, if the receptor can adopt more than two states, then it should be possible to identify compounds that bias signaling toward selected pathways in a ligand-specific manner (5).
It is axiomatic that such ligand behaviors can have a profound impact on allosteric drug discovery (6). Arguably, this is most evident in the field of GPCRs, which represent the largest superfamily of receptors in the genome and account for nearly 30% of all drug targets (5, 7). Indeed, GPCRs fulfill many of the criteria for allosteric proteins as proposed by the MWC model (5), but in contrast to other protein families, detailed mechanistic and structural studies of allostery at this important receptor family are currently lacking. Surprisingly and despite some theoretical treatises (e.g. Ref. 8), there have been no studies to systematically evaluate the properties of a GPCR allosteric modulator in terms of the key predictions of the MWC model. The rapid rate with which novel GPCR allosteric ligands are being discovered, however, necessitates a defined chemical biological framework within which it is possible to assess the mechanism of action of these ligands to facilitate classification, structure-activity studies, and eventual compound progression. In the absence of such a framework, mechanistic differences in the actions of novel allosteric drug candidates may be misconstrued with cellular and assay system-dependent variables.
The current study presents a comprehensive approach for the assessment of allosteric modulation of GPCRs in terms of the MWC model. As an exemplar system we have utilized the recently described M1 mAChR allosteric modulator, BQCA (Fig. 1), which exhibits both in vitro and in vivo efficacy as a potentiator of acetylcholine (ACh) signaling (9, 10). We propose that the interaction between BQCA and the M1 mAChR represents an unprecedented example of GPCR allosteric modulation that is entirely consistent with a strict, two-state MWC mechanism. It is envisaged that the approaches outlined herein can be broadly applicable in the evaluation of mechanism of action of various allosteric ligands of other GPCR families.
FIGURE 1.

BQCA shows hM1mAChR state dependence. A, CCh (filled circles) or BQCA (open circles) inhibit the equilibrium binding of the inverse agonist, [3H]NMS, in membranes of FlpIn CHO hM1 cells. Data points represent the mean ± S.E. of three independent experiments. The dashed line denotes curve fit assuming full inhibition of [3H]NMS binding by BQCA. B, BQCA potentiates CCh-mediated inhibition of the equilibrium binding of [3H]NMS. Data points represent the mean ± S.E. of four independent experiments performed in triplicate. Curves drawn through the points represent the best fit of an allosteric ternary complex model (Equation 1).
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium, Flp-In Chinese hamster ovary (CHO) cells, fluorescein di(β-d-galactopyranoside), Saccharomyces cerevisiae EasyComp transformation kit, and hygromycin B were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from ThermoTrace (Melbourne, VIC, Australia). N-[3H]Methylscopolamine ([3H]NMS) was purchased from GE Healthcare. [35S]GTPγS (1000 Ci/mmol), AlphaScreen reagents, and Ultima gold scintillation mixture were from PerkinElmer Life Sciences. Fluo-4-AM, Hoechst 33342, and AlexaTM 568-conjugated phalloidin were purchased from Molecular Probes (Carlsbad, CA). pcDNA3L-His-CAMYEL was purchased from ATCC, and polyethyleneimine was from Polysciences (Warrington, PA). All other reagents were purchased from Sigma. Benzyl quinolone carboxylic acid (BQCA, Fig. 1) was synthesized in-house.
Cell Culture and Transfections
FlpIn CHO cells stably expressing wild type (WT) or DREADD (Y106C/A196G) hM1 mAChR were generated and cultured as described previously (11). Transient transfections were performed using linear polyethyleneimine (molecular mass 25 kDa) (12).
Receptor Mutagenesis
Y106C/A196G and L116A hM1 mAChR mutants were generated by site-directed mutagenesis using the QuikChange kit (Agilent Technologies) and following the manufacturer's instructions.
Membrane Preparation and Radioligand Binding
Membrane preparation of CHO FlpIn hM1 cells was performed as described previously (11). Competition binding assays were performed in membranes derived from CHO FlpIn hM1 WT cells or Y106C/A196G hM1. 15 μg of membranes were incubated in binding buffer (20 mm HEPES, 100 mm NaCl, 10 mm MgCl2, pH 7.4) containing increasing concentrations of orthosteric ligands and/or BQCA for 3 h at 37 °C in the presence of a [3H]NMS concentration approximately equal to its equilibrium dissociation constant. Nonspecific binding was defined in the presence of 10 μm atropine. Termination of the assay and radioactivity measurements were performed as described in Ref. 11.
Yeast Transformations and Signaling Assay
S. cerevisiae strains expressing chimeras consisting of the five C-terminal amino acids of the relevant human Gα protein with Gpa1 (amino acids 1–467) have been previously described (13). The yeast strains were further transformed with a p416GPD vector containing the gene encoding the human M1 mAChR with an intracellular third loop deletion (hM1Δi3 mAChR) using the S. cerevisiae EasyComp transformation kit in accordance with manufacturer's instructions. The conditions for the signaling component of the assay have also been described (13). Briefly, single colonies were cultured overnight at 30 °C, and cells were pelleted and diluted to 0.02 A600 ml−1 in SC medium lacking amino acids for plasmid maintenance but supplemented with 0–15 mm 3-aminotriazole, 1 μm fluorescein di(β-d-galactopyranoside), and 0.1 m sodium phosphate, pH 7.3. Cell suspension was diluted into 96-well plates with appropriate ligand dilutions and incubated for 24 h at 30 °C. Fluorescence was measured in a FlexstationTM (Molecular Devices) plate reader using 485 excitation and 515 emission wavelengths.
