

Abstract
Mutations in Cu,Zn superoxide dismutase (SOD1) are associated with familial amyotrophic lateral sclerosis (ALS). Mutant SOD1 causes a complex array of pathological events, through toxic gain of function mechanisms, leading to selective motor neuron degeneration. Mitochondrial dysfunction is among the well established toxic effects of mutant SOD1, but its mechanisms are just starting to be elucidated. A portion of mutant SOD1 is localized in mitochondria, where it accumulates mostly on the outer membrane and inside the intermembrane space (IMS). Evidence in cultured cells suggests that mutant SOD1 in the IMS causes mitochondrial dysfunction and compromises cell viability. Therefore, to test its pathogenic role in vivo we generated transgenic mice expressing G93A mutant or wild-type (WT) human SOD1 targeted selectively to the mitochondrial IMS (mito-SOD1). We show that mito-SOD1 is correctly localized in the IMS, where it oligomerizes and acquires enzymatic activity. Mito-G93ASOD1 mice, but not mito-WTSOD1 mice, develop a progressive disease characterized by body weight loss, muscle weakness, brain atrophy, and motor impairment, which is more severe in females. These symptoms are associated with reduced spinal motor neuron counts and impaired mitochondrial bioenergetics, characterized by decreased cytochrome oxidase activity and defective calcium handling. However, there is no evidence of muscle denervation, a cardinal pathological feature of ALS. Together, our findings indicate that mutant SOD1 in the mitochondrial IMS causes mitochondrial dysfunction and neurodegeneration, but per se it is not sufficient to cause a full-fledged ALS phenotype, which requires the participation of mutant SOD1 localized in other cellular compartments.
Introduction
Cu,Zn superoxide dismutase (SOD1) is an abundant soluble enzyme that catalyzes the formation of an oxygen and a hydrogen peroxide during its cycle of copper reduction and reoxidation (Klug et al., 1972). The majority of SOD1 is localized in the cytosol, but a portion is associated with various organelles, including the nucleus (Crapo et al., 1992), endoplasmic reticulum (Kikuchi et al., 2006), and mitochondria (Okado-Matsumoto and Fridovich, 2001; Sturtz et al., 2001; Vijayvergiya et al., 2005). SOD1 is imported inside mitochondria through a regulated mechanism involving the cellular redox state (Kawamata and Manfredi, 2008), and it concentrates in the intermembrane space (IMS), where it dismutates superoxide released on the outer side of the inner membrane (Sturtz et al., 2001). In agreement with this function, we demonstrated that SOD1 in the IMS protects mitochondria from oxidative damage and contributes to maintaining neuronal viability (Fischer et al., 2011).
Mutations in SOD1 are responsible for ∼20% of familial amyotrophic lateral sclerosis (ALS) cases (Rosen et al., 1993). The pathophysiology of SOD1-ALS is not completely understood, and different mechanisms may participate in pathogenesis (Ilieva et al., 2009), including mitochondrial dysfunction (Hervias et al., 2006; Shi et al., 2010). Mutant SOD1 accumulates on the mitochondrial outer membrane (Vande Velde et al., 2008) and interacts with some crucial proteins, such as Bcl2 (Pedrini et al., 2010) and VDAC (Israelson et al., 2010). It is difficult to differentiate the pathogenic role of mutant SOD1 on the outer membrane from that of cytosolic SOD1, because both pools of SOD1 are outside of mitochondria. However, mutant SOD1 also localizes inside the mitochondrial IMS, where it accumulates and misfolds, potentially interfering with the assembly and maturation of mitochondrial proteins (Vijayvergiya et al., 2005; Ferri et al., 2006; Kawamata and Manfredi, 2008).
The pathogenic role of the IMS pool of mutant SOD1 is supported by evidence form cultured motorneuronal cells, where it causes mitochondrial functional, morphological, and axonal transport abnormalities (Cozzolino et al., 2009; Magrané et al., 2009). However, the pathogenic effects of mutant SOD1 localized in the IMS have not been tested in vivo. Therefore, to conclusively address this issue, we generated transgenic mice expressing G93A mutant or wild-type (WT) human SOD1 targeted to the mitochondrial IMS (mito-SOD1 mouse), in the nervous system and muscle. Mito-SOD1 was anchored to the mitochondrial inner membrane facing the IMS to ensure that its localization was restricted to this compartment. Mito-SOD1 accumulated in the IMS at levels comparable to those found in transgenic mice expressing untargeted G93A mutant SOD1.
Mito-G93ASOD1, but not mito-WTSOD1, resulted in progressive weight loss, motor dysfunction, motor neuron loss, muscle atrophy, and mitochondrial biochemical and structural defects, in neural and muscle cells. Despite these abnormalities, no overt muscle denervation was observed. Together, these findings indicate that mutant SOD1 in the mitochondrial IMS is pathogenic, but in the absence of mutant SOD1 localized in other cell compartments it is not sufficient to fully reproduce the clinical and pathological features typical of ALS.
Materials and Methods
Transgenic mice.
All the animal procedures were approved by the Animal Care and Use Committee of the Weill Medical College of Cornell University. The generation of mito-SOD1 transgenic mice was described previously (Fischer et al., 2011). Briefly, WT or G93A human SOD1 cDNA was inserted in-frame in the mouse prion promoter vector (Borchelt et al., 1996), downstream of the cDNA encoding the first 187 aa of mouse mitofilin (GenBank: NM_029673) (John et al., 2005). Positive male founders (mito-WTSOD1 or mito-G93ASOD1) were bred with non-transgenic B6SJLF1/J females (The Jackson Laboratory). To generate homozygous mito-SOD1 mice, hemizygous males were crossed with hemizygous females.
Genotyping and quantitative real-time PCR.
For genotyping of mito-SOD1 transgenic mice, PCR was performed on DNA from tail biopsies, using the primers 5′-CCG CTC GAG ATG CTG CGG GCG TGT CAG-3′ (forward) and 5′-CCG CTC GAG TTA TTG GGC GAT CCC AAT-3′ (reverse).
Homozygosity and number of genomic insertions of mito-SOD1 transgenic constructs were determined by quantitative real-time PCR on tail DNA extracted with Wizard Genomic DNA purification kit (Promega), using the primers 5′-AGT TAT CCG GCG TGA C-3′ and 5′-GGA ACA GTA TAA GGC GCA-3′, and the LightCycler FastStart DNA Master SYBR Green I kit (Roche). PCR cycling parameters in the LightCycler were as follows: 95°C for 10 min for one cycle, followed by 95°C for 15 s, 55°C for 15 s, and 72°C for 15 s on single acquisition mode for 40 cycles. β-actin was used as internal reference gene, using primers 5′-GGC CAA CCG TGA AAA GAT GA-3′ and 5′-GCC TGG ATG GCT ACG TAC ATG-3′. After amplification, a melting curve analysis was performed, and the specificity of the PCR products was confirmed by gel electrophoresis. Calculations of threshold cycle and difference were analyzed with LightCycler analysis software (Roche). The amounts of mito-SOD1 and β-actin DNA were calculated using linear regression analysis from standard curves. Successive dilutions of mito-WTSOD1 plasmid were prepared in non-transgenic genomic DNA and used to build a standard curve for transgene insertion quantification.
Mitochondrial isolation from mouse tissues.
Brain or spinal cord tissues were homogenized in a Dounce glass/glass homogenizer in MS-EGTA buffer (containing, in mm: 225 mannitol, 75 sucrose, 5 HEPES, 1 EGTA, pH 7.4). The homogenate was centrifuged at 2000 × g for 5 min at 4°C. The supernatant was centrifuged at 15,000 × g for 20 min at 4°C. The supernatant is referred to as cytosolic fraction, and the pellet as crude mitochondrial fraction. The cytosolic fraction was then centrifuged twice at 22,000 × g for 20 min at 4°C. To prepare purified mitochondria, the crude mitochondrial pellet was washed twice in MS-EGTA and layered on top of 9 ml of chilled 23% Percoll made in MS-EGTA. The gradient was centrifuged at 25,000 × g for 11 min at 4°C, the pellet resuspended in MS-EGTA and centrifuged at 14,000 × g for 14 min at 4°C. The final step was repeated 3 times. The purified mitochondrial pellet was resuspended in MS-EGTA at ∼10 mg/ml. All reagents used were from Sigma-Aldrich.
Primary cortical neurons and astrocytic cultures.
Primary cortical neurons were isolated and cultured from E16 mouse embryos as previously described (Magrané et al., 2004).
Primary astrocytes were isolated from newborn pups (P2) as previously described (Nagai et al., 2007). After 2 weeks in culture, flasks were shaken at 230 rpm, overnight at 37°C to separate nonadherent microglial cells from astrocytic primary cultures.
Topology of mito-SOD1.
Mitoplasts preparations, proteinase K treatments and proteins alkaline extraction were performed to assess intramitochondrial mito-SOD1 localization, as previously described (Fischer et al., 2011).
Western blot analyses.
Proteins in total tissue homogenates (10 μg), cytosol (20 μg), mitochondria (20 μg), and primary cultures lysates (20 μg) were separated on a 12% SDS-PAGE, transferred to PVDF membranes (Bio-Rad) and immunoblotted. The following primary antibodies were used (1:5000 dilution): polyclonal antibodies against SOD1 (sheep; Calbiochem), GFAP (rabbit; DakoCytomation), Akt1 (goat; Santa Cruz Biotechnology), and HSP60 (goat; Enzo Life Sciences); and monoclonal mouse antibodies against NeuN (Millipore), Tim23 (Stressgen), Cytochrome c (BD PharMingen), β-actin (Sigma), and VDAC (Invitrogen). All secondary antibodies were from Jackson ImmunoResearch and used at 1:10,000 dilution. Immunoreactive bands were quantified using NIH ImageJ.
