

Background: Sulfiredoxin catalyzes the reactivation of hyperoxidized peroxiredoxins, which are peroxidases.
Results: Under low steady-state levels of H2O2 ([H2O2]ss), loss of function of sulfiredoxin leads to redox imbalance and sensitizes cells to apoptosis.
Conclusion: Sulfiredoxin functions as a critical antioxidant for redox balance and survival of cells exposed to low [H2O2]ss.
Significance: Sulfiredoxin may serve as a cell survival factor even in the presence of low [H2O2]ss.
Keywords: Peroxiredoxin, Reactive Oxygen Species (ROS), Redox, Redox Regulation, Redox Signaling, Hydrogen Peroxide, Sulfiredoxin
Abstract
Sulfiredoxin (Srx) is an enzyme that catalyzes the reduction of cysteine sulfinic acid of hyperoxidized peroxiredoxins (Prxs). Having high affinity toward H2O2, 2-Cys Prxs can efficiently reduce H2O2 at low concentration. We previously showed that Prx I is hyperoxidized at a rate of 0.072% per turnover even in the presence of low steady-state levels of H2O2. Here we examine the novel role of Srx in cells exposed to low steady-state levels of H2O2, which can be achieved by using glucose oxidase. Exposure of low steady-state levels of H2O2 (10–20 μm) to A549 or wild-type mouse embryonic fibroblast (MEF) cells does not lead to any significant change in oxidative injury because of the maintenance of balance between H2O2 production and elimination. In contrast, loss-of-function studies using Srx-depleted A549 and Srx−/− MEF cells demonstrate a dramatic increase in extra- and intracellular H2O2, sulfinic 2-Cys Prxs, and apoptosis. Concomitant with hyperoxidation of mitochondrial Prx III, Srx-depleted cells show an activation of mitochondria-mediated apoptotic pathways including mitochondria membrane potential collapse, cytochrome c release, and caspase activation. Furthermore, adenoviral re-expression of Srx in Srx-depleted A549 or Srx−/− MEF cells promotes the reactivation of sulfinic 2-Cys Prxs and results in cellular resistance to apoptosis, with enhanced removal of H2O2. These results indicate that Srx functions as a novel component to maintain the balance between H2O2 production and elimination and then protects cells from apoptosis even in the presence of low steady-state levels of H2O2.
Introduction
Reactive oxygen species (ROS)2 are highly reactive molecules that are generated by various sources, including mitochondria, peroxisomes, and NADPH oxidase as endogenous sources and ultraviolet light, ionizing radiation, and chemotherapeutics as exogenous sources (1). ROS are potent inducers of oxidative damage that consequently cause cell and tissue dysfunction. An imbalanced ratio between ROS production and detoxification may induce cell death such as apoptosis or necrosis, ultimately increasing the risk of disease (2).
Mammalian cells have H2O2-eliminating enzymes such as catalase, glutathione peroxidase, and peroxiredoxin (Prx). Prxs play an important role in peroxide detoxification and regulation of redox-dependent signaling pathways (3–5). All Prx enzymes contain a conserved cysteine residue at the NH2-terminal region, which is the primary site of oxidation by H2O2. The mammalian Prxs exist in six isoforms, which can be divided into three subgroups, namely, 2-Cys, atypical 2-Cys, and 1-Cys subgroups (6). Prx isoforms are differently distributed in most cellular compartments, including cytosol, mitochondria, nucleus, endoplasmic reticulum, and peroxisomes. In the normal catalytic cycle, catalytic cysteine of 2-Cys Prxs (Prx I–IV) is oxidized to sulfenic acid (Cys-SOH), and then 2-Cys Prxs are converted back to a reduced active state by thioredoxin. In the eukaryotic system, increasing H2O2 flux leads to overoxidation of the peroxidatic cysteine of several Prxs to sulfinic acid (Cys-SO2H), resulting in loss of peroxidase activity (7, 8). This oxidation had been thought to be irreversible, but it was demonstrated that the sulfinic form of Prx is rapidly reduced to the catalytically active thiol form in mammalian cells (9). Sulfiredoxin (Srx) was subsequently identified as an enzyme responsible for the regeneration of hyperoxidized Prx in mammals as well as yeast (10, 11). This reduction of sulfinic acid by Srx is specific to 2-Cys Prxs (12).
Because the distance between two sulfur atoms in the two disulfide-forming cysteine residues of 2-Cys Prxs is ∼13 Å (13), the formation of an intermolecular disulfide between these residues is a slow process, and the catalytic cysteine residue of 2-Cys Prxs is occasionally overoxidized to sulfinic acid. Indeed, our kinetic analysis has shown that Prx I is hyperoxidized at a rate of 0.072% per turnover at 30 °C in the presence of low steady-state levels of H2O2 (7). However, Prxs are unusually efficient for removing H2O2 at low concentrations because the hydrogen-bonding network created by several amino acid residues at their active site not only provides a binding pocket site for H2O2 but also activates the bound peroxide for the reaction with the active site thiol group (14). We then assumed that Srx in cells exposed to low steady-state levels of H2O2 may play an important role in maintaining the H2O2-eliminating ability via the regeneration of the peroxidase activity of 2-Cys Prxs.
Here we demonstrate that exposure of low steady-state levels of H2O2 (10–20 μm) to A549 or wild-type mouse embryonic fibroblast (MEF) cells did not lead to any significant change in oxidative stress-induced damage, whereas Srx-depleted A549 or Srx-null (Srx−/−) MEF cells showed accumulation of H2O2 in the medium, increase in cellular ROS level and hyperoxidation of 2-Cys Prxs, and activation of apoptotic cell death pathways including mitochondria membrane potential (ΔΨm) disruption, cytochrome c release, and activation of caspase-9 and -3. Furthermore, ectopic re-expression of Srx in Srx-depleted A549 or Srx−/− MEF cells was sufficient to restore the ability to resist oxidative damage induced by low steady-state levels of H2O2, suggesting that Srx is a novel component to maintain the redox balance in cells exposed to low steady-state levels of H2O2.