Intracellular Calcium Mobilization
FlpIn CHO hM1 cells were cultured overnight in 96-well plates at 37 °C in 5% CO2. Cells were washed twice in Ca2+ assay buffer (150 mm NaCl, 2.6 mm KCl, 1.2 mm MgCl2, 10 mm dextrose, 10 mm HEPES, 2.2 mm CaCl2, 0.5% (w/v) BSA, and 4 mm probenecid), then replaced with assay buffer containing 1 μm Fluo-4-AM and incubated for 1 h at 37 °C in 5% CO2. Cells were washed twice more and replaced with warm assay buffer. The addition of the drugs and fluorescence measurements was performed in a FlexstationTM (Molecular Devices) using 485 excitation and 520 emission wavelengths. Peak fluorescence was measured as a marker for Ca2+ mobilization and used in further analyses. Co-addition of drugs was performed for allosteric interaction studies.
Bioluminescence Resonance Energy Transfer cAMP
FlpIn CHO hM1 cells were transfected with 2 μg of pcDNA3L-His-CAMYEL. 24 h post-transfection cells were plated in 96-well assay plates and grown overnight. The assay was started by adding 10 μl of the cell-permeant substrate specific for Renilla luciferase (Rluc), coelenterazine h, to the well to yield a final concentration of 5 μm. The agonist activity of the compounds was measured by adding those 5 min after the Rluc substrate. Co-addition of drugs was performed for allosteric interaction studies. Reads of the plates started 5 min after the addition of the agonists. Bioluminescence resonance energy transfer readings were collected using a LumiSTAR Omega instrument that allows the sequential integration of the signals detected in the 465–505- and 515–555-nm windows using filters with the appropriate band pass.
ERK1/2 Phosphorylation
FlpIn CHO hM1 cells were seeded into transparent 96-well plates at 25,000 cells per well and grown overnight. The cells were then washed twice with phosphate-buffered saline (PBS) and incubated in serum-free DMEM at 37 °C for at least 4 h. Cells were stimulated for 5 min and incubated at 37 °C in 5% CO2. For interaction studies, increasing concentrations of orthosteric ligand and BQCA were added simultaneously, and in all cases 10% (v/v) FBS was used as a positive control. The reaction was terminated by removal of media and drugs, and sample processing using the AlphaScreen SureFire p-ERK1/2 kit was performed following the manufacturer's instructions. The fluorescence signal was measured using a Fusion-α plate reader (PerkinElmer Life Sciences). Data were normalized to the maximal response elicited by 10% (v/v) FBS at the same time point.
[35S]GTPγS Binding Assay
Cell membranes (5 μg) were equilibrated for 1 h at 30 °C with varying concentrations of ligands in binding buffer (20 mm HEPES, 100 mm NaCl, 10 mm MgCl2, pH 7.4) containing 1 μm GDP. [35S]GTPγS (0.1 nm) was added to a final volume of 0.2 ml, and membranes were incubated for a further 30 min at 30 °C. Termination of [35S]GTPγS binding was by rapid filtration with a Packard plate harvester onto 96-well GF/C filter plates followed by 3 washes with ice-cold 0.9% NaCl. Bound radioactivity was measured in a TopCount microplate scintillation counter (Packard Instrument Co.).
Cytoskeletal Rearrangement Assay and Image Analysis
Membrane ruffling in FlpIn CHO hM1 cells was performed as described previously (13). Briefly, cells were cultured overnight in 96-well plates at 37 °C in 5% CO2. Samples were serum-starved 4 h before assaying then treated with ligand at indicated time points. Samples were fixed in 4% paraformaldehyde and permeabilized in 0.3% (v/v) Tween 20 in PBS. Samples were stained with 0.2 μg ml−1 Hoechst 33342 and 2 units ml−1 Alexa 568-phalloidin and imaged using an INCell analyzer 1000 (GE Healthcare). For the cytoskeletal analysis component, the images were randomized and blinded and analyzed manually to detect the number of cells that exhibited membrane ruffling. That number was subsequently normalized to the nuclei content per image, which were counted using INCell Developer software. Each concentration-response curve data point represents one image performed in duplicate over the number of times indicated in the figure legends. On average, ∼200 cells were present in each image.
Data Analysis
Competition binding curves between [3H]NMS and CCh in the absence or presence of BQCA were fitted to the following allosteric ternary complex model (14),
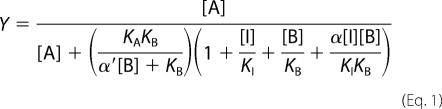 |
where Y is percentage (vehicle control) binding, [A], [B], and [I] are the concentrations of [3H]NMS, BQCA, and CCh respectively, KA and KBb are the equilibrium dissociation constants of [3H]NMS and BQCA, respectively, KB is the equilibrium dissociation constant of CCh, and α′ and α are the cooperativities between BQCA and [3H]NMS or CCh, respectively. Values of α (or α′) >1 denote positive cooperativity; values <1 (but >0) denote negative cooperativity, and values = 1 denote neutral cooperativity.
Functional data were analyzed using an operational model of allosterism and agonism (15) according to Equation 2,
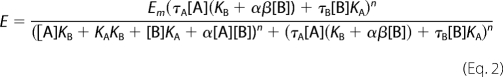 |
where Em is the maximum possible cellular response, and [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, KA and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, respectively, τA and τB are operational measures of orthosteric and allosteric ligand efficacy (which incorporate both signal efficiency and receptor density), respectively, α is the binding cooperativity parameter between the orthosteric and allosteric ligand, and β denotes the magnitude of the allosteric effect of the modulator on the efficacy of the orthosteric agonist. In all instances, the equilibrium dissociation constant of each agonist and BQCA was fixed to that determined from the binding assays. In the application of the operational model, it is assumed that the interconversion between active and inactive receptor states occurs freely and is at equilibrium, in which case the conformational selection (MWC) model is quantitatively equivalent to a conformational induction model in its treatment of efficacy parameters (16).