Blue native gel electrophoresis.
Brain purified mitochondria (50 μg) were solubilized with n-dodecyl β-d-maltoside (1.6 g per g of proteins) for 5 min on ice, then centrifuged at 20,000 × g for 30 min at 4°C. Proteins contained in the supernatant were separated on a nondenaturing 10–16% gradient blue native gel, containing 50 mm BisTris and 500 mm 6-aminocaproic acid, pH 7.0, as previously described (Schägger and von Jagow, 1991). After electrophoresis, proteins were transferred to PVDF membranes and immunoblotted with SOD1 antibody and core II subunit of complex III mouse monoclonal antibody (CIII, 1:5000, Invitrogen). A high molecular weight calibration kit (GE Healthcare) was used to determine protein sizes.
SOD1 activity.
SOD1 activity was assessed on purified brain mitochondria (50 μg) by spectrophotometry, as previously described (Fischer et al., 2011). One unit of SOD activity was defined as the amount of enzyme required to inhibit the rate of reduction of cytochrome c by 50% (Vives-Bauza et al., 2007).
Survival analyses, body weight and behavioral assessments.
Mice were monitored for survival and body weight. The ability to maintain balance on the rotarod (Economex accelerating rotarod; Columbus Instruments) accelerating from 4 to 28 rpm over 2 min was tested. Briefly, the animals were placed on the rod for 4 trials per day for 4 consecutive days, and latency to fall was scored. The average of 4 trials of the fourth day was used for the comparisons. Mice were also tested for their limb strength (hang test) by placing them on a grid ∼30 cm above a padded surface and allowing them to grip with four paws; the grid was then turned upside down, and the time to fall recorded.
Mitochondrial bioenergetic assays.
For mitochondrial respiration, 100 μg of brain purified mitochondria were resuspended in 0.5 ml of respiration buffer containing (in mm): 125 KCl, 20 HEPES, 4 K2HPO4, 0.1 mg/ml BSA, pH 7.2, and 1 ADP. Glutamate (5 mm) and malate (2 mm) were used as oxidative substrates. Oxygen consumption was recorded with an oxygraph equipped with a Clark electrode (Hansatech), as described previously (Hofhaus et al., 1996). State 4 (nonphosphorylating) respiration was induced by the addition of 3.2 μm carboxyatractylate. Successive additions of 2 nm SF6847 were used to uncouple the mitochondrial respiratory chain and evaluate the sensitivity of the mitochondria to the uncoupler. The uncoupled respiration rates were plotted against the added amount of uncoupler and the slope of the linear portion of the curve was calculated by linear regression using an MS Excel in-build algorithm. The slope (nmol O2/min/pmol of SF6847) was taken as a measure of the sensitivity of mitochondrial respiration to workload. Data are presented as percentage of matched non-transgenic controls for mito-WTSOD1 and mito-G93ASOD1.
Brain mitochondrial Ca2+ uptake dynamics were measured with the fluorescent ratiometric dye Fura 6F, as previously described (Damiano et al., 2006).
Cytochrome oxidase (COX) and citrate synthase (CS) enzymatic activities in spinal cord lysates (10 μg of proteins) were measured spectrophotometrically as described previously (Birch-Machin and Turnbull, 2001) and expressed as a percentage of non-transgenic mitochondria.
Brain and spinal cord histology and analysis.
For histological studies mice were perfused with cold PBS for 1 min, followed by cold 4% paraformaldehyde in PBS for 3 min. Brains were dissected and weighted using a precise scale. Images of whole brains were taken using a digital camera. Spinal cords were dissected, and lumbar segments were identified using the ribs and the vertebrae as spatial references. Tissues were postfixed in 4% paraformaldehyde overnight, and then cryoprotected in 30% sucrose for 48 h. Brains and lumbar spinal cords were cryosectioned in serial coronal sections (40 and 50 μm thick, respectively), mounted on slides and stained with 0.25% cresyl violet in 6% acetic acid (Nissl staining). Pictures of brain sections were taken in an area including the retrosplenial/motor cortex at the level of bregma −1.94 mm (low magnification) and mid-way between the edge of the cortex and the cingulum (high magnification), using mouse brain stereotaxic atlas as a guide and a Q-imaging camera (MicroBrightField). For stereological analysis of spinal motor neurons, cell counts were made on lumbar hemi-sections as previously described (Kiaei et al., 2006). Only anterior horn neurons with diameter equal or larger than 30 μm were included in the count.
Immunohistochemistry and confocal microscopy.
The following antibodies were used: mouse anti-SOD1 antibody (1:100, Sigma), rabbit anti-SOD2 (MnSOD; 1:100, Assay Designs), rabbit anti-GFAP (1:500, DakoCytomation), and cy2- and cy3-conjugated secondary antibodies (1:200, Jackson ImmunoResearch). Lumbar spinal cord sections (20 μm thick) were collected as free-floating sections and processed for immunofluorescence. Sections were incubated overnight in the primary antibody mixture containing 2% NGS/TBS. After washing in 1% NGS/TBS, sections were incubated for 2 h with secondary antibodies in 1.5% NGS/TBS.
Images were collected using a Leica TCS SP5 spectral confocal microscope (Leica Microsystems Inc.), using either a 10× dry [numerical aperture (NA) 0.30] or a 63× oil-immersion (NA 1.4) lens, and a standard pinhole of 1 A.U. For 10×, 15 μm z-stack image was taken (5 μm interval). For 63×, single z-sections were acquired.
Neuromuscular junction morphology.
Fixed, frozen muscle sections were prepared for neuromuscular junction analysis as previously described (Fischer et al., 2011). Motor axons were labeled with rabbit anti-PGP9.5 (1:500, UltraClone) overnight at room temperature, followed by FITC-conjugated secondary antibody (1:150, Jackson ImmunoResearch). Motor endplates were labeled with Alexa 555-conjugated α-bungarotoxin (Invitrogen). Confocal images were obtained as z-stacks on a Zeiss LSM 510 NLO META system, and compressed with LSM Image Examiner software (Zeiss). Totally innervated, partially innervated and denervated endplates were defined by complete, partial or absent overlap between nerve terminal and endplate, respectively.
Muscle analysis and histochemistry.
Tibialis anterior muscles were dissected and snap frozen in dry ice-cooled isopentane. Ten μm cross sections were obtained using a cryostat, mounted on slides and kept overnight at 4°C. For succinate dehydrogenase (SDH) staining, sections were incubated at 37°C for 30 min in 0.1 m Tris buffer containing 0.2 g of sodium succinate, 1 mg of phenazine methosulfate, and 20 mg of nitroblue tetrazolium, followed by extraction in 30, 60, 90, 60, and 30% ethanol in sequence. After 3 washes in PBS, sections were mounted in Cytoseal 60 mounting medium (Thermo Fisher Scientific). Images were captured using a Nikon Optiphot-2 light microscope equipped with an Olympus DP25 digital camera. Oxidative (type I) and glycolytic (type II) fibers were distinguished based on their SDH intensity. Area of fibers was estimated using Image-Pro Plus software (Media Cybernetics).
Electron microscopy.
Mice were perfused with a fixative solution containing 4% PFA, 2.5% glutaraldehyde, and 0.02% saturated picric acid. Spinal cord and sciatic nerve were harvested and further processed, as previously described with minor modifications (Damiano et al., 2006). Digital images were recorded using a Veleta 2Kx2K camera (Olympus Soft Imaging Solutions, GmbH). Pictures were taken at magnifications of 14,000× and 48,000× (spinal cord and sciatic nerve, respectively). Tibialis anterioris muscles were immersion fixed in 5% buffered glutaraldehyde, treated with 1% osmium tetroxide, and embedded in Epon plastic according to standard techniques. Ultrathin sections were mounted on uncoated mesh copper grids, and imaged with a Hitachi H-7500 electron microscope attached to a Gatan BioScan CCD camera. To estimate the frequency of mitochondrial abnormalities (cristae disorganization, swelling, and vacuolization), 12–24 random fields were analyzed. The fields containing at least one mitochondrial structural abnormality were counted, in at least 2 mice per group.
Cardiac analyses.
M-mode and 2D echocardiography were performed on anesthetized non-transgenic and homozygous mito-G93ASOD1 mice, as previously described (Veugelers et al., 2004).
Statistical analyses.
Kaplan–Meier survival analysis was used for survival comparisons. ANOVA with repeated measurements followed by Fisher post hoc was used for multiple comparisons. Two-tailed, unpaired, Student's t test was used to compare two groups, unless otherwise indicated. Data are represented as means ± SEM. For comparisons of mitochondrial bioenergetic parameters paired Student's t test was used on mito-WTSOD1 or mito-G93ASOD1 matched to non-transgenic controls.
Results
Mito-WTSOD1 and mito-G93ASOD1 expression and localization in transgenic mice
To direct the import of SOD1 to mitochondria, we fused the N-terminal first 187 aa of mouse mitofilin, a mitochondrial inner membrane (IM) protein (John et al., 2005), to human SOD1. The resulting chimeric protein of ∼38 kDa was termed mito-SOD1 (Fischer et al., 2011).
For the generation of transgenic mice the construct was cloned downstream of the mouse prion promoter (MoPrP), which drives high expression in brain, spinal cord, skeletal muscle, heart and kidney, low expression in lung and spleen, and virtually no expression in liver (Wang et al., 2005; Fischer et al. 2011). We used one line of mito-G93ASOD1 and one line of mito-WTSOD1 (previously described by Fischer et al., 2011). In both lines, the transgene was transmitted with mendelian frequency, both through the maternal and the paternal lineages, indicating that the mito-WTSOD1 and mito-G93ASOD1 transgenes were inserted in autosomal chromosomes. As we previously showed in mito-WTSOD1 mice (Fischer et al., 2011), also the mito-G93ASOD1 transgene was highly expressed in brain, spinal cord and skeletal muscle (data not shown). Both neurons and astrocytes expressed mito-SOD1, as shown by Western blot of cell homogenates from primary cultures of isolated cortical neurons and astrocytes (Fig. 1A).