EXPERIMENTAL PROCEDURES
Materials
Glucose oxidase (GOX), perchloric acid, ammonium iron(II) sulfate hexahydrate, puromycin, trichloroacetic acid, and xylenol orange were from Sigma; acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC) and acetyl-Leu-Glu-His-Asp-7-amino-4-trifluoromethyl coumarin (Ac-LEHD-AFC) were from BIOMOL; (5-(and 6-)chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), 10-N-nonylacridine orange (NAO), propidium iodide, and tetramethylrhodamine ethyl ester (TMRE) were from Molecular Probes; and pSUPERIOR-puro vector was from Oligoengine. Monoclonal antibodies to cytochrome c, β-actin, and α-tubulin were from BD Pharmingen, Abcam, and Santa Cruz Biotechnology, respectively. Rabbit polyclonal antibodies to human Prx I, Prx III (15), Prx-SO2 (9), and Srx (16) were described previously.
Generation of Srx Knock-out Mice
To generate conditional Srx knock-out mice, we constructed a targeting vector that is flanked by exon 2 of Srx gene with the loxP site and the PGK-neo selection cassette that is flanked by the loxP site is inserted downstream of exon 2 of Srx gene (17). The existence of ES cells with floxed Srx-targeted locus was confirmed by Southern blot analysis of ScaI-digested tail genomic DNA. Systemic Srx knock-out mice were achieved by crossing the floxed Srx mice with an EIIa-Cre transgenic line (The Jackson Laboratory, Bar Harbor, ME). Srxflox/flox:EIIa-Cre mice were designated as Srx−/− mice. To confirm deleted Srx allele, HindIII-digested tail genomic DNA was subjected to Southern blot analysis. All animal experiments were approved by the Animal Care and Use Committee of Ewha Womans University.
MEF Cells from Srx+/+ and Srx−/− Mice
MEF cells were prepared at embryonic day 13.5 from embryos obtained by mating the Srx+/− mice (17) and were maintained in Dulbecco's minimum essential medium supplemented with 10% fetal bovine serum and penicillin-streptomycin.
Cell Culture and Treatment
A549 (human lung carcinoma) cells were maintained in Ham's F-12K medium and supplemented with 10% fetal bovine serum and penicillin-streptomycin. A549 cells (6 ×105) were exposed for the indicated times to GOX.
Generation and Maintenance of A549 Cells Expressing Small Hairpin RNA Targeting Srx
To construct a pSUPER-siSrx for expression of small hairpin RNA targeting human Srx, oligonucleotides containing the small interfering RNA sequences targeting Srx (5′-GGAGGUGACUACUUCUACU-3′) (16) were purchased from Genotec (Daejeon, Korea), annealed, and cloned into the pSUPERIOR-puro (pSUPER) vector (Oligoengine). A549 cells were transfected with pSUPER-siSrx vectors using the Nucleofector instrument (Lonza Cologne GmbH, Cologne, Germany). pSUPER empty vector was used as the negative control vector. Following selection with 1.5 μg/ml puromycin, single clones were expanded and characterized for knockdown of Srx protein expression.
Flow Cytometry
A FACSCalibur flow cytometer (BD Biosciences) was used for analyses, with a minimum of 6 × 105 cells per sample for each measurement. The excitation wavelength was 488 nm, and the observation wavelength was 530 nm for green fluorescence and 585 nm for red fluorescence. Relative change in fluorescence was analyzed with the WinMDI software. For analysis of apoptosis, cells were labeled with annexin V and propidium iodide using an Annexin-V-FLUOS staining kit (Roche Applied Science, Basel, Switzerland) according to the manufacturer's protocol or stained with propidium iodide (25 μg/ml), and the percentage of hypodiploid (apoptotic) cells was determined. For evaluation of cardiolipin peroxidation in mitochondria, cells were labeled with 5 μm NAO for 30 min. For evaluation of changes in the ΔΨm, cells were incubated with TMRE for 20 min at 37 °C.
Measurement of ROS
For measurement of intracellular ROS, detached cells were loaded with 5 μm CM-H2DCFDA for 30 min at 37 °C, washed, and then analyzed immediately by flow cytometry. For measurement of H2O2, the amount of H2O2 in medium was determined by the ferrous xylenol orange in perchloric acid (PCA-FOX) assay (18). Portions of medium (0.2 ml) were collected, mixed with 0.05 ml of 30% trichloroacetic acid, and then centrifuged at 13,000 × g for 10 min. The resulting supernatant (0.1 ml) was mixed with 0.9 ml of PCA-FOX solution (0.25 mm xylenol orange and 0.25 mm ferrous ammonium sulfate in 0.11 m perchloric acid) and incubated for 1 h at room temperature, after which the absorbance at 560 nm was measured and compared with a standard curve generated with dilutions of a standard H2O2 solution.
Preparation of Cell Lysates
A549 cells were washed twice with ice-cold phosphate-buffered saline and then lysed at 4 °C in a solution containing 20 mm HEPES-NaOH (pH 7.0), 2 mm EGTA, 1 mm EDTA, 20 mm β-glycerophosphate, 1% Triton X-100, 10% glycerol, 1 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, aprotinin (10 μg/ml), and leupeptin (10 μg/ml). The lysates were centrifuged at 12,500 × g for 10 min, and the protein concentration of the resulting supernatant was measured with a Bradford assay before experiments.