All affinity, potency, and cooperativity values were estimated as logarithms (17), and statistical comparisons between values were by Student's t test or one way analysis of variance using a Tukey's multiple comparison post-test, as appropriate. A value of p < 0.05 was considered statistically significant.
RESULTS
BQCA Displays Positive Cooperativity with Agonists but Negative Cooperativity with Inverse Agonist
The MWC model predicts that allosteric modulators should have differential affinities for receptor states that can manifest as opposing effects depending on whether the ligand is combined with an agonist or an inverse agonist (1, 5). However, initial reports of the actions of BQCA at the M1 mAChR suggested that the modulator had neutral cooperativity with the inverse agonist, [3H]NMS (9, 10). We hypothesized that this “neutral” cooperativity may reflect a negative cooperativity that was not detected in prior studies due to a hitherto unappreciated low binding affinity of BQCA. To confirm this, we performed [3H]NMS equilibrium binding studies in CHO FlpIn cells stably expressing the hM1 mAChR using concentrations of BQCA up to its solubility limit of 100 μm. As shown in Fig. 1A, the orthosteric agonist, CCh, competitively inhibited the binding of the inverse agonist with a pKI of 3.71 ± 0.03 (n = 3). Interestingly, 100 μm BQCA also inhibited [3H]NMS binding, revealing a low affinity, negative cooperativity. To accurately determine BQCA affinity, we performed interaction studies between CCh and BQCA against a fixed concentration of [3H]NMS. As expected, BQCA caused a substantial increase in the affinity of CCh to compete with [3H]NMS (Fig. 1B). Application of an allosteric ternary complex model to these data (Equation 1 and Ref. 18) yielded an estimate of the dissociation constant of BQCA for the allosteric site on the free M1 mAChR of 105 μm (pKB = 3.98; Table 1), thus explaining why prior studies utilizing radioligand binding may have missed the negative cooperativity with inverse agonists (9, 10). In contrast, the positive cooperativity with CCh was very high (α = 324; Table 1), indicating that the modulator has a marked preference for the active receptor state. Essentially identical results were obtained when these experiments were repeated using ACh instead of CCh (supplemental Fig. S1 and Table S1). These findings reveal that BQCA is one of the lowest affinity yet most positively cooperative (with agonists) allosteric modulators thus far identified for a GPCR.
TABLE 1.
Allosteric ternary complex model (Equation 1) binding parameters for the interaction between BQCA, CCh, and [3H]NMS at the hM1 mAChR
Estimated parameter values represent the mean ± S.E. of three experiments performed in duplicate.
a Negative logarithm of the equilibrium dissociation constant of BQCA.
b Negative logarithm of the equilibrium dissociation constant of CCh.
c Logarithm of the binding cooperativity factor between BQCA and CCh.
d Logarithm of the binding cooperativity factor between BQCA and [3H]NMS; this parameter was constrained to an arbitrarily low value, consistent with very high negative cooperativity between the modulator and radioligand.
The Functional Allosteric Modulation Mediated by BQCA Is Linked to Stimulus-Response Coupling Efficiency and Orthosteric Ligand Efficacy
A second prediction of the MWC model is that all allosteric ligands should display some agonism or inverse agonism depending on whether they stabilize active or inactive receptor states. The degree of modulation should thus vary with the magnitude of positive or negative efficacy promoted by the orthosteric and/or allosteric ligand (1, 5, 19). In practice, however, this is not always observed and may be because the bioassay used cannot detect these effects due to signal threshold considerations (5). We addressed this by investigating the impact of signal pathway or orthosteric ligand efficacy on modulator behavior. Using [35S]GTPγS binding as a surrogate assay for a proximal step in GPCR signal transduction, we found that BQCA robustly potentiated CCh-mediated G protein activation while having essentially no effect on basal M1 mAChR activity (Fig. 2A). We then determined the direct effects of BQCA at three other pathways linked to M1 mAChR activation: cAMP accumulation, ERK1/2 phosphorylation, and intracellular Ca2+ mobilization (Fig. 2B). The three highest concentrations of modulator mediated a small but significant effect above basal for cAMP accumulation (p < 0.05; two-way ANOVA with Bonferroni multiple comparisons test); this modest effect is unsurprising given that the pathway is not efficiently coupled to M1 mAChR activation (20). However, striking effects were observed for the more efficiently coupled ERK1/2 phosphorylation and Ca2+ mobilization pathways, where BQCA behaved as a full agonist. Our results thus suggest that the detection, or lack thereof, of direct agonism in the actions of BQCA is primarily a function of cellular stimulus-response coupling efficiency.
FIGURE 2.