Figure 1.
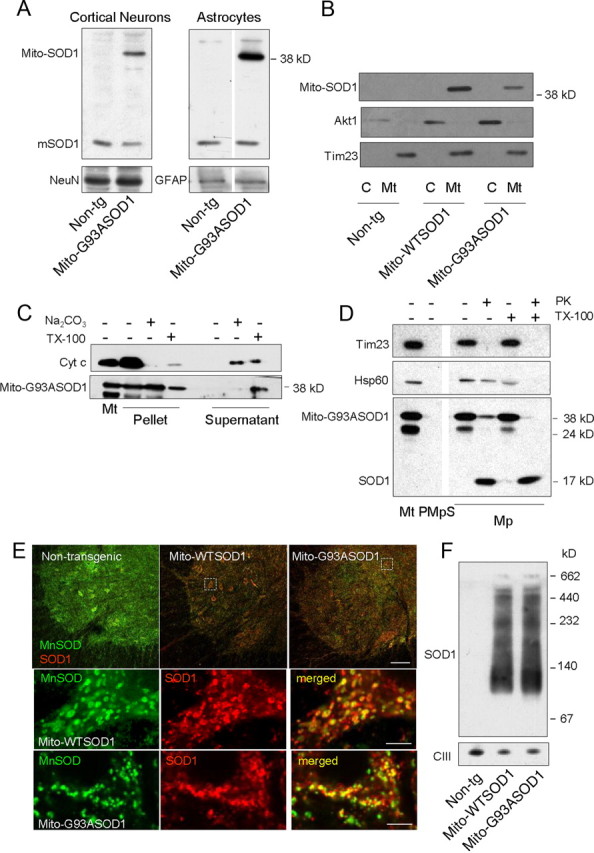
Mito-SOD1 expression and localization in cells and tissues of mito-SOD1 transgenic mice. A, Detection of mito-SOD1 by Western blot in primary cortical neurons and astrocytes isolated from mito-G93ASOD1 and non-transgenic (Non-tg) mice. A sheep polyclonal anti-SOD1 antibody that recognizes both human (mito-SOD1) and endogenous mouse SOD1 (mSOD1) was used. A total of 20 μg of proteins were loaded. NeuN and GFAP were used as neuronal and astrocytic markers, respectively. B, Detection of mito-SOD1 by Western blot in purified mitochondria (Mt) and cytosolic fractions (C) derived from spinal cords of mito-SOD1 mice, using the same antibody as above. A total of 20 μg of proteins were loaded. Note that mito-SOD1 was only detectable in the mitochondrial fraction. Akt1 and Tim23 were used as cytosolic and mitochondrial markers, respectively. C, Protein alkaline extraction. Western blot of the pellet and supernatant of 50 μg mito-G93ASOD1 brain mitochondria ± 0.1 m Na2CO3 (pH 11.5) or 1% Triton X-100 (TX-100). D, Proteinase K (PK) digestion. Western blot of 25 μg mitoplasts (Mp) ± 20 μg/ml PK or 0.1% TX-100. Mitoplasts were prepared from mito-G93ASOD1 brain mitochondria (PMt). PMpS, post-mitoplast supernatant. Tim23, mitochondrial inner membrane protein; Hsp60, soluble mitochondrial matrix protein. E, Immunohistochemistry on spinal cord sections from 12-month-old non-transgenic, hemizygous mito-WTSOD1 and mito-G93ASOD1 mice. Antibodies against SOD1 (in red) and the mitochondrial matrix protein MnSOD (in green) were used. Top shows low-magnification merged images of lumbar ventral horns. Note the increased expression of SOD1 in mito-SOD1 mice. Scale bar, 100 μm. Bottom shows high magnifications of dashed areas from mito-WTSOD1 and mito-G93ASOD1 images. Colocalization between the mitochondrial marker MnSOD (in green) and SOD1 transgene (in red) was observed in the merged images (in yellow). Scale bar, 5 μm. F, Analysis of mito-SOD1 oligomerization. Blue native gel electrophoresis and Western blot with anti-SOD1 antibodies detected high molecular weight protein complexes (multimers) in permeabilized mitochondria isolated from brains of non-transgenic, mito-WTSOD1 and mito-G93ASOD1 mice. A total of 50 μg of proteins were loaded. The mitochondrial respiratory chain Complex III (CIII) was used as a loading control.
Mito-SOD1 localized exclusively in the mitochondrial fraction from spinal cords, and it was undetectable in the cytosol (Fischer et al., 2011) (Fig. 1B). Protein alkaline extraction of brain mitochondria showed mito-G93ASOD1 protein almost exclusively in the insoluble pellet, whereas cytochrome c, a soluble protein of the intermembrane space, was found in the supernatant (Fig. 1C). Most mito-G93ASOD1 was released in the soluble fraction after treatment with Triton X-100. Thus, mito-G93ASOD1 was anchored to the mitochondrial inner membrane through its transmembrane domain. To study the intramitochondrial localization of mito-G93ASOD1, mitoplasts were prepared by removing the mitochondrial outer membrane from brain mitochondria. Mito-G93ASOD1 was associated with mitoplasts and absent from post-mitoplast supernatant containing the outer membrane and intermembrane space fractions (Fig. 1D). Following digestion of mitoplast surface proteins with proteinase K, most of the mito-G93ASOD1 was degraded to a smaller ∼19 kDa fragment. This fragment displayed gel migration properties similar to those of human SOD1, which is proteinase K resistant (Vijayvergiya et al., 2005), indicating that proteinase K digested the portion of the protein anchoring SOD1 to the inner membrane. This result was reproduced by solubilization of mitoplasts with Triton X-100 followed by proteinase K digestion.
The localization of mito-SOD1 was further investigated by confocal microscopy of lumbar spinal cord sections using a monoclonal antibody against SOD1. A strong signal was detected in both transgenic mito-WTSOD1 and mito-G93ASOD1 spinal cord motor neurons (Fig. 1E, top); low SOD1 fluorescence in non-transgenic controls resulted from endogenous mouse SOD1 expression. At higher magnification, colocalization of SOD1 signal with that of the mitochondrial protein MnSOD could be appreciated (Fig. 1E, bottom), confirming the mitochondrial targeting of mito-SOD1.
Together, these results showed that mito-G93ASOD1 localizes in the mitochondria, where it is anchored to the IM through the transmembrane domain of mitofilin and projects into the IMS. Tethering to the IM prevents the escape of mito-SOD1 into the cytosol, while the flexible arm of mitofilin allows for SOD1 folding and enzymatic activity, as we previously described for mito-WTSOD1 (Fischer et al., 2011).
To assess the molecular conformation of mito-SOD1 we investigated its oligomerization state by blue native gel electrophoresis (BNGE) and Western blot of purified brain mitochondria. In this experiment, the protein complexes in their native state are separated in a nondenaturing gel and then detected by specific antibodies. Both in mito-WTSOD1 and mito-G93ASOD1 mitochondria we detected a major SOD1-reactive band with an estimated size of 100–120 kDa (Fig. 1F). In addition, there was a smear of high molecular weight protein complexes that migrated above the major band. We did not detect a putative 76 kDa dimer of mito-SOD1, suggesting that the protein formed higher order complexes. Note that the BNGE separation conditions did not allow for resolving putative mitoSOD1 monomers (38 kDa) and the endogenous SOD1 monomers or mito-SOD1 dimers (17 kDa and 34 kDa, respectively).
Together, these results showed that transgenic WT and G93A mito-SOD1 form similar high-order oligomers in mitochondria.
Characterization of hemizygous mito-SOD1 mice
To assess whether the expression of mito-SOD1 in hemizygous mice was associated with pathological phenotypes we studied mito-G93ASOD1 and mito-WTSOD1 mice, and compared them to age and gender matched non-transgenic littermates. The three cohorts of mice included approximately equal numbers of males and females. There was no significant change in the lifespan of mito-G93ASOD1 (557 ± 74 d) or mito-WTSOD1 mice (565 ± 98 d), compared with non-transgenic controls (510 ± 50 d) (Fig. 2A). Motor competence, assessed by accelerating rotarod from 2 months until 18 months of age, did not reveal significant impairments in transgenic mice (Fig. 2B). However, the body weight of mito-G93ASOD1 mice was significantly reduced, compared with mito-WTSOD1 and non-transgenic controls (p = 0.05 vs non-transgenic; p = 0.004 vs mito-WTSOD1; ANOVA with repeated measurements, followed by Fisher post hoc test; n = 21–33 mice per group) (Fig. 2C). No significant differences in body weight were found between mito-WTSOD1 and non-transgenic controls.
Figure 2.