Subcellular Fractionation
Cytosolic and mitochondria-enriched fractions were prepared from A549 cells with the use of a subcellular proteome extraction kit (Calbiochem) according to the manufacturer's protocol.
Assay of Caspase Activity
Caspase-3 and -9 activities were assayed by incubating cell lysate (20 μg of protein) with 100 μl of reaction buffer (50 mm HEPES-NaOH (pH 7.4), 10% (w/v) sucrose, 0.1% CHAPS, 10 mm dithiothreitol) containing 25 μm Ac-DEVD-AMC or 50 μm Ac-LEHD-AFC, respectively. The fluorescence generated by cleavage of the artificial substrate was measured every 1 min for 10 min by using a DTX880 instrument (Beckman Coulter) at excitation and emission wavelengths of 360 and 465 nm for caspase-3 and 380 and 505 nm for caspase-9, respectively.
Adenovirus-mediated Gene Expression
A pShuttle-AdEasy system (Stratagene) was used to create adenovirus vectors. Inserting full-length rat Srx (11) into the KpnI/XhoI sites of pShuttle-CMV, we generated pShuttle-CMV-Srx. Then pShuttle-CMV-Srx was linearized by PmeI digestion and was recombined with pAdEasy-1 in BJ5183 cells. This generated recombinant adenoviral plasmid pAd-CMV-Srx by homologous recombination. The pAd-CMV-Srx vector was then amplified in XL10-Gold ultracompetent cells, linearized by PacI, and transfected into adenovirus E1 gene complementing AD-293 packaging cells to obtain replication-defective recombinant Ad-Srx viral particles. The control recombinant virus, Ad-LacZ, containing the bacterial β-galactosidase gene was generated from pShuttle-CMV-LacZ (Stratagene). Recombinant viruses were propagated in AD-293 cells and purified. Adenoviral stocks were titered with the use of standard viral plaque assays, and A549 cells were infected at a multiplicity of infection of 50 plaque-forming units per cell.
Data Analysis
All experiments were repeated at least three times, and quantitative data are presented as means ± S.D. of triplicate determinations from representative experiments.
RESULTS
Srx Inhibits Continuous Accumulation of ROS in Cells Exposed to Low Steady-state Levels of H2O2
To selectively deplete Srx from A549 cells, we first subjected the cells to stable transfection with a pSUPER-siSrx vector that produces small interfering RNAs specific to Srx. As a control, cells were transfected with a pSUPER empty vector. A549 cells stably transfected with the pSUPER and pSUPER-siSrx vectors are hereafter designated pSUPER and pSUPER-siSrx cells, respectively. Three clones each of pSUPER (clone numbers A, B, and C) and pSUPER-siSrx (clone numbers 1, 2, and 3) cells were chosen for use in subsequent experiments. Expression of Srx was greatly reduced in pSUPER-siSrx cells but was not affected in pSUPER cells (Fig. 1A).
FIGURE 1.

Effects of Srx depletion on ROS accumulation and reduction of sulfinic Prxs. A549 cells were stably transfected with a pSUPER control vector or an expression plasmid pSUPER-siSrx. A, expression level of Srx and tubulin in several clonal cell lines. The cell lysates were subjected to immunoblot with the indicated antibodies. hSrx, human Srx. B, pSUPER and pSUPER-siSrx clonal cell lines were exposed to 15 milliunits/ml GOX for the indicated times, and the amount of H2O2 in the culture medium was measured by the PCA-FOX assay as described under “Experimental Procedures.” C and D, three individual clones of the control or Srx-depleted cell lines were exposed to 15 milliunits/ml of GOX. After the indicated times, the intracellular ROS level was measured with CM-H2DCFDA dye (CM-DCF) and flow cytometry (C), and the cell lysates were subjected to immunoblot with the specific antibodies to Prx-SO2, Srx, Prx I, and tubulin (D). In C, data are means ± S.D. of independent experiments (*, p < 0.05 and **, p < 0.01 versus pSUPER; n = 3).
To study the role of Srx in cells exposed to low steady-state levels of H2O2, we incubated pSUPER cells with GOX, which catalyzes the oxidation of β-d-glucose in the presence of oxygen to produce d-gluconic acid and H2O2, and measured the concentration of H2O2 in the medium. In the presence of GOX at a concentration of 15 milliunits/ml, the concentration of H2O2 in the medium reached a plateau after 12 h and was then maintained in the range of 10–20 μm (Fig. 1B), implying that the cells were exposed to low steady-state levels of H2O2 as the result of the balance between consumption by the cells and production by GOX. In contrast, Srx depletion resulted in a dramatic accumulation of H2O2 in the medium from 12 h after GOX treatment, indicating that the H2O2-eliminating ability of pSUPER-siSrx cells was decreased. We then investigated whether Srx depletion also affects the increase in the level of ROS in cells exposed to GOX. The ROS level in pSUPER cells exposed to low steady-state levels was not significantly changed at all, whereas that of pSUPER-siSrx cells was strikingly increased after GOX treatment in a time-dependent manner (Fig. 1C), indicating that the Srx depletion causes a pronounced decrease in cellular ability to eliminate H2O2 and then accumulation of intracellular H2O2.
Next, the question of whether the hyperoxidation of 2-Cys Prxs is increased in accordance with the accumulation of intracellular H2O2 was addressed. To do this, we determined the hyperoxidation of 2-Cys Prxs in cells by using a specific antibody that recognizes both sulfinic and sulfonic forms of 2-Cys Prxs (19). In accordance with the increase in cellular ROS level, the hyperoxidation of Prx I, II, and III was markedly increased in the pSUPER-siSrx cells as compared with what was barely detectable in the pSUPER cells exposed to low steady-state levels of H2O2 (Fig. 1D), indicating that the Srx depletion causes an accumulation of inactivated sulfinic 2-Cys Prxs, which can lead to an even greater increase in cellular H2O2. Collectively, the results indicate that even in the presence of low steady-state levels of H2O2 (10–20 μm), Srx plays a critical role in maintaining the H2O2-eliminating capacity of cells through the restoration of the peroxidase activity of 2-Cys Prxs.