BQCA exhibits direct agonism, modulation, and probe dependence in a pathway- and ligand-dependent manner. A, CCh-mediated [35S]GTPγS binding in the absence or presence of BQCA. Data represent the mean ± S.E. of three independent experiments. Curves drawn through the points are the best fit of an operational model of allosterism (Equation 2). B, direct BQCA-mediated signaling at hM1 mAChR-linked pathways is shown. Data represent the mean ± S.E. of four independent experiments. The asterisks indicate significantly different (p < 0.05) above basal response. C, shown are the effects of BQCA on cAMP accumulation by different orthosteric hM1 agonists. The ability of increasing concentrations of BQCA to modulate the response generated by an EC50 concentration of ACh, CCh, pilocarpine, or xanomeline was determined. Results are presented as the increase over the signal generated by the EC50 of each ligand in the absence of BQCA (ΔEC50) and represent the mean ± S.E. of four independent experiments. D, shown are the effects of orthosteric hM1 antagonists/inverse agonists on BQCA (EC80 concentration)-mediated intracellular Ca2+ mobilization. Data represent the mean ± S.E. of three independent experiments. QNB, 3-quinuclydinyl benzylate; 4-DAMP, 1,1-Dimethyl-4-diphenylacetoxypiper idinium iodide.
According to a two-state MWC mechanism, we also expected the modulation by BQCA to vary with the efficacy of the orthosteric ligand; this could represent a mechanistic basis for the phenomenon of “probe dependence” that is commonly used to refer to changes in the magnitude and direction of the allosteric effect depending on the orthosteric ligand used to probe receptor function (15). To confirm this we chose a weakly coupled pathway (cAMP accumulation) to ensure an appropriate detection window that can differentiate strong from weak positive agonists and a strongly coupled pathway (Ca2+ mobilization) to ensure a robust response to BQCA as an agonist when interacting it with inverse agonists. As shown in Fig. 2C, the combination of an EC50 concentration of orthosteric agonists with BQCA resulted in the greatest positive enhancement being noted with the full agonists, ACh and CCh, whereas a lower degree of potentiation was seen with the partial agonists, pilocarpine and xanomeline. The difference in the magnitude of maximal potentiation noted between the two partial agonists suggests that pilocarpine must possess higher intrinsic efficacy than xanomeline (although lower than ACh or CCh). Fig. 2D shows the results of the interaction between an EC80 concentration of BQCA and various antagonists/inverse agonists. In all instances complete inhibition of the BQCA response was noted, confirming the results of our binding assay that BQCA displays high negative cooperativity with ligands that prefer an inactive receptor state. The subsequent determination of detailed interaction experiments between NMS and CCh or BQCA indicated that the negative cooperativity between the modulator and NMS is essentially indistinguishable from a competitive interaction (supplemental Fig. S2).
Finally, we hypothesized that the degree of allosteric modulation should track with the degree of agonism/stimulus-response coupling efficiency observed at each pathway. Thus, we performed interaction studies between CCh and BQCA at each of the cAMP, ERK1/2, and Ca2+ mobilization assays. The results of these experiments are shown in Fig. 3, and a global analysis of the entire family of curves across the four pathways, according to an operational model of allosterism and agonism (15), is summarized in Table 2. From this analysis it can be seen that the cooperativity (quantified by the logαβ parameter in the operational model) is indeed positively associated with the degree of direct agonism displayed by BQCA (quantified by the logτB parameter); for both the direct agonistic effect of BQCA and the positive modulation of CCh signaling, the following rank order of pathway preferences was retained: ERK1/2 > Ca2+ ≫ cAMP > [35S]GTPγS.
FIGURE 3.

The degree of positive allosteric modulation by BQCA of CCh signaling varies with stimulus-response coupling efficiency. Shown is the interaction between BQCA and CCh in intracellular Ca2+ mobilization (A), cAMP accumulation (B), or ERK1/2 (C) phosphorylation. Data points represent the mean ± S.E. of three to five experiments. Curves drawn through the points are the best fit of an operational allosteric model (Equation 2; Table 2).
TABLE 2.
Operational model parameters for the functional allosteric interaction between CCh and BQCA at the hM1 mAChR
Parameter values represent the mean ± S.E. from three experiments performed in duplicate and analyzed according to Equation 2.
| Parameter | [35S]GTPγS | cAMP | ERK1/2 | Ca2+ |
|---|---|---|---|---|
| pKBa | 3.98 | |||
| Log αβb | 2.02 ± 0.05 (αβ = 105) | 3.60 ± 0.08 (αβ = 5400) | 4.06 ± 0.06 (αβ = 12000) | 4.26 ± 0.14 (αβ = 19000) |
| logτBc | 0.07 ± 0.10 | 0.71 ± 0.13 | 2.67 ± 0.02 | 3.69 ± 0.04 |
a Negative logarithm of the equilibrium dissociation constant of BQCA; value was fixed to that determined from radioligand binding assays at the wild type M1 mAChR (Table 1).
b Logarithm of the product of the binding cooperativity (α) and activation modulation (β) factors between CCh and BQCA. Antilogarithm shown in parentheses.
c Logarithm of the operational efficacy parameter of BQCA as an allosteric agonist.
Activating and Inactivating Mutations of M1 mAChR Have Opposite Effects on BQCA Activity
The mutation of L116A (L3.43A using the Ballesteros/Weinstein convention (21)) has previously been demonstrated to stabilize an active state of the rat M1 receptor (22); we thus generated the equivalent mutant at the human M1 mAChR. Although the mutation impairs cell surface expression, this can be rescued by 48 h of treatment (followed by extensive washout) with the inverse agonist, atropine (22). Fig. 4A illustrates the effect of this mutation on the binding of CCh or BQCA when tested against [3H]NMS. Strikingly, both the orthosteric agonist (pKI 3.62 ± 0.09 (WT) versus 6.13 ± 0.11 (L116A); n = 3) and allosteric modulator (pKI 3.50 ± 0.43 (WT) versus 5.88 ± 0.11 (L116A); n = 3) displayed significantly enhanced (p < 0.01) affinities for the mutant receptor. Importantly, this also facilitated the definitive demonstration that BQCA can completely inhibit [3H]NMS binding once the affinity of the modulator is substantially higher than solubility-imposed limits. Furthermore, BQCA displayed a significant increase (p < 0.01; n = 4) in its signaling efficacy at the L116A mutant M1 mAChR (Fig. 4B), as predicted.