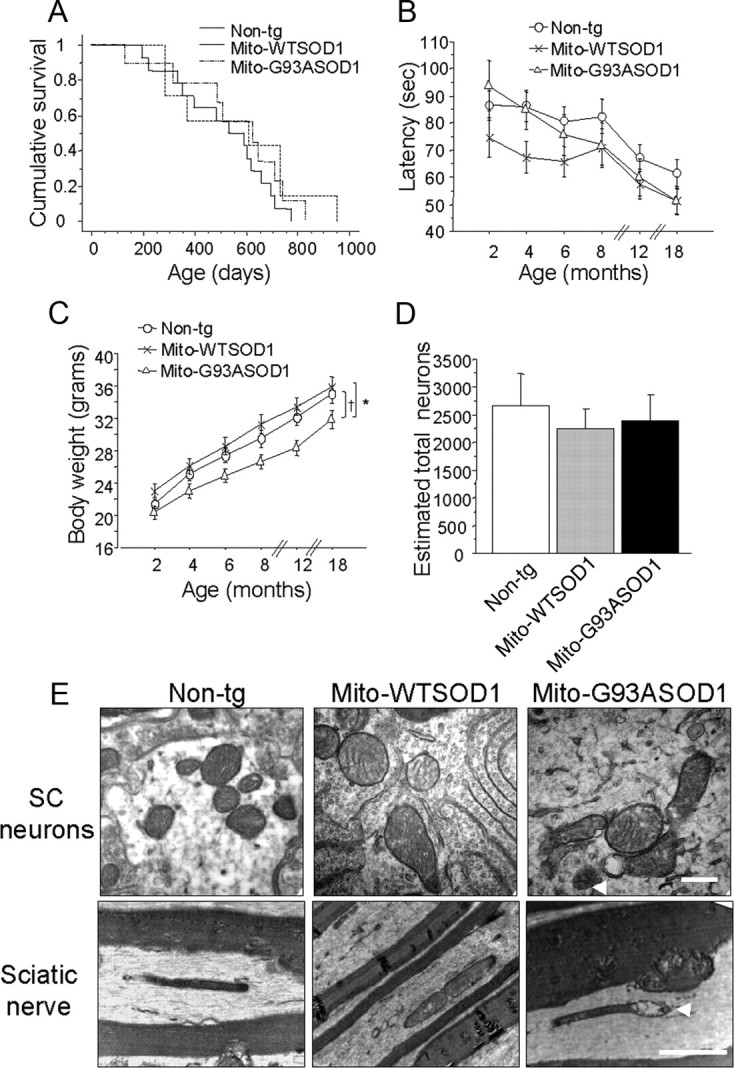
Characterization of hemizygous mito-WTSOD1 and mito-G93ASOD1 mice. A, Kaplan–Meier cumulative survival plot demonstrated that lifespan of mito-G93ASOD1 mice was unchanged compared with control mice. Equal numbers of males and females were studied in each group. n = 4–10 mice per group. B, Absence of significant differences in rotarod performance over time of non-transgenic (n = 33 mice), mito-WTSOD1 (n = 21 mice), and mito-G93ASOD1 (n = 22 mice). C, Analysis of body weight over time of non-transgenic (n = 33 mice), mito-WTSOD1 (n = 21 mice), and mito-G93ASOD1 (n = 22 mice). Mito-G93ASOD1 mice were significantly smaller than non-transgenic and mito-WTSOD1 control mice. *p = 0.0443 versus non-transgenic group, †p = 0.0088 versus mito-WTSOD1, after ANOVA with repeated measurements followed by Fisher post hoc test. D, Neuron cell counts after Nissl staining of lumbar spinal cord sections from 12-month-old non-transgenic, mito-WTSOD1, and mito-G93ASOD1 mice. n = 6–11 mice per group. No significant differences were found. E, Ultrastructural analysis performed by transmission electronic microscopy on spinal cord transversal sections (scale bar, 0.5 μm) and sciatic nerve longitudinal sections (scale bar, 2 μm) from 12-month-old non-transgenic, mito-WTSOD1 and mito-G93ASOD1 mice. Note that structural mitochondrial abnormalities, including cristae disorganization, mitochondrial swelling, and vacuolization (arrowheads), were present in mito-G93ASOD1, but not in non-transgenic and mito-WTSOD1 mice.
Consistent with the lack of a motor phenotype, we did not observe significant changes in the motor neuron counts in the lumbar spinal cord of 12-month-old mito-G93ASOD1 mice (Fig. 2D), compared with non-transgenic and mito-WTSOD1 controls.
Next, to determine whether the presence of mito-SOD1 resulted in mitochondrial ultrastructural changes we studied spinal cord transversal sections and sciatic nerve longitudinal sections from mice at 12 months of age by transmission electron microscopy. Mito-G93ASOD1 mitochondria presented mitochondria cristae disorganization, swelling, and vacuolization, in both spinal cord neurons and sciatic nerve (Fig. 2E, arrowheads). In mito-G93ASOD1 mice, 73% and 79% of the fields analyzed in spinal cord and sciatic nerve, respectively, contained at least one mitochondrial structural abnormality, while non-transgenic and mito-WTSOD1 contained abnormalities in only ∼30% of the fields.
Generation of homozygous mito-G93ASOD1 mice
Since we found no overt indications of neurodegeneration in hemizygous mito-G93ASOD1 mice, we hypothesized that the expression levels of the transgene may be insufficient to cause mitochondrial dysfunction and neurodegeneration, within the timeframe of the lifespan of the mice. This is a common occurrence in mutant SOD1 rodent models, where disease phenotype is highly dependent on the expression of the transgene. Therefore, to increase protein levels, we generated homozygous mito-G93ASOD1 mice.
In homozygotes, quantitative real-time PCR demonstrated an approximately twofold increase of the ratio between mito-G93ASOD1 transgene and β-actin reference gene (hemizygous 0.86 ± 0.073 vs homozygous 1.491 ± 0.145; p = 0.0002 by Student's t test; n = 17–33). Quantitative real-time PCR also determined that there were 45 ± 5, 23 ± 4 and 42 ± 3 copies of the transgenic constructs in mito-WTSOD1, mito-G93ASOD1 hemizygous and homozygous DNA, respectively.
Consistently with the DNA results, we estimated that the amount of mito-G93ASOD1 protein in spinal cord homogenates of homozygous mice, normalized to β-actin, was approximately twice that in the hemizygous, but similar to that in mito-WTSOD1 spinal cords (ratios are indicated at the bottom of each lane in Fig. 3A). We also estimated the protein levels of mito-G93ASOD1 in homozygous and hemizygous purified spinal cord mitochondria, and compared them to the mutant SOD1 content in the mitochondria of high-expressor hemizygous (untargeted) G93A SOD1 mice (strain B6SLJ-Tg-(SOD1*G93A)1Gur/J from The Jackson Laboratory) (Gurney et al., 1994). Since VDAC1 has recently been shown to interact with mutant SOD1 (Israelson et al., 2010), and may be involved in mitochondrial dysfunction, we compared both the amounts of transgenic SOD1 and VDAC, which were similar in homozygous mito-G93ASOD1 and G93A SOD1 mice (Fig. 3B). Finally, there were no differences in the amount of mito-G93ASOD1 expressed in the spinal cord between male and female mice (data not shown).
Figure 3.
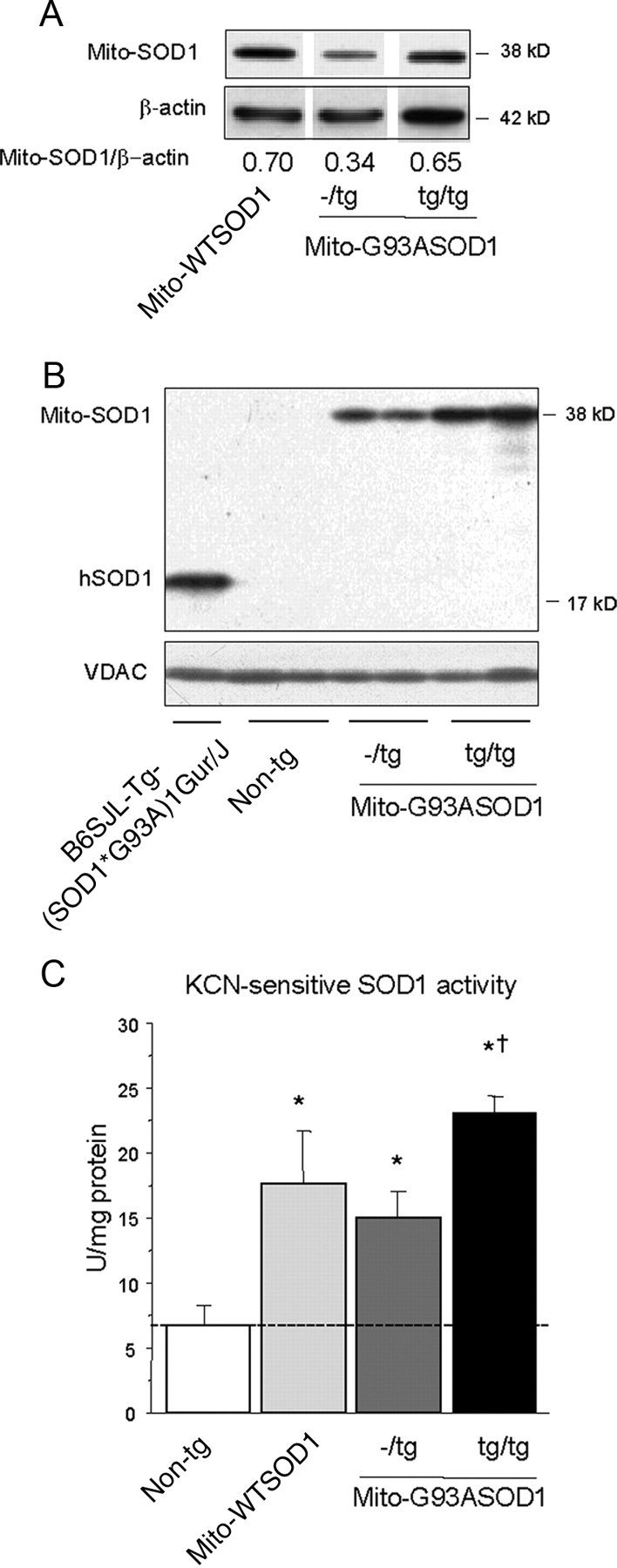
Generation of homozygous mito-G93ASOD1 mice. A, Western blot of transgenic mouse spinal cord lysates (10 μg of protein), using a sheep polyclonal anti-SOD1 and mouse monoclonal anti-β-actin, as normalizing control. All samples were run on the same gel. Mito-SOD1/β-actin intensity ratios are indicated. Note an approximately twofold increase in mito-SOD1 expression in homozygous mice compared with heterozygotes. Homozygous mito-G93ASOD1 levels are similar to hemizygous mito-WTSOD1 controls. B, Western blot of non-transgenic, mito-G93ASOD1 (hemizygous and homozygous), and G93A SOD1 mouse spinal cord mitochondria (20 μg), using a sheep polyclonal anti-SOD1 antibody and a mouse monoclonal anti-VDAC antibody. Note an increase in mito-SOD1 expression in homozygous mice compared with hemizygous. hSOD1, human G93A SOD1. C, KCN-sensitive SOD1 activity in brain mitochondria. n = 3–8 mice per group; versus non-transgenic, *p = 0.001 mito-WTSOD1, *p = 0.005 mito-G93ASOD1−/tg, *p = 0.0001 mito-G93ASOD1tg/tg, respectively; versus mito-G93ASOD1−/tg, †p = 0.006 mito-G93ASOD1tg/tg. The dashed line denotes endogenous SOD activity in mitochondria.