Srx Protects A549 Cells from Apoptosis Induced by Low Steady-state Levels of H2O2
Because the increase in cellular ROS levels leads to cell death, we further examined the effect of Srx depletion on cell death under low steady-state levels of H2O2. Srx depletion increased the number of dead cells in a time-dependent manner after exposure to GOX, as revealed by phase-contrast microscopy (Fig. 2A) and trypan blue dye staining (Fig. 2B). We also quantified apoptotic cells by flow cytometry after staining cells with both propidium iodide and annexin V. When cells were exposed to GOX, the pSUPER-siSrx cells evidenced a dramatic increase of apoptotic death in a time-dependent manner (Fig. 2, C and D). In contrast, there was no detectable cell death in the pSUPER cells exposed to low steady-state levels of H2O2 achieved by GOX treatment (Fig. 2, A–D). The results collectively indicate that Srx plays a crucial role in protecting cells from apoptosis under low steady-state levels of H2O2 (10–20 μm).
FIGURE 2.

Effects of Srx depletion on cell death. Control (pSUPER) or Srx-depleted (pSUPER-siSrx) cell lines were exposed to 15 milliunits/ml of GOX for the indicated times. A, the cytotoxicity was corroborated by observing cell morphology. B, the cells were then stained with trypan blue, and dye-positive cells were counted. The number of dead cells was determined as a percentage of the total cell number. Data are means ± S.D. of a representative experiment (**, p < 0.01 versus pSUPER; n = 3). C and D, 18 h after GOX treatment, the cells were labeled with Annexin-V-FLUOS and propidium iodide. In C, dot plots are representative of three independent experiments. In D, data are means ± S.D. of the percentage of cells in annexin V-positive fractions (including both propidium iodide-negative and propidium iodide-positive fractions) obtained from independent experiments (**, p < 0.01 versus pSUPER; n = 3).
Srx Negatively Regulates Apoptotic Pathways in A549 Cells Exposed to Low Steady-state Levels of H2O2
As shown in Fig. 1D, like cytosolic Prx I and Prx II, mitochondrial Prx III is hyperoxidized in pSUPER-siSrx cells. The increased level of sulfinic inactivated Prx III results in an accumulation of mitochondrial H2O2. Because mitochondrial ROS-induced apoptosis is associated with a reduction in the ΔΨm, we therefore examined the effects of Srx depletion on this event with the use of TMRE, which shows a red shift in its fluorescence spectra upon ΔΨm-driven mitochondrial uptake (20). Flow cytometric analysis of red fluorescence indicated that the ΔΨm of pSUPER cells exposed to low steady-state levels was not significantly changed at all, whereas that of pSUPER-siSrx cells was distinctly dissipated after GOX treatment in a time-dependent manner (Fig. 3A).
FIGURE 3.

Effects of Srx depletion on mitochondria-mediated apoptotic signaling. Control (pSUPER) or Srx-depleted (pSUPER-siSrx) cell lines were exposed to 15 milliunits/ml of GOX for the indicated times. A and B, cells were then stained with TMRE (A) or NAO (B), respectively, and analyzed by flow cytometry. Data are means ± S.D. of the percentage of cells with low ΔΨm (A) and of the NAO fluorescence (B) obtained from independent experiments (**, p < 0.01 versus pSUPER; n = 3). C, the cells were separated into cytosolic and mitochondrial fractions by subcellular fractionation, and cytochrome c was detected in cytosolic fractions. D and E, the activity of caspase-9 (D) or caspase-3 (E) in cell lysates was measured as described under “Experimental Procedures.” Data are means ± S.D. of the relative fluorescence units (RFU) per minute obtained from independent experiments (**, p < 0.01 versus pSUPER; n = 4).
Because cytochrome c is bound to the mitochondrial membrane through interaction with cardiolipin and its dissociation is facilitated by mitochondrial ROS-mediated oxidation of cardiolipin (21), we measured cardiolipin oxidation by flow cytometric analysis with NAO-loaded cells that had been exposed to GOX treatment. The oxidation of cardiolipin markedly increased in the pSUPER-siSrx cells as compared with what was hardly detectable in the pSUPER cells exposed to low steady-state levels of H2O2 (Fig. 3B).
Cytochrome c is an important factor in the apoptosis pathway. If the release of cytochrome c is increased in the cytosol under apoptosis, it activates various proapoptotic molecules and cascade of caspases. We therefore measured the release of cytochrome c into the cytosol and activities of caspases. Caspase activity was measured using peptide conjugated to fluorophores. Consistent with the increase in oxidation of cardiolipin, cytochrome c release and activation of caspase-3 and caspase-9 were subsequently enhanced in pSUPER-siSrx cells. In contrast, there was neither detectable cytochrome c in the cytosol fractions nor activation of these caspases in the pSUPER cells exposed to low steady-state levels of H2O2 achieved by GOX treatment (Fig. 3, C–E). The results collectively indicate that Srx functions as a suppressor of mitochondria-mediated apoptotic pathways in cells under low steady-state levels of H2O2.