FIGURE 4.

BQCA displays opposing sensitivities to activating and inactivating hM1 mAChR mutations. A, shown are the effects of CCh or BQCA on the equilibrium binding of [3H]NMS in cells transiently transfected with WT (filled symbols) or L116A (open symbols) hM1 mAChR. Data points represent the mean ± S.E. of four independent experiments. B, BQCA-induced cAMP accumulation in the WT, L116A (L116A) or the DREADD (Y106C/A196G) hM1 mAChRs is shown. Data represent the mean ± S.E. of three independent experiments.
For our choice of inactivating mutation, we and others have shown that the double mutation of the conserved residues, Y113C/A203G (Y3.33C/A5.46G; also referred to as a DREADD) at the M4 mAChR results in a mutant receptor for which ACh, CCh, and other prototypical orthosteric ligands lose both affinity and signaling efficacy (23, 24). The corresponding mutations in the hM1 subtype (Y106C/A196G) yielded a receptor with similar characteristics (supplemental Fig. S3). Unfortunately, [3H]NMS displayed reduced affinity at the DREADD such that direct binding against CCh or BQCA could not be determined. However, and in agreement with our hypothesis, the mutation abolished BQCA agonism (Fig. 4B). To ensure that this was not due to a global loss of functionality or expression of the DREADD, we confirmed that the mutant was robustly activated by clozapine N-oxide, an otherwise inert ligand that gains signaling efficacy specifically at DREADDs (supplemental Fig. S3 and Ref. 23).
BQCA Does Not Engender Signal Pathway-biased Agonism
The increasing identification of biased agonists, i.e. ligands displaying divergent efficacies at a common GPCR in a pathway-specific manner, indicates that GPCRs likely adopt multiple biologically relevant states (25, 26). Allosteric GPCR modulators should thus also promote pathway-biased allosteric modulation if they differentially stabilize such states (15). In contrast, if the allosteric modulation is adequately described by a two-state mechanism, then there should be no pathway-biased allosteric effects. We utilized different genetically engineered yeast (S. cerevisiae) strains, each expressing a specific human Gα/yeast Gpa1 protein chimera linked to a luciferase reporter gene readout as facile surrogates for G protein-biased GPCR signaling, a system previously validated for identifying biased agonism in the actions of GPCR ligands (13). We transformed each strain with a hM1Δi3 mAChR (that lacks most of intracellular loop 3 to allow better expression in yeast while retaining G protein coupling specificity) and then determined the response to CCh in the absence and presence of BQCA (Fig. 5A). An additional advantage of this system is a lack of stimulus-response amplification, allowing potencies obtained in this assay to be closely aligned with the affinity of the ligand for the receptor-G protein complex (27). Analysis of the data with our operational model revealed that the cooperativity between CCh and BQCA was not significantly different (p > 0.05) across the four G protein strains (Table 3), indicating a lack of pathway bias with respect to the action of BQCA; the low estimates for the τB values (0–6; Table 3) also indicated that any direct agonistic activity of the BQCA is unlikely to contribute substantially to the observed modulation. This absence of signaling bias is illustrated graphically (Fig. 5B) through the generation of “bias plots” (25, 28), which compare the normalized responses to equimolar concentrations of CCh between any two pathways. If the agonist has an equal preference (equivalent potency) for each pathway, then the bias plot would follow the line of identity; otherwise, the plot would skew toward the pathway for which the agonist has higher potency (e.g. Gαq). Furthermore, if the allosteric modulator imposes bias on this signaling preference, then the degree of modulation would also vary in a pathway-dependent manner, and the resulting bias plot for the agonist in the presence of modulator would diverge dramatically for that in its absence. As shown in Fig. 5B, however, the CCh bias plot in the presence of 10 μm BQCA closely follows the plot in its absence. We also determined the difference in potencies (ΔpEC50 values) of CCh across each of the yeast strains (comparing the most potent pathway, Gαq, to each of the others) in the absence or presence of 10 μm BQCA (Table 4), which confirmed that there was no significant difference in the pathway preferences in the absence or presence of modulator. An additional novel finding from the yeast studies was the prediction that BQCA can robustly potentiate CCh response via a G12-linked pathway. To validate this prediction, we determined the effect of a single concentration (10 μm) of BQCA on CCh-mediated membrane ruffling in our hM1 mAChR CHO FlpIn cells as a surrogate of Gα12/13 protein coupling (13, 29). Supplemental Fig. S4 shows that BQCA promoted a significant (p < 0.05) potentiation of CCh-mediated membrane ruffling without exhibiting any allosteric agonism, as noted in the yeast assay (pEC50 = 6.74 ± 0.21 versus 7.64 ± 0.18; n = 4).
FIGURE 5.

BQCA does not engender signal pathway-bias at the hM1 mAChR. A, shown is the interaction between CCh and BQCA in S. cerevisiae strains expressing hM1Δi3 and a Gα/Gpa1 protein chimera corresponding to Gαq, Gαs, Gαi1/2, or Gα12. Curves drawn through the data points represent the best fit of an operational model of allosterism (Equation 2). Data points represent the mean ± S.E. of three independent experiments. B, bias plots show the responses to equimolar concentrations of CCh between the various pathways in the absence or presence of 10 μm BQCA.
TABLE 3.