To determine whether the increased amount of transgenic protein in mitochondria corresponded to an increase in enzymatic activity we measured SOD1 activity in purified brain mitochondria. After subtracting the activity present in the mitochondria of non-transgenic brains (6.66 ± 1.52 U/mg protein), which derives from endogenous mouse SOD1, homozygous mito-G93ASOD1 mice showed approximately a twofold increase in SOD1 activity (absolute value 23.17 ± 1.17 U/mg protein; p = 0.0001 vs non-transgenic, p = 0.006 vs hemizygous) compared with hemizygous (14.93 ± 1.99 U/mg protein; p = 0.005 vs non-transgenic), and a 1.5-fold increase compared with mito-WTSOD1 (17.71 ± 3.98 U/mg protein; p = 0.001 vs non-transgenic) (Fig. 3C).
Together, these results showed that in homozygous mito-G93ASOD1 mice mitochondrial SOD1 content was approximately twofold higher than in hemizygous mice, but comparable to the levels of mito-WTSOD1 mice and the untargeted G93A SOD1 mice.
Mitochondrial dysfunction in homozygous mito-G93ASOD1 mice
To determine whether homozygous mito-G93ASOD1 mice (henceforth defined as mito-G93ASOD1) had oxidative phosphorylation defects we measured the rates of oxygen consumption in isolated brain mitochondria, using glutamate and malate as substrates. Mito-G93ASOD1 brain mitochondria from 12-month-old mice did not show significant differences in the rates of resting (state 4) and phosphorylating (state 3) respiration (data not shown).
To evaluate the overall respiratory capacity of mitochondria, we measured the sensitivity of their respiration to uncoupling. Figure 4A shows a representative graph of mitochondrial respiration rate after successive additions of SF6847, in one non-transgenic and one mito-G93ASOD1 mitochondria preparations, respectively. Experiments were paired, comparing mito-WTSOD1 and mito-G93ASOD1 to their respective non-transgenic mitochondria. Upon titration of the uncoupler SF6847, mito-G93ASOD1 mitochondria required significantly less SF6847 to become fully uncoupled than mitochondria from non-transgenic controls (sensitivity in mito-G93ASOD1 151.9 ± 23.1% of non-transgenic; p = 0.0387 after one-tailed, unpaired, Student's t test; n = 8 measurements from 4 independent preparations per group) (Fig. 4B). The higher sensitivity to uncoupling strongly indicated a reduced respiratory capacity of mito-G93A-SOD1 mitochondria. Mito-WTSOD1 mitochondria did not show significant change of sensitivity to uncoupler when compared with non-transgenic mitochondria.
Figure 4.
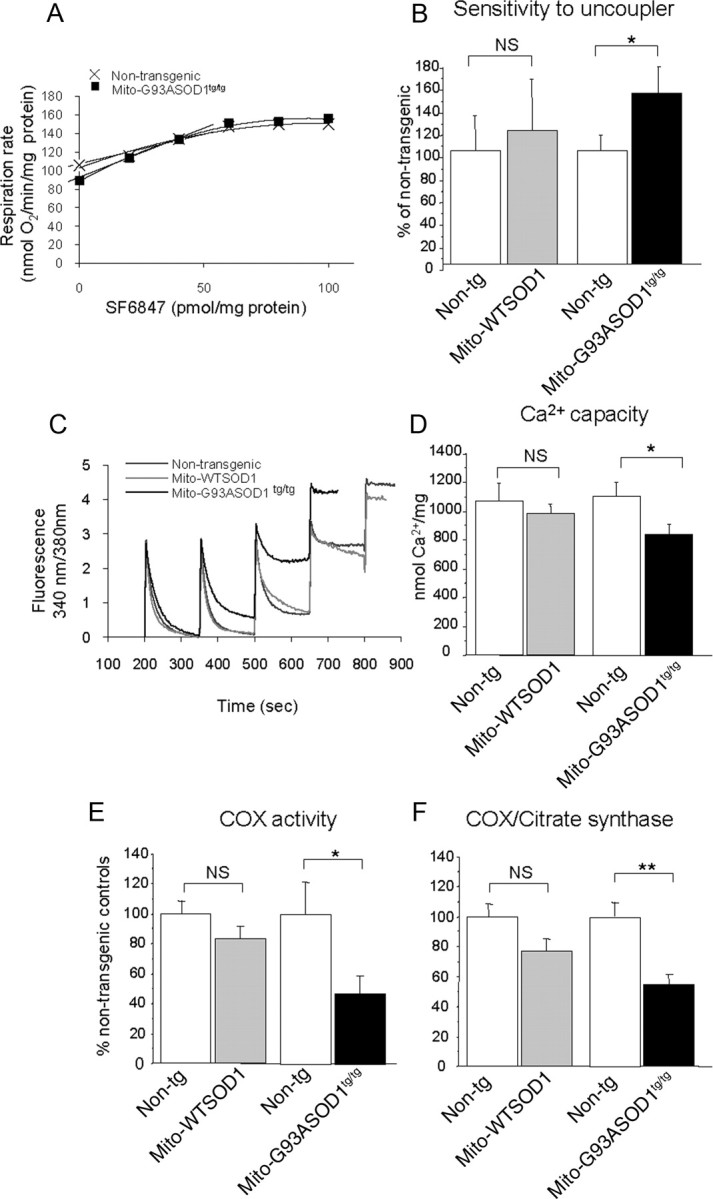
Biochemical assessments on brain purified mitochondria from control and homozygous mito-G93ASOD1 mice. A, Representative graph showing the sensitivity of brain mitochondria respiration to successive additions of 2 nm SF6847, in one non-transgenic and one mito-G93ASOD1 mouse. B, Graph representing the amount of SF6847 required to totally uncouple mitochondria respiration, determined by the respiration rate, in nanomoles of O2 per minute per picomole of SF6847. Means ± SEM are shown for 4 individual brain mitochondria preparations per group. Samples were run in duplicates. NS, not significant; *p = 0.0387 after one-tailed, unpaired, Student's t test versus non-transgenic mitochondria. Mito-G93ASOD1 brain mitochondria are more sensitive to the uncoupling effect of SF6847 than are non-transgenic ones. C, Kinetic traces of mitochondrial Ca2+ uptake in brain mitochondria from 12-month-old mice monitored by Fura 6 fluorescence ratio (340/380 nm excitation, 510 nm emission). The peaks correspond to sequential bolus additions of 25 nmol of CaCl2 (Ca2+). The downward deflections reflect mitochondria Ca2+ uptake. Non-transgenic and mito-WTSOD1 mitochondria took up 4 additions of Ca2+, whereas mito-G93ASOD1 showed loss of Ca2+ uptake after 3 additions. D, Brain mitochondrial Ca2+ capacity expressed in nanomoles of Ca2+ per milligram of protein. Mean ± SEM is shown for 3–7 independent brain mitochondrial preparations. Samples were run in duplicates. *p = 0.019 versus non-transgenic mitochondria. Mito-G93ASOD1 brain mitochondria take up less Ca2+ than mito-WTSOD1 and non-transgenic mitochondria. E, F, COX and CS enzymatic activities measured in spinal cord lysates from 12-month-old non-transgenic and mito-G93ASOD1 mice. Means ± SEM are shown for n = 7–8 mice per group. Samples were run in duplicates or triplicates. *p = 0.05 and 0.007 for COX and COX/CS, respectively, versus non-transgenic mice. Mito-G93ASOD1tg/tg mitochondria have significantly lower COX activity than non-transgenic ones. Mito-WTSOD1 and mito-G93ASOD1 mitochondria were compared with their respective matched non-transgenic controls.
We then investigated the Ca2+ handling capacity in brain mitochondria isolated from 12-month-old mice. A representative experiment of a mitochondrial Ca2+ capacity measurement is shown in Figure 4C. The Ca2+ accumulation and retention capacity of mito-G93ASOD1 was lower than in matched non-transgenic controls (Ca2+ capacity in non-transgenic 1111.10 ± 91.00 nmol/mg protein, mito-G93ASOD1 835.50 ± 72.45; p = 0.03 by ANOVA followed by Fisher post hoc test; n = 8 mitochondrial preparations from 4 individual brains per group) (Fig. 4D). Ca2+ capacity in mito-WTSOD1 brain mitochondria was not different from controls (non-transgenic 1070.50 ± 128.25 nmol/mg protein, mito-WTSOD1 987.50 ± 61.52 nmol/mg protein; n = 6–8 mitochondria preparations from 4 individual brains per group).