Ectopic Re-expression of Srx in pSUPER-siSrx Cells Is Sufficient to Restore Ability to Resist Low Steady-state Levels of H2O2
To examine whether re-expression of Srx in Srx-depleted cells reverses the phenotypes under low steady-state levels of H2O2, we carried out an “add-back” rescue experiment by adenoviral expression of rat Srx in pSUPER-siSrx cells. The adenovirus-induced expression level of Srx in pSUPER-siSrx cells was ∼80% of that of endogenous Srx in pSUPER cells (Fig. 4A). Reintroducing Srx via adenoviral expression appreciably suppressed hyperoxidation of Prx I, II, and III (Fig. 4A). Furthermore, adenoviral expression of Srx reduced an accumulation of cellular ROS as well as apoptotic cell death under low steady-state levels of H2O2 (Fig. 4, B and C). Collectively, these results indicate that Srx is a novel component to maintain the redox balance in cells exposed to low steady-state levels of H2O2.
FIGURE 4.
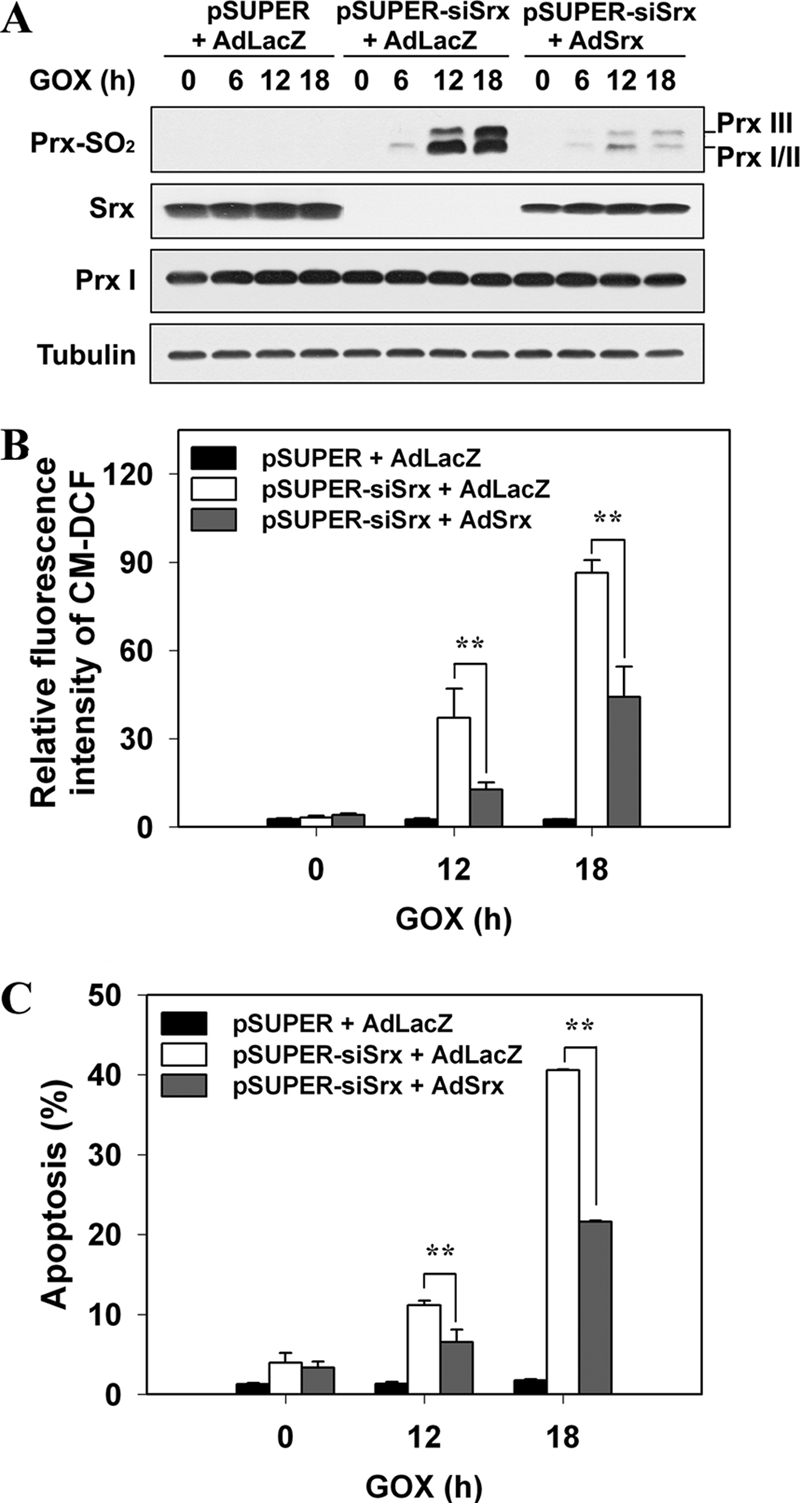
Ectopic re-expression of Srx in pSUPER-siSrx cells is sufficient to restore ability to resist low steady-state levels of H2O2. Control (pSUPER) or Srx-depleted (pSUPER-siSrx) cell lines were infected with adenovirus encoding LacZ (AdLacZ) or rat Srx (AdSrx) as respectively indicated. 24 h after infection, cells were exposed to 15 milliunits/ml of GOX for the indicated times. A, cell lysates were subjected to immunoblot with the specific antibodies to Prx-SO2, Srx, Prx I, and tubulin. The blots are representative of three independent experiments. B, the intracellular ROS level was measured with CM-H2DCFDA dye (CM-DCF) and flow cytometry. Data are means ± S.D. of fluorescence values obtained from independent experiments (**, p < 0.01; n = 3). C, cells were analyzed for DNA content using flow cytometry following staining with propidium iodide. Apoptotic cells were defined as those with DNA content less than that typically present in cells during the G1 phase of the cell cycle. Data are means ± S.D. of the percentage of apoptotic cells obtained from independent experiments (**, p < 0.01; n = 3).