Operational model parameters for the functional allosteric interaction between CCh and BQCA at the hM1 mAChR in S. cerevisiae
Parameter values represent the mean ± S.E. from three experiments performed in duplicate and analyzed according to Equation 2. Values of log αβ were compared using one-way variance with a Tukey's multiple comparison post test; no significant difference (p > 0.05) was determined.
| Parameter | Gq | Gi1/2 | Gs | G12 |
|---|---|---|---|---|
| pKBa | 3.97 | |||
| logαβb | 2.62 ± 0.15 (αβ = 417) | 2.57 ± 0.03 (αβ = 371) | 2.59 ± 0.10 (αβ = 389) | 2.74 ± 0.07 (αβ = 550) |
| logτBc | 0.78 ± 0.02 | 0.23 ± 0.04 | −100 | −100 |
a Negative logarithm of the equilibrium dissociation constant of BQCA; value was fixed to that determined from radioligand binding assays at the wild type M1 mAChR (Table 1).
b Logarithm of the product of the binding cooperativity (α) and activation modulation (β) factors between CCh and BQCA. Antilogarithm is shown in parentheses.
c Logarithm of the operational efficacy parameter of BQCA as an allosteric agonist. Estimates for the Gs and G12 strains were constrained at an arbitrarily low value due to lack of discernible signaling efficacy.
TABLE 4.
Comparison of CCh potency differences (ΔpEC50) across different yeast strains in the absence or presence of 10 μm BQCA
Values were compared using Student's t test; no significant difference (p > 0.05) was determined.
| Pathways compared | ΔpEC50 (control) | ΔpEC50 (+10 μm BQCA) |
|---|---|---|
| Gαq − Gαi1/2 | 0.69 ± 0.10 | 0.73 ± 0.16 |
| Gαq − Gαs | 1.84 ± 0.09 | 1.63 ± 0.12 |
| Gαq − Gα12 | 1.83 ± 0.08 | 1.61 ± 0.13 |
DISCUSSION
Our study has evaluated BQCA at the M1 mAChR in terms of the key predictions of a two-state MWC model for allosteric proteins. We propose a chemical biological framework with the minimal expectations that all allosteric ligands should be GPCR state-selective, possess the potential for direct agonism (or inverse agonism), and modulate the actions of orthosteric ligands in a manner that correlates with signaling efficacy and stimulus-response coupling. The work may serve as a guide for evaluating novel allosteric compounds, and deviations from these expected behaviors can provide presumptive evidence for the existence of more complex (e.g. multi-state) or mixed mode (e.g. orthosteric/allosteric) mechanisms of action (see below).
The increasing identification of GPCR allosteric modulators has resulted in numerous behaviors ascribed to such ligands (19). This poses a challenge for modulator classification, rational medicinal chemistry, and drug candidate optimization if mechanisms of modulator action at the level of the target GPCR are not divorced from host-system influences. There are at least four characteristics that may be associated with phenotypic behavior of allosteric modulators; (a) saturability to their effect, (b) probe dependence, (c) potential for different effects on orthosteric ligand affinity versus signaling efficacy, and (d) pathway-biased agonism. The first behavior is linked to the cooperativity between orthosteric and allosteric ligands (19, 30), which itself should be linked to the efficacy of the interacting ligands according to MWC (1). Similarly, the most parsimonious mechanism to account for probe dependence is that the degree of modulation will be greater depending on the degree of efficacy of the orthosteric ligand. As noted, the interaction between BQCA and either positive or inverse agonists satisfied both of these predictions and has implications for the interpretation of prior studies. For instance, if BQCA were neutrally cooperative with NMS (9, 10), then this would imply equal affinities for both active and inactive receptor states, which is not the case. Rather, any apparent neutral cooperativity reflects a lack of appreciable allosteric-site occupancy due to the very low affinity of BQCA. This is not an issue when BQCA is examined in the presence of agonists, due to positive cooperativity (Tables 1 and 2), or at an M1 mAChR harboring an activating mutation, due to increased BQCA affinity (Fig. 4). Furthermore, the dependence of the cooperative effect on the intrinsic efficacy of the orthosteric ligand raises important considerations for potential combination therapy involving BQCA and agonists such as xanomeline, which on its own can improve cognitive deficits in schizophrenia (31). Although this may be deemed desirable, the modest potentiation noted in our study for this particular combination suggests that any benefits gained are likely to be minor, at least at the level of the M1 mAChR as the target. It is also very common to see mAChR antagonists, such as scopolamine, used to promote cognitive deficits in a variety of animal models (32). Our finding of high negative cooperativity between BQCA and structurally related antagonists now brings this approach into question if the aims of such experiments are to differentiate mAChR orthosteric agonism from allosteric modulation as the mechanism underlying putative in vivo efficacy.
The extension of our studies to multiple signaling pathways provided additional insights into other phenotypic behaviors of GPCR modulators. The first relates to the dependence of the agonism exhibited by BQCA on the strength of pathway stimulus-response coupling (Fig. 2). This is something we have noted previously with allosteric ligands at the M4 mAChR and adenosine A1 receptor (18, 24, 33), indicating that the phenomenon is likely to be widespread. Importantly, it also suggests that “pure” positive or negative allosteric modulators, which are typically sought after in most allosteric drug discovery programs, are unlikely to exist. Rather, the absence or presence of an (allosteric) agonistic effect is probably determined by cellular stimulus-response coupling and/or receptor density. This is not to say that the therapeutic effect of an allosteric drug will not primarily reflect its ability to modulate the endogenous orthosteric ligand, but it remains important to ascertain the properties of any candidate allosteric molecules across a range of signaling pathways and/or expression levels given that a common aim in pursuing modulators (rather than agonists) is the potential to maintain spatial and temporal characteristics of endogenous physiological signaling (30).