We also determined that the activity of COX, the terminal enzyme of the respiratory chain, was reduced by ∼50% in the spinal cord of 12-month-old mito-G93ASOD1 mice, compared with non-transgenic controls (non-transgenic 0.57 ± 0.12 IU/mg protein, mito-G93ASOD1 0.27 ± 0.07; p = 0.05 by Student's t test; n = 7–8 per group) (Fig. 4E). The COX defect was confirmed also when the activity was normalized by the matrix enzyme CS, which was normal in all groups (data not shown), indicating that the loss of COX was not attributable to a decline in mitochondrial mass (COX/CS mito-G93ASOD1 55.0 ± 6.4% of non-transgenic group, p = 0.007 by Student's t test; n = 7–8 per group) (Fig. 4F). COX activity was not impaired in mito-WTSOD1 spinal cord tissues.
These results indicated that mice with targeted mito-G93ASOD1 share similar biochemical impairments with untargeted G93A SOD1 mice, expressing G93A mutant SOD1, which have impaired respiratory (Mattiazzi et al., 2002) and Ca2+ capacity (Damiano et al., 2006), and suggest that mutant SOD1 localized in the IMS is responsible for these bioenergetic defects.
Disease phenotype in mito-G93ASOD1 mice
Figure 5A shows the appearance of an 8-month-old mito-G93ASOD1 female (right) in comparison with a non-transgenic control female (left). At this age, mito-G93ASOD1 mice had a clear loss of body mass accompanied by muscle atrophy, especially in the hind limbs. They also displayed a hunched-back posture, severe ulcerative dermatitis with tail scabbing in females, and penis prolapse in males. The severity of these symptoms did not allow us to follow the evolution of the disease animals for longer than one year, when the animals had to be killed according to Institutional Animal Care Committee guidelines.
Figure 5.
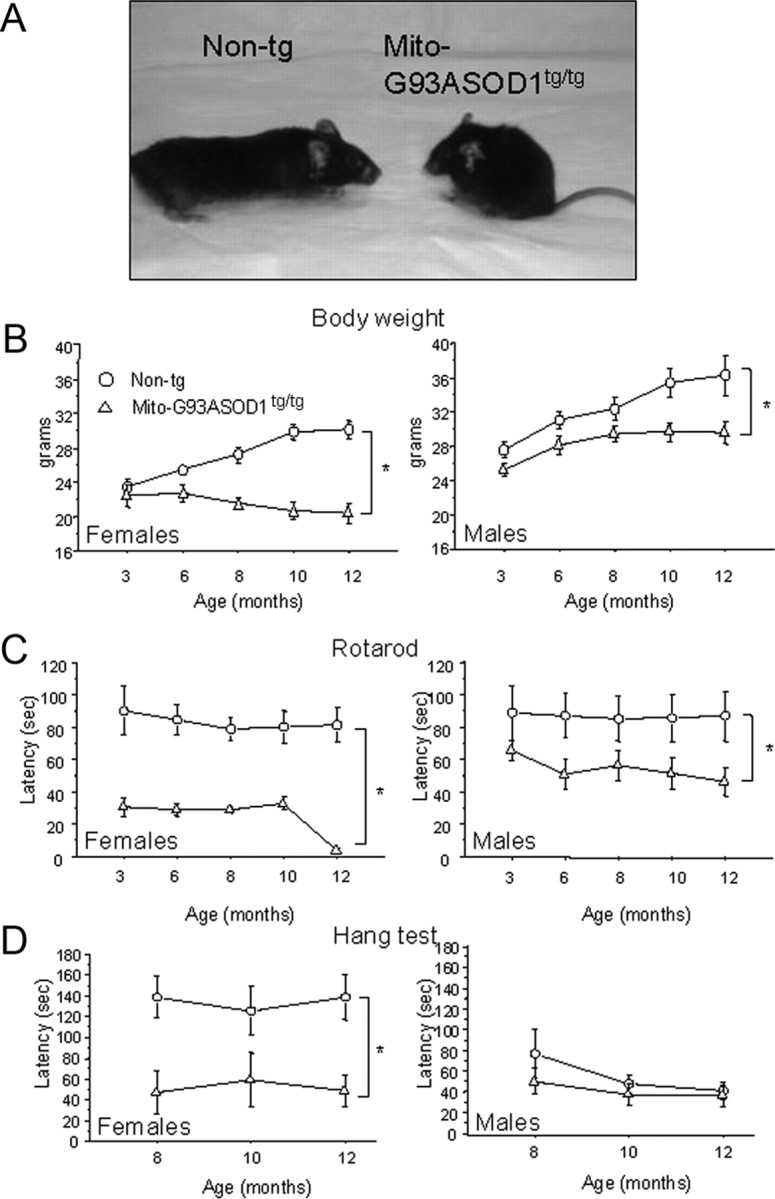
Abnormalities of body weight, motor performance, and grip strength in homozygous mito-G93ASOD1 mice. A, Representative image of 8-month-old mito-G93ASOD1 and non-transgenic female mice. Note the smaller and hunched appearance of the mito-G93ASOD1 mouse. B, Changes over time in body weight of non-transgenic and mito-G93ASOD1 mice. n = 5–7 females and 6–11 males per group. *p = 0.0005 and 0.0185 versus non-transgenic females and males, respectively. C, Changes over time in motor performance of non-transgenic and mito-G93ASOD1 mice. n = 5–7 females and 6–11 males per group. *p = 0.0006 and 0.049 versus non-transgenic females and males, respectively. D, Changes over time in limb muscle strength of non-transgenic and mito-G93ASOD1 mice. n = 6 females and 5–11 males per group. *p = 0.015 versus non-transgenic females. ANOVA with repeated measurements followed by Fisher post hoc test was used.
The disease phenotype was analyzed in male and female mito-G93ASOD1 mice. Both genders presented a progressive decrease in body weight compared with non-transgenic controls (p = 0.0005 and 0.0185, respectively, by ANOVA with repeated measurements followed by Fisher post hoc test; n = 5–7 females and 6–11 males per group) (Fig. 5B), but there were an earlier onset and more severe course in females. Moreover, as early as 3 months of age (females) and 6 months of age (males), mito-G93ASOD1 mice revealed a motor impairment at the accelerating rotarod test, compared with non-transgenic littermates (p = 0.0006 and 0.049, for females and males, respectively, by ANOVA with repeated measurements followed by Fisher post hoc test; n = 5–7 females and 6–11 males per group) (Fig. 5C). In addition, female mutant mice, but not males, had decreased limb muscle strength, as determined by hang test performance, compared with non-transgenic controls (p = 0.015 by ANOVA with repeated measurements followed by Fisher post hoc test) (Fig. 5D).
These results indicated that mito-G93ASOD1 mice developed a motor disease, which was more severe in females, and was accompanied by systemic symptoms.
Neurodegeneration in mito-G93ASOD1
To determine whether the biochemical and phenotypical abnormalities observed in mito-G93ASOD1 mice were associated with CNS abnormalities, we studied symptomatic mice at 8 months of age. We found that brains from mito-G93ASOD1 mice were smaller (Fig. 6A), weighing ∼20% less than non-transgenic controls (non-transgenic 528.9 ± 19.2 mg, mito-G93ASOD1 418 ± 26.6 mg; p = 0.015 by Student's t test; n = 4 per group) (Fig. 6B). Brains from mito-WTSOD1 mice did not show significant weight difference compared with respective non-transgenic brains. Nissl staining of coronal brain sections from mito-G93SOD1 mice showed a decrease in the density of neurons in the cortex region that included the motor cortex (Fig. 6C). No obvious abnormalities were observed in the striatum and cerebellum (data not shown). These findings suggested neuronal loss in the cortex of mito-G93ASOD1 brains. Mito-WTSOD1 coronal brain sections did not appear different from non-transgenic ones.
Figure 6.

Brain abnormalities in mito-G93ASOD1 mice. A, Representative images of the brains of 8-month-old non-transgenic and mito-G93ASOD1 males. Scale bar, 0.5 cm. B, Brain weight quantification of 8-month-old mito-WTSOD1, mito-G93ASOD1, and relative matching non-transgenic controls. Data are shown as percentage of non-transgenic controls. n = 4–7 brains per group. *p = 0.015 versus non-transgenic group. C, Representative images of Nissl-stained brain coronal sections. Top shows low magnification of an area including the retrosplenial/motor cortex. Scale bar, 100 μm. Bottom shows higher magnification of a region located midway between the edge of the cortex and the cingulum (dashed square). Scale bar, 25 μm.
We next assessed alterations in the spinal cord of mito-G93ASOD1 mice. Nissl staining revealed a decrease in motor neuron cell bodies in mito-G93ASOD1 mice (Fig. 7A). Cell quantification revealed a significant decrease (by ∼35%) in the counts of motor neurons with diameter larger than 30 μm, in the anterior horn of lumbar spinal cords of mito-G93ASOD1 mice, compared with non-transgenic controls (2306 ± 196 non-transgenic, 1518 ± 111 mito-G93ASOD1; p = 0.008 Student's t test; n = 5 per group) (Fig. 7B). Spinal cord motor neuron counts in mito-WTSOD1 mice did not show significant difference compared with non-transgenic mice. We also observed a moderate increase in GFAP staining in the lumbar spinal cord of mito-G93ASOD1 mice, compared with non-transgenic and mito-WTSOD1 tissues (Fig. 7C), suggestive of mild reactive astrocytosis.
Figure 7.