Ectopic Re-expression of Srx in Srx−/− MEF Cells Is Sufficient to Restore Ability to Resist Low Steady-state Levels of H2O2
Next, we examined the role of Srx using MEF cells from Srx+/+ and Srx−/− embryos obtained by mating the Srx+/− mice (17). When the Srx+/+ MEF cells were exposed to GOX (4 milliunits/ml), the concentration of H2O2 in the medium reached a plateau after 6 h and was then maintained in the range of 10–20 μm, implying that the cells were exposed to low steady-state levels of H2O2 (Fig. 5B). In contrast, Srx−/− MEF cells showed an accumulation of H2O2 in the medium from 6 h after GOX treatment, indicating that the H2O2-eliminating capacity of Srx−/− MEF cells was reduced. The ROS level in Srx+/+ MEF cells exposed to low steady-state levels of H2O2 was not significantly changed, whereas that of Srx−/− MEF cells was strikingly increased after GOX treatment (Fig. 5C). In accordance with the increase in cellular ROS level, the hyperoxidation of Prx I, II, and III was markedly increased in Srx−/− MEF cells as compared with what was barely detectable in Srx+/+ MEF cells exposed to low steady-state levels of H2O2 (Fig. 5D). We also examined apoptotic cell death in Srx+/+ and Srx−/− MEF cells. When cells were exposed to GOX, Srx−/− MEF cells evidenced a dramatic increase of apoptosis at 24 h after GOX treatment (Fig. 5E). In contrast, there was no detectable apoptosis in Srx+/+ MEF cells exposed to low steady-state levels of H2O2. Furthermore, overexpressed Srx converted Srx−/− MEF cells to be more resistant to accumulation of cellular ROS, hyperoxidation of 2-Cys Prxs, and apoptosis under low steady-state levels of H2O2 (Fig. 5, C–E). Together, these data strongly suggest that Srx functions as a negative regulator of apoptosis under low steady-state levels of H2O2 (10–20 μm) by regenerating the hyperoxidized 2-Cys Prxs.
FIGURE 5.

Ectopic re-expression of Srx in Srx−/− MEF cells is sufficient to restore ability to resist low steady-state levels of H2O2. A, lysates of Srx+/+ and Srx−/− MEF cells were subjected to immunoblot with the specific antibodies to Srx and β-actin. B, Srx+/+ and Srx−/− MEF cells were cultured in the presence of GOX (4 milliunits/ml) for the indicated times, and the amount of H2O2 in the culture medium was measured by the PCA-FOX assay as described under “Experimental Procedures.” C–E, Srx+/+ and Srx−/− MEF cells were infected with adenovirus encoding LacZ (AdLacZ) or rat Srx (AdSrx) as respectively indicated. 24 h after infection, cells were exposed to 4 milliunits/ml of GOX for an additional 24 h. C, the intracellular ROS level was measured with CM-H2DCFDA dye (CM-DCF) and flow cytometry. Data are means ± S.D. of fluorescence values obtained from independent experiments (**, p < 0.01; n = 3). D, cell lysates were subjected to immunoblot with the specific antibodies to Prx-SO2, Srx, Prx I, and β-actin. The blots are representative of three independent experiments. E, cells were analyzed for DNA content using flow cytometry following staining with propidium iodide. Apoptotic cells were defined as those with DNA content less than that typically present in cells during the G1 phase of the cell cycle. Data are means ± S.D. of the percentage of apoptotic cells obtained from independent experiments (**, p < 0.01; n = 3).
DISCUSSION
In the present study, we show that Srx has a functional role in protecting cells from apoptosis under low steady-state levels of H2O2 due to its ability to inhibit the accumulation of hyperoxidized 2-Cys Prxs. These findings further confirm and extend the results of in vitro steady-state kinetic studies on the turnover rate of Prx I hyperoxidation during catalysis (7). ROS are formed upon incomplete reduction of oxygen and include the superoxide anion (O2˙̄), the hydroxyl radical (OH•), and the hydrogen peroxide (H2O2). Not all ROS are equally implicated in signaling functions. For example, because H2O2 is a nonpolar and nonradical molecule, it is membrane-permeable and diffusible, less reactive, and longer lived than other ROS. These characteristics of H2O2 make it function as an efficient signaling molecule (22–25). H2O2 exerts different functions depending on its concentration. When H2O2 exists at a low concentration of about 10−8-10−7 m, it acts as a mitogen to help the growth of cell in vivo. When H2O2 concentration becomes denser at about 10−6-10−5 m, it suppresses the proliferation of the cell or stops the cell cycle. Also, at a high concentration of about 10−4-10−3 m, finally, H2O2 induces cell death such as apoptosis or necrosis (2). Therefore, the intracellular defense mechanism for maintaining a constant intracellular H2O2 concentration is an important factor in deciding whether to go to cell growth or death. In mammalian system, intracellular H2O2 is removed mostly by catalase, glutathione peroxidase, and Prx. Prxs are unusually efficient for the reduction of H2O2 at low concentrations because the hydrogen-bonding network created by several amino acid residues at their active site provides a binding site for H2O2 and at the same time activates the bound peroxide for the reaction with the active site thiol group (14). On the other hand, the active site cysteine residue of 2-Cys Prxs (Prx I–IV) is hyperoxidized to cysteine sulfinic acid (Cys-SO2) during catalysis with concomitant loss of peroxidase activity (12), and Prx I is hyperoxidized at a rate of 0.072% per turnover at 30 °C in the presence of a low steady-state level of H2O2 (7). Reactivation of the hyperoxidized Prx is catalyzed by Srx (10). Although exposing cells to a steady-state low concentration of H2O2 mimics the situation in vivo better and is more representative of oxidative pathophysiologic conditions than of a high bolus