A second key issue relates to the concept of pathway-biased modulation. It is now clear that GPCRs can adopt multiple biologically active states that are differentially selected by orthosteric ligands (25, 34), and this has been extended to allosteric modulators (15). However, we now highlight a cellular mechanism that can also account for such observations, namely, that the degree of modulation will depend on coupling efficiency, such that amplified responses will appear to be modulated to a greater extent than less amplified responses. In this regard, the use of the genetically modified yeast platform may prove particularly useful in delineating the potential for G protein-biased allosteric modulation. The application of the operational model also provides an analytical method for assessing whether the degree of allosteric modulation tracks with the degree of direct agonism (Tables 2 and 3). Only if this criterion is addressed (and excluded) can one more confidently ascribe a multistate (i.e. >2) mechanism to the molecular actions of putative allosteric ligands.
Our findings also raise fundamental questions about BQCA itself. To our knowledge this compound represents the most striking example of a GPCR allosteric ligand displaying pharmacological behavior strictly consistent with a two-state MWC mechanism. Other well characterized allosteric modulators of the mAChRs, such as gallamine, alcuronium, and C7/3-phth are different in that they inhibit the actions of both orthosteric agonists and antagonists (35–37). One obvious explanation is that more than two GPCR conformational states are required to account for the behavior of such ligands. However, it is also possible that deviations from a two-state MWC mechanism actually reflect mixed modes of allosteric modulation and steric inhibition in the actions of such ligands; there is kinetic evidence to support both allosteric and direct steric hindrance in the actions of numerous mAChR modulators (38–40). These issues may be resolved once a high resolution structure of a GPCR in complex with an allosteric and orthosteric ligand is solved.
Finally, it was of interest that the affinity of BQCA for the M1 mAChR was found to be very low, yet the positive cooperativity exhibited with agonists was remarkably high. Such low (micromolar to millimolar) affinity is commonly seen in fragment-based drug discovery, and thus an additional implication of our results is that it may be possible to identify novel allosteric modulators among fragment libraries using traditional cell-based assays rather than the current paradigm that relies on biophysical and structural approaches (41). Although speculative, it may also be that the “fragment-like” nature of BQCA imposes an inherent limit to the number of receptor conformations that such a simple molecule can sample, thus providing an additional mechanism to account for its two-state behavior. Irrespective, it is envisaged that the approaches described herein can be used more broadly to explain the pharmacological properties of various classes of GPCR allosteric modulators as well as further validating the applicability of the MWC model to this important receptor family.
Supplementary Material
Acknowledgments
We thank TGR BioSciences for generously providing the SureFire ERK1/2 kits, Dr. Jürgen Wess (NIDDK, National Institutes of Health) for the p416GPD hM1Δi3 mAChR DNA, and Dr. Simon Dowell, GlaxoSmithKline, United Kingdom, for the yeast strains.
This work was funded in part by National Health and Medical Research Council Program Grants 519461 (to A. C. and P. M. S.), APP1011796 (to M. C.), and APP1011920 (to J. R. L.) by and Australian Research Council Discovery Grant DP110100687 (A. C.).

This article contains supplemental Table S1 and Figs. 1–4.
- MWC
- Monod-Wyman-Changeux
- Ach
- acetylcholine
- mAChR
- M1 muscarinic acetylcholine receptor
- hM1
- human M1
- ([3H]NMS
- N-[3H]methylscopolamine
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- BQCA
- benzyl quinolone carboxylic acid
- CCh
- carbachol.
REFERENCES
- 1. Monod J., Wyman J., Changeux J. P. (1965) J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 2. Changeux J. P., Edelstein S. J. (2005) Science 308, 1424–1428 [DOI] [PubMed] [Google Scholar]
- 3. Karlin A. (1967) J. Theor. Biol. 16, 306–320 [DOI] [PubMed] [Google Scholar]
- 4. Leff P. (1995) Trends Pharmacol. Sci. 16, 89–97 [DOI] [PubMed] [Google Scholar]
- 5. Canals M., Sexton P. M., Christopoulos A. (2011) Trends Biochem. Sci. 36, 663–672 [DOI] [PubMed] [Google Scholar]
- 6. Christopoulos A. (2002) Nat. Rev. Drug Discov. 1, 198–210 [DOI] [PubMed] [Google Scholar]
- 7. Conn P. J., Christopoulos A., Lindsley C. W. (2009) Nat. Rev. Drug Discov. 8, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hall D. A. (2000) Mol. Pharmacol. 58, 1412–1423 [DOI] [PubMed] [Google Scholar]