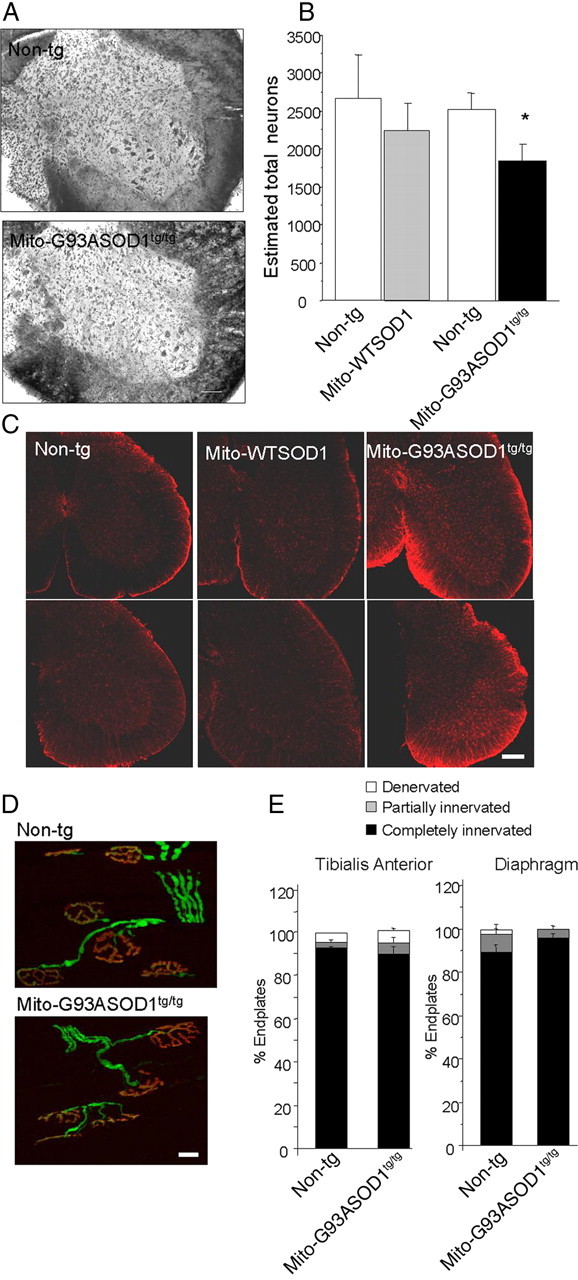
Abnormalities in spinal cord of mito-G93ASOD1 mice, in the absence of muscle denervation. A, Representative images for Nissl-stained lumbar spinal cord coronal sections. Scale bar, 100 μm. B, Quantification of the number of neurons in serial sections of the lumbar spinal cord sections. Neurons larger than 30 μm were counted on every sixth half sections. n = 6–11 mice per group. *p = 0.008, mito-G93ASOD1 versus non-transgenic group. C, Increased astrogliosis in mito-G93ASOD1 mice. Spinal cord sections from 8-month-old mice were stained with an anti-GFAP antibody. Images from two different mice in each group are shown. Note an increase in GFAP staining in mito-G93ASOD1 mice. Scale bar, 200 μm. D, Representative confocal microscopy projections of tibialis anterior neuromuscular junctions of 8-month-old non-transgenic and mito-G93ASOD1 mice. Motor axons (PGP 9.5) are labeled in red and endplates (α-bungarotoxin) in green. Scale bar, 20 μm. E, Quantification of totally innervated, partially innervated, and denervated endplates in tibialis anterior and diaphragm muscles. n = 3 mice per group.
Finally, to determine whether the decrease in the number of spinal cord motor neurons was associated with muscle denervation the neuromuscular junctions in the tibialis anterior and diaphragm muscles were evaluated by the overlap between the presynaptic marker PGP9.5 and the postsynaptic α-bungarotoxin fluorescent labeling (Fig. 7D). Surprisingly, quantification of the innervation in both muscles revealed no significant differences between mito-G93ASOD1 (89.2 ± 2.9% innervated) and non-transgenic littermate mice (93 ± 1.2% innervated) (Fig. 7E).
These findings indicated that, despite the moderate loss of motor neurons in the spinal cord, there was no overt loss of neuromuscular junctions, at a time when the mice exhibited motor symptoms and muscle atrophy, suggesting that the motor impairment was not associated with a pattern of muscle denervation, which is typically observed in ALS.
Skeletal muscle and cardiac abnormalities in mito-G93ASOD1
Since mito-SOD1 transgene driven by the prion promoter was expressed also in skeletal muscle (Fischer et al., 2011), and mito-G93ASOD1 mice had muscle weakness in the absence of denervation, we investigated skeletal muscle pathology in the tibialis anterior. This muscle contains a high proportion of oxidative fibers rich in mitochondria, and was shown to be an early site of denervation in the G93A SOD1 mouse (Fischer and Glass, 2007). At 8 months of age, muscles from both female and male mito-G93ASOD1 mice were significantly smaller, weighing ∼30 and 40% less than non-transgenic and mito-WTSOD1 muscles, respectively (females and males, respectively: 53.4 ± 2.2 mg and 68.5 ± 3.8 mg of non-transgenic, 62.7 ± 11.9 mg and 81.5 ± 6.4 mg of mito-WTSOD1, 34.5 ± 6.5 mg and 47.0 ± 4.8 mg of mito-G93ASOD1; p = 0.0276 and 0.0047 vs non-transgenic and p = 0.0101 and 0.0002 vs mito-WTSOD1, by ANOVA followed by Fisher post hoc test; n = 3–7 females and 6–11 males per group) (Fig. 8A).
Figure 8.

Muscle abnormalities in mito-G93ASOD1 mice. A, Decrease in the weight of tibialis anterior muscles from 12-month-old mito-G93ASOD1 mice compared with non-transgenic and mito-WTSOD1 mice. n = 3–7 females and 6–11 males per group. *p = 0.0276 and 0.047 versus non-transgenic female and non-transgenic male groups, respectively. †p = 0.0101 and 0.0002 versus mito-WTSOD1 female and mito-WTSOD1 male groups, respectively. B, Representative images of succinate dehydrogenase staining of tibialis anterior sections from non-transgenic and mito-G93ASOD1 8-month-old mice. Arrows indicate strongly stained oxidative fibers (type I); asterisks indicate weakly stained glycolytic fibers (type II). Scale bar, 50 μm. C, Quantification of the area of oxidative and glycolytic fibers from non-transgenic and mito-G93ASOD1 mice. n = 2510 ± 646 oxidative fibers and 1231 ± 342 glycolytic fibers, from 2 mice per group. *,†p = 0.0001 versus non-transgenic group and mito-WTSOD1, respectively. D, Quantification of the total number of oxidative and glycolytic fibers from non-transgenic and mito-G93ASOD1 mice. n = 2510 ± 646 oxidative fibers and 1231 ± 342 glycolytic fibers, from 2 mice per group. Note that the total number of fibers was unchanged, although the muscle fiber areas were smaller in mito-G93ASOD1 mice compared with both non-transgenic and mito-WTSOD1 groups. E, Transmission electronic microscopy images of the tibialis anterior of 8-month-old non-transgenic and homozygous mito-G93ASOD1 mice. Note the presence of mitochondrial abnormalities (arrows), which included cristae disorganization and moderate mitochondrial swelling, in mito-G93ASOD1 muscles. Scale bar, 0.5 μm.
Muscle H&E staining did not reveal evidence of muscle fiber necrosis or regeneration, and no evidence of chronic neuropathic changes, including angular fibers or fiber type grouping, was observed, consistent with the finding that mito-G93ASOD1 muscles were normally innervated.
We then examined SDH histoenzymatic activity in the tibialis anterior (Fig. 8B). SDH is a mitochondrial enzyme involved in oxidative phosphorylation, which can be used to assess whether muscle fibers are more oxidative (type I fibers, strong SDH staining) or more glycolytic (type II fibers, weak SDH staining). By measuring the fiber areas in muscle cross sections, we found that both oxidative and glycolytic fibers were significantly smaller in mito-G93ASOD1 muscles, compared with non-transgenic and mito-WTSOD1 ones (Fig. 8C; for oxidative fibers: 1888.9 ± 15.4 μm2 non-transgenic, 2018.2 ± 18.9 μm2 mito-WTSOD1, and 1716.9 ± 19.3 μm2 mito-G93ASOD1, n = 1864–3157; for glycolytic fibers: 2632.8 ± 20.1 μm2 non-transgenic, 2723.0 ± 30.8 μm2 mito-WTSOD1, and 2288.0 ± 24.6 μm2 mito-G93ASOD1, n = 966–1615; p < 0.0001 by ANOVA followed by Fisher post hoc test). No significant differences in the number of fibers were found for either fiber type (Fig. 8D).
Electron microscopy studies of the tibialis anterior from mito-G93ASOD1 8-month-old females revealed mitochondrial abnormalities (Fig. 8E, arrows), including cristae disorganization and mild mitochondria swelling, suggesting that mutant SOD1 targeted to the IMS causes disruption of mitochondrial structure also in muscle cells.
COX activity in muscle homogenates was unaffected in mito-G93ASOD1 mice (data not shown), indicating that mitochondrial morphological abnormalities in the muscle of these mice were not associated with COX deficiency.
Because the prion promoter drives the expression of mito-SOD1 also in the heart, we studied heart function in a small subset of 12-month-old mice (n = 2 males per group). The left ventricular ejection fraction was decreased in mito-G93ASOD1 mice (73.7 ± 6% non-transgenic, 42.7 ± 14.6% mito-G93ASOD1) and the heart size was also reduced (117.5 ± 9.6 mg of non-transgenic, 58 ± 6.6 mg of mito-G93ASOD1). Although the heart studies need to be extended to a larger number of animals, they suggest that cardiac dysfunction may participate in causing the disease phenotype in mito-G93ASOD1 mice.
Together, these findings suggest that mito-G93ASOD1 induces skeletal muscle atrophy with mitochondrial abnormalities and muscle heart dysfunction, which may contribute to the muscle weakness and the body weight loss observed in the mutant mice.