concentration of H2O2, the role of Srx in cells exposed to low steady-state levels of H2O2 had not been studied yet. We therefore hypothesize that even in the presence of low steady-state levels of H2O2, Srx may play a critical role in maintaining the H2O2-eliminating capacity of cells through the restoration of the peroxidase activity of 2-Cys Prxs. Indeed, we show here that Srx is a novel component to maintain the redox balance in cells exposed to low steady-state levels of H2O2 by using Srx-depleted cell lines and Srx−/− MEF cells. In this study, the expression of Srx gene was depleted by an RNA interference system, but this approach has sometimes been found to silence nontargeted genes (26). To alleviate this concern about RNA interference and to emphasize the critical role of Srx under low steady-state levels of H2O2, we herein used Srx+/+ and Srx−/− MEF cells. When the Srx+/+ MEF cells were treated with GOX (4 milliunits/ml), low steady-state levels of H2O2 (10–20 μm) were maintained in the extracellular space. However, after GOX treatment, the H2O2-eliminating capacity of Srx−/− MEF cells was so reduced that H2O2 was continuously accumulated in the medium. Given that the accumulated H2O2 in the medium can diffuse freely across biological membranes, the intracellular ROS level of Srx−/− MEF cells was dramatically increased, which in turn resulted in a marked increase in the hyperoxidation of 2-Cys Prxs in Srx−/− MEF cells. These results demonstrate that even in the presence of low levels of H2O2, Srx plays a pivotal role in maintaining a balance between H2O2 production and the cellular peroxidase capacity through promoting the regeneration of 2-Cys Prxs. When there is not enough reduced 2-Cys Prxs to handle intracellular H2O2, accumulation of sulfinic 2-Cys Prxs takes place, which could lead to an even greater accumulation in intracellular H2O2 and oxidative stress-induced cell death. We therefore wondered whether Srx could also protect MEF cells against cell death induced by low steady-state levels of H2O2 through restoration of the peroxidase activity of 2-Cys Prxs. Our results now provide support for this scenario. When cells were exposed to GOX, Srx−/− MEF cells evidenced a dramatic apoptosis. In contrast, there was merely detectable apoptosis in Srx+/+ MEF cells exposed to low steady-state levels of H2O2. Furthermore, re-expression of Srx in Srx−/− MEF cells efficiently promoted the regeneration of sulfinic 2-Cys Prxs and contributed to the elimination of intracellular H2O2. The decreased accumulation of sulfinic 2-Cys Prxs that resulted from Srx re-expression thus decreased the rate of apoptosis in Srx−/− MEF cells exposed to low steady-state levels of H2O2. Although the Srx expression level in Srx−/− MEF cells after re-expression was higher than that of Srx+/+ MEF cells, the levels of ROS accumulation, Prx hyperoxidation, and apoptosis of Srx−/− MEF cells were not completely reverted to that of Srx+/+ MEF cells. These results imply that recombinant adenovirus Srx may not be expressed in each cell at an equal amount. However, these data now indicate that Srx is a novel component to maintain the balance between H2O2 production and elimination and that it protects cells from apoptosis under low steady-state levels of H2O2.
Recent findings demonstrate that phorbol ester, a tumor-promoting agent, induces Srx expression in wild-type mice (27) and that Srx is highly expressed in certain human skin malignancies (28), breast adenocarcinoma (29), and lung adenocarcinoma (30), implicating a role for Srx in the regulation of cancer maintenance and metastasis via lowering sensitivity to ROS-generating anticancer drugs or enhancing cell proliferation rates. These results suggest that Srx is a functionally novel target in cancer prevention and treatment. Several studies have demonstrated that Srx expression is induced in lung tissue exposed to cigarette smoke (27) or hyperoxia (31) and that defective Srx expression potentiates oxidative damage on macrophages under inflammatory conditions (32). The physiological role of Srx induction is probably as a self-defense mechanism against oxidative stress, which suggests therapeutic implications for augmenting the activity of Srx in cell survival. For instance, treatments that promote expression of Srx may reduce the effects of a prolonged exposure to relatively low levels of H2O2 in pathological conditions such as chronic inflammation and hyperoxia.
In this study, the key antioxidant functions of Srx, the guardian of 2-Cys Prxs, is evident from the marked oxidative damage induced by low steady-state levels of H2O2 in Srx−/− MEF cells. It seems that Srx is a bridge among multiple key redox-regulating systems, and further understanding of its cellular functions could implicate it as a plausible drug target.
This work was supported by Bio R&D Program Grant M10642040002-07N4204-00210, National Core Research Center Program Grant 2011-0006244 through the National Research Foundation of Korea funded by the Korea government (Ministry of Education, Science and Technology (MEST)), and the Brain Korea 21 Scholars Program (to J. Y. B., S. H. H., and Y. H. N.).
This article was selected as a Paper of the Week.
- ROS
- reactive oxygen species
- Prx
- peroxiredoxin
- Srx
- sulfiredoxin
- MEF
- mouse embryonic fibroblast
- ΔΨm
- mitochondria membrane potential
- GOX
- glucose oxidase
- Ac-DEVD-AMC
- acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin
- Ac-LEHD-AFC
- acetyl-Leu-Glu-His-Asp-7-amino-4-trifluoromethyl coumarin
- CM-H2DCFDA
- 5-(and 6-)chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- NAO
- 10-N-nonylacridine orange
- PCA-FOX
- ferrous xylenol orange in perchloric acid
- TMRE
- tetramethylrhodamine ethyl ester.