- 9. Ma L., Seager M. A., Seager M., Wittmann M., Jacobson M., Bickel D., Burno M., Jones K., Graufelds V. K., Xu G., Pearson M., McCampbell A., Gaspar R., Shughrue P., Danziger A., Regan C., Flick R., Pascarella D., Garson S., Doran S., Kreatsoulas C., Veng L., Lindsley C. W., Shipe W., Kuduk S., Sur C., Kinney G., Seabrook G. R., Ray W. J. (2009) Proc. Natl. Acad. Sci. U. S. A. 106, 15950–15955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shirey J. K., Brady A. E., Jones P. J., Davis A. A., Bridges T. M., Kennedy J. P., Jadhav S. B., Menon U. N., Xiang Z., Watson M. L., Christian E. P., Doherty J. J., Quirk M. C., Snyder D. H., Lah J. J., Levey A. I., Nicolle M. M., Lindsley C. W., Conn P. J. (2009) J. Neurosci. 29, 14271–14286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Avlani V. A., Langmead C. J., Guida E., Wood M. D., Tehan B. G., Herdon H. J., Watson J. M., Sexton P. M., Christopoulos A. (2010) Mol. Pharmacol. 78, 94–104 [DOI] [PubMed] [Google Scholar]
- 12. Scholten D., Canals M., Wijtmans M., de Munnik S., Nguyen P., Verzijl D., de Esch I., Vischer H., Smit M., Leurs R. (2011) Br. J. Pharmacol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart G. D., Sexton P. M., Christopoulos A. (2010) ACS Chem. Biol. 5, 365–375 [DOI] [PubMed] [Google Scholar]
- 14. Ehlert F. J. (1988) Mol. Pharmacol. 33, 187–194 [PubMed] [Google Scholar]
- 15. Leach K., Sexton P. M., Christopoulos A. (2007) Trends Pharmacol. Sci. 28, 382–389 [DOI] [PubMed] [Google Scholar]
- 16. Giraldo J. (2004) FEBS Lett. 556, 13–18 [DOI] [PubMed] [Google Scholar]
- 17. Christopoulos A. (1998) Trends Pharmacol. Sci. 19, 351–357 [DOI] [PubMed] [Google Scholar]
- 18. Leach K., Loiacono R. E., Felder C. C., McKinzie D. L., Mogg A., Shaw D. B., Sexton P. M., Christopoulos A. (2010) Neuropsychopharmacology 35, 855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keov P., Sexton P. M., Christopoulos A. (2011) Neuropharmacology 60, 24–35 [DOI] [PubMed] [Google Scholar]
- 20. Thomas R. L., Mistry R., Langmead C. J., Wood M. D., Challiss R. A. J. (2008) J. Pharmacol. Exp. Ther. 327, 365–374 [DOI] [PubMed] [Google Scholar]
- 21. Ballesteros J. A., Weinstein H. (1995) Methods Neurosci. 25, 366–428 [Google Scholar]
- 22. Lu Z. L., Hulme E. C. (1999) J. Biol. Chem. 274, 7309–7315 [DOI] [PubMed] [Google Scholar]
- 23. Armbruster B. N., Li X., Pausch M. H., Herlitze S., Roth B. L. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5163–5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nawaratne V., Leach K., Suratman N., Loiacono R. E., Felder C. C., Armbruster B. N., Roth B. L., Sexton P. M., Christopoulos A. (2008) Mol. Pharmacol. 74, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 25. Stallaert W., Christopoulos A., Bouvier M. (2011) Expert Opin. Drug Discov. 6, 811–825 [DOI] [PubMed] [Google Scholar]
- 26. Whalen E. J., Rajagopal S., Lefkowitz R. J. (2011) Trends Mol. Med. 17, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu R. C., Pesce C. G., Colman-Lerner A., Lok L., Pincus D., Serra E., Holl M., Benjamin K., Gordon A., Brent R. (2008) Nature 456, 755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gregory K. J., Hall N. E., Tobin A. B., Sexton P. M., Christopoulos A. (2010) J. Biol. Chem. 285, 7459–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pertz O., Hodgson L., Klemke R. L., Hahn K. M. (2006) Nature 440, 1069–1072 [DOI] [PubMed] [Google Scholar]
- 30. May L. T., Leach K., Sexton P. M., Christopoulos A. (2007) Annu. Rev. Pharmacol. Toxicol. 47, 1–51 [DOI] [PubMed] [Google Scholar]
- 31. Shekhar A., Potter W. Z., Lightfoot J., Lienemann J., Dubé S., Mallinckrodt C., Bymaster F. P., McKinzie D. L., Felder C. C. (2008) Am. J. Psychiatry 165, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 32. Klinkenberg I., Blokland A. (2010) Neurosci. Biobehav. Rev. 34, 1307–1350 [DOI] [PubMed] [Google Scholar]
- 33. Valant C., Aurelio L., Urmaliya V. B., White P., Scammells P. J., Sexton P. M., Christopoulos A. (2010) Mol. Pharmacol. 78, 444–455 [DOI] [PubMed] [Google Scholar]
- 34. Wisler J. W., DeWire S. M., Whalen E. J., Violin J. D., Drake M. T., Ahn S., Shenoy S. K., Lefkowitz R. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stockton J. M., Birdsall N. J., Burgen A. S., Hulme E. C. (1983) Mol. Pharmacol. 23, 551–557 [PubMed] [Google Scholar]
- 36. Tucek S., Musílková J., Nedoma J., Proska J., Shelkovnikov S., Vorlícek J. (1990) Mol. Pharmacol. 38, 674–680 [PubMed] [Google Scholar]
- 37. Lanzafame A., Christopoulos A., Mitchelson F. (1996) Eur. J. Pharmacol. 316, 27–32 [DOI] [PubMed] [Google Scholar]
- 38. Proska J., Tucek S. (1994) Mol. Pharmacol. 45, 709–717 [PubMed] [Google Scholar]
- 39. Avlani V. A., Gregory K. J., Morton C. J., Parker M. W., Sexton P. M., Christopoulos A. (2007) J. Biol. Chem. 282, 25677–25686 [DOI] [PubMed] [Google Scholar]
- 40. Avlani V., May L. T., Sexton P. M., Christopoulos A. (2004) J. Pharmacol. Exp. Ther. 308, 1062–1072 [DOI] [PubMed] [Google Scholar]
- 41. Murray C. W., Rees D. C. (2009) Nat. Chem. 1, 187–192 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.