Discussion
A portion of cellular SOD1 localizes in mitochondria (Okado-Matsumoto and Fridovich, 2001; Sturtz et al., 2001; Vijayvergiya et al., 2005), and it was proposed that mutant SOD1 in the mitochondrial IMS could be responsible for mitochondrial alterations and contribute to motor neuron toxicity in SOD1 familial ALS (Xu et al., 2004; Hervias et al., 2006; Magrané et al., 2009). Our group and others showed that mutant SOD1 expressed in neuronal mitochondria induced toxic effects and mitochondrial dynamics abnormalities in in vitro models (Cozzolino et al., 2009; Magrané et al., 2009). Thus, the goal of this study was to assess the pathogenic role of mutant SOD1 localized in the IMS in vivo.
The strategy to target human SOD1 selectively to the IMS had already been tested successfully with the WT SOD1 (Fischer et al., 2011). Here, we demonstrated that mito-G93ASOD1 was abundant in cell types where the prion promoter drives its expression, notably neurons, astrocytes, skeletal muscle, and heart, and correctly targeted to mitochondria. We also confirmed that mito-G93ASOD1 was exclusively localized in the IMS and anchored to the outer face of the inner membrane. Mito-G93ASOD1, similar to mito-WTSOD1 (Fischer et al., 2011), was enzymatically active in mitochondria, where it formed high-order multimers.
The pathogenic effects of mito-G93ASOD1 were dependent on expression levels. Thus, hemizygous mice had mitochondrial ultrastructural abnormalities in neurons, but did not develop an overt disease phenotype, except for a mild reduction in body weight, and had a normal lifespan. On the other hand, mito-G93ASOD1 mice, which contained approximately twice as much mutant SOD1 protein in mitochondria, displayed a progressive disease, characterized by motor impairment, muscle weakness and atrophy, brain cortex atrophy, cardiac insufficiency, and body weight loss.
The clinical phenotype was more severe in females. The reasons for the gender differences, despite similar levels of mito-G93ASOD1 expression, remain to be elucidated. However, the observation is intriguing, because several mouse lines expressing mutant SOD1 present a more severe phenotype with shorter lifespan in males (Veldink et al., 2003; Heiman-Patterson et al., 2005), suggesting that IMS SOD1 may affect genders in the opposite way than the cytoplasmic counterpart.
Mito-WTSOD1 mice contained mitochondrial levels of the protein comparable to the mito-G93ASOD1 mice and did not manifest a disease phenotype, indicating that the pathology did not simply derive from expression of SOD1 targeted to mitochondria. Furthermore, mito-G93ASOD1 mice contained levels of mitochondrial SOD1 in the CNS comparable to those found in “untargeted” G93A SOD1 mice, allowing for a comparison of the mitochondrial pathogenic effects in the two models. Mito-G93ASOD1 CNS mitochondria had impaired respiratory chain function and decreased Ca2+ capacity, like in G93A SOD1 mitochondria (Mattiazzi et al., 2002; Damiano et al., 2006; Son et al., 2008), suggesting that similar molecular mechanisms leading to mitochondrial bioenergetic dysfunction may be involved. In addition, mito-G93ASOD1 mice showed a decreased number of spinal cord motor neurons, albeit not as severe as the G93A SOD1 mice. On the other hand, there were significant phenotypic differences between the two disease models. Mito-G93ASOD1 had a much longer lifespan than high-expressor G93A SOD1 mice (Gurney et al., 1994), and despite the presence of a motor impairment did not die of paralysis. Instead, mito-G93ASOD1 mice developed symptoms affecting different tissues, such as skin and genital organs, which are not commonly observed in G93A SOD1 mice.
Muscle denervation is a typical pathological feature that develops early on in the disease course of G93A SOD1 mice, before the animals start displaying motor defects (Fischer et al., 2004). Instead, mito-G93ASOD1 mice did not show muscle denervation, even at a time when they had motor impairment and muscle weakness. This remarkable difference suggests that the presence of mutant SOD1 in the mitochondrial IMS is detrimental for the animals, but does not cause some of the typical pathological features of ALS. Our results also suggest that the mitochondrial IMS component of mutant SOD1 affects various tissues in ways that could not be phenotypically appreciated in the G93A SOD1 mouse. For example, the effects of mutant SOD1 localized in the IMS of skeletal muscle mitochondria could be masked by the progressive denervation that takes place in G93A SOD1, but not in mito-G93ASOD1 mice.
A caveat of comparing the two models is in the different tissue expression pattern of the transgenes. In the G93A SOD1 models, the transgene is usually expressed under the control of the endogenous promoter, while mito-G93ASOD1 is driven by the prion promoter. Although at least one mouse model of SOD1 familial ALS has been generated using the prion promoter, it did not carry the G93A mutation (Wang et al., 2005). It is well known that mutant SOD1 pathogenesis is non-cell autonomous (Ilieva et al., 2009); thus, it is possible that different modulation of the expression of the prion promoter in certain cell types has contributed to modifying the pathogenic process, in the mito-G93ASOD1 mouse.
The interpretation of the findings reported in this study allows us to draw some conclusions on the effects of G93A mutant SOD1 in the mitochondrial IMS. First, G93A mutant, but not WT, SOD1 in the IMS is sufficient to cause mitochondrial bioenergetic impairments. COX activity defects may result from the interactions of mutant SOD1 in the IMS with assembly factors and prosthetic groups biosynthesis, which are necessary for the correct function of the enzymatic complexes (Son et al., 2008). Ca2+ capacity can be decreased by impaired bioenergetics and by interactions with key components of Ca2+ uptake regulation that may affect the susceptibility to mitochondrial permeability transition. However, because of its internal localization, it is likely that mutant SOD1 in the IMS does not affect a set of mitochondrial component of the outer membrane that have been recently been shown to be affected by interactions with mutant SOD1, such as Bcl2 (Pedrini et al., 2010) and VDAC (Israelson et al., 2010). Furthermore, extra-mitochondrial (cytosolic) mutant SOD1 may also affect mitochondria by interfering with the mechanisms regulating mitochondrial transport, such as kinesin and dynein motors (Ström et al., 2008; Magrané and Manfredi, 2009).
The loss of motor neurons in the spinal cord and the muscle, heart, and brain abnormalities accompanied by mitochondrial bioenergetic dysfunction indicate that the mitochondrial damage induced by G93A mutant SOD1 in the IMS is sufficient to cause widespread pathology per se. Therefore, the evidences emerging from our novel mitoSOD1 disease model suggest that the mitochondrial IMS component of mutant SOD1 may contribute to the pathogenesis of neurodegeneration in SOD1 familial ALS. However, the lack of denervation and the relatively slow disease course, even in homozygous mice, indicate that other, extra-mitochondrial, toxic effects of mutant SOD1 are necessary to cause the full-fledged ALS phenotype.
Footnotes
This work was supported by grants from The Robert Packard Center for ALS Research (to G.M. and J.M.), the Muscular Dystrophy Association (to J.M.), and the National Institutes of Health (Grants RO1-NS051419 and RO1-NS062055 to G.M.). We are grateful to Dr. Noel Y. Calingasan (Weill Cornell Medical College) for his help on the stereological analysis of spinal motor neurons.
The authors declare no competing financial interests.
References
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Pesaresi MG, Amori I, Crosio C, Ferri A, Nencini M, Carrì MT. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid Redox Signal. 2009;11:1547–1558. doi: 10.1089/ars.2009.2545. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc Natl Acad Sci U S A. 1992;89:10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carrì MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Igoudjil A, Magrané J, Li Y, Hansen JM, Manfredi G, Glass JD. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011;134:196–209. doi: 10.1093/brain/awq314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1–G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hervias I, Beal MF, Manfredi G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve. 2006;33:598–608. doi: 10.1002/mus.20489. [DOI] [PubMed] [Google Scholar]
- Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet. 2008;17:3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guégan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug D, Rabani J, Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- Magrané J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1615–1626. doi: 10.1089/ars.2009.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009;18:4552–4564. doi: 10.1093/hmg/ddp421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19:2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2010;1802:45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Leary SC, Romain N, Pierrel F, Winge DR, Haller RG, Elliott JL. Isolated cytochrome c oxidase deficiency in G93A SOD1 mice over-expressing CCS protein. J Biol Chem. 2008;283:12267–12275. doi: 10.1074/jbc.M708523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ström AL, Gal J, Shi P, Kasarskis EJ, Hayward LJ, Zhu H. Retrograde axonal transport and motor neuron disease. J Neurochem. 2008;106:495–505. doi: 10.1111/j.1471-4159.2008.05393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast cu,zn-superoxide dismutase and its metallochaperone, ccs, localize to the intermembrane space of mitochondria. a physiological role for sod1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldink JH, Bär PR, Joosten EA, Otten M, Wokke JH, van den Berg LH. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord. 2003;13:737–743. doi: 10.1016/s0960-8966(03)00104-4. [DOI] [PubMed] [Google Scholar]
- Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM, La Perle K, Vaughan CJ, O'Hagan A, Bennett KR, Meyer BJ, Legius E, Karttunen M, Norio R, Kaariainen H, Lavyne M, Neau JP, Richter G, Kirali K, Farnsworth A, et al. Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc Natl Acad Sci U S A. 2004;101:14222–14227. doi: 10.1073/pnas.0405535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Yang L, Manfredi G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007;80:155–171. doi: 10.1016/S0091-679X(06)80007-5. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C, Copeland NG, Jenkins NA, Borchelt DR. Somatodendritic accumulation of misfolded SOD1–L126Z in motor neurons mediates degeneration: {alpha}B-crystallin modulates aggregation. Hum Mol Genet. 2005;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jung C, Higgins C, Levine J, Kong J. Mitochondrial degeneration in amyotrophic lateral sclerosis. J Bioenerg Biomembr. 2004;36:395–399. doi: 10.1023/B:JOBB.0000041774.12654.e1. [DOI] [PubMed] [Google Scholar]