REFERENCES
- 1. Finkel T., Holbrook N. J. (2000) Nature 408, 239–247 [DOI] [PubMed] [Google Scholar]
- 2. Giorgio M., Trinei M., Migliaccio E., Pelicci P. G. (2007) Nat. Rev. Mol. Cell Biol. 8, 722–728 [DOI] [PubMed] [Google Scholar]
- 3. Rhee S. G., Kang S. W., Jeong W., Chang T. S., Yang K. S., Woo H. A. (2005) Curr. Opin. Cell Biol. 17, 183–189 [DOI] [PubMed] [Google Scholar]
- 4. Poole L. B., Nelson K. J. (2008) Curr. Opin. Chem. Biol. 12, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flohé L., Harris J. R. (2007) Subcell. Biochem. 44, 1–25 [PubMed] [Google Scholar]
- 6. Rhee S. G., Kang S. W., Chang T. S., Jeong W., Kim K. (2001) IUBMB Life 52, 35–41 [DOI] [PubMed] [Google Scholar]
- 7. Yang K. S., Kang S. W., Woo H. A., Hwang S. C., Chae H. Z., Kim K., Rhee S. G. (2002) J. Biol. Chem. 277, 38029–38036 [DOI] [PubMed] [Google Scholar]
- 8. Rabilloud T., Heller M., Gasnier F., Luche S., Rey C., Aebersold R., Benahmed M., Louisot P., Lunardi J. (2002) J. Biol. Chem. 277, 19396–19401 [DOI] [PubMed] [Google Scholar]
- 9. Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., Rhee S. G. (2003) Science 300, 653–656 [DOI] [PubMed] [Google Scholar]
- 10. Biteau B., Labarre J., Toledano M. B. (2003) Nature 425, 980–984 [DOI] [PubMed] [Google Scholar]
- 11. Chang T. S., Jeong W., Woo H. A., Lee S. M., Park S., Rhee S. G. (2004) J. Biol. Chem. 279, 50994–51001 [DOI] [PubMed] [Google Scholar]
- 12. Woo H. A., Jeong W., Chang T. S., Park K. J., Park S. J., Yang J. S., Rhee S. G. (2005) J. Biol. Chem. 280, 3125–3128 [DOI] [PubMed] [Google Scholar]
- 13. Schröder E., Littlechild J. A., Lebedev A. A., Errington N., Vagin A. A., Isupov M. N. (2000) Structure 8, 605–615 [DOI] [PubMed] [Google Scholar]
- 14. Hall A., Nelson K., Poole L. B., Karplus P. A. (2011) Antioxid. Redox Signal. 15, 795–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang S. W., Chae H. Z., Seo M. S., Kim K., Baines I. C., Rhee S. G. (1998) J. Biol. Chem. 273, 6297–6302 [DOI] [PubMed] [Google Scholar]
- 16. Chang T. S., Cho C. S., Park S., Yu S., Kang S. W., Rhee S. G. (2004) J. Biol. Chem. 279, 41975–41984 [DOI] [PubMed] [Google Scholar]
- 17. Bae S. H., Sung S. H., Cho E. J., Lee S. K., Lee H. E., Woo H. A., Yu D. Y., Kil I. S., Rhee S. G. (2011) Hepatology 53, 945–953 [DOI] [PubMed] [Google Scholar]
- 18. Gay C. A., Gebicki J. M. (2002) Anal. Biochem. 304, 42–46 [DOI] [PubMed] [Google Scholar]
- 19. Woo H. A., Kang S. W., Kim H. K., Yang K. S., Chae H. Z., Rhee S. G. (2003) J. Biol. Chem. 278, 47361–47364 [DOI] [PubMed] [Google Scholar]
- 20. Scaduto R. C., Jr., Grotyohann L. W. (1999) Biophys. J. 76, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ott M., Robertson J. D., Gogvadze V., Zhivotovsky B., Orrenius S. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1259–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Finkel T. (1998) Curr. Opin. Cell Biol. 10, 248–253 [DOI] [PubMed] [Google Scholar]
- 23. Rhee S. G. (1999) Exp. Mol. Med. 31, 53–59 [DOI] [PubMed] [Google Scholar]
- 24. Rhee S. G., Bae Y. S., Lee S. R., Kwon J. (2000) Sci. STKE 2000, pe1. [DOI] [PubMed] [Google Scholar]
- 25. Suzuki Y. J., Ford G. D. (1999) J. Mol. Cell. Cardiol. 31, 345–353 [DOI] [PubMed] [Google Scholar]
- 26. Jackson A. L., Bartz S. R., Schelter J., Kobayashi S. V., Burchard J., Mao M., Li B., Cavet G., Linsley P. S. (2003) Nat. Biotechnol. 21, 635–637 [DOI] [PubMed] [Google Scholar]
- 27. Singh A., Ling G., Suhasini A. N., Zhang P., Yamamoto M., Navas-Acien A., Cosgrove G., Tuder R. M., Kensler T. W., Watson W. H., Biswal S. (2009) Free Radic. Biol. Med. 46, 376–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei Q., Jiang H., Matthews C. P., Colburn N. H. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19738–19743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei K., Townsend D. M., Tew K. D. (2008) Oncogene 27, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei Q., Jiang H., Xiao Z., Baker A., Young M. R., Veenstra T. D., Colburn N. H. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 7004–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bae S. H., Woo H. A., Sung S. H., Lee H. E., Lee S. K., Kil I. S., Rhee S. G. (2009) Antioxid. Redox Signal. 11, 937–948 [DOI] [PubMed] [Google Scholar]
- 32. Kim H., Jung Y., Shin B. S., Kim H., Song H., Bae S. H., Rhee S. G., Jeong W. (2010) J. Biol. Chem. 285, 34419–34428 [DOI] [PMC free article] [PubMed] [Google Scholar]