

Abstract
Chemotaxis is essential for shaping immune responses and chemokine-receptor antagonists are now being evaluated as therapies for various inflammatory and autoimmune diseases. However, the dysregulation of chemotaxis in autoimmune disease may involve both promotion and inhibition of B-cell migration. This review focuses on the disparate mechanisms by which two inflammatory cytokines that have been associated with autoimmune disease, namely interferon-α (IFNα) and interleukin-17 (IL-17), may regulate B-cell migratory responses. Chemotactic responses play a key role in orchestrating the cell-cell interactions in the germinal centers (GCs). This process involves ongoing shuttling of the antigen-carrying B cells between the marginal zone and the GCs. We have shown that in autoimmune BXD2 mice, the migration of marginal zone precursor B cells is promoted by high levels of interferon (IFN)-α produced by plasmacytoid dendritic cells in the marginal sinus that antagonize the activity of the S1P1 chemokine receptor. In contrast, within the GCs, interleukin-17A (IL-17A) upregulates the expression of regulators of G protein signaling (RGS) in B cells to desensitize the G protein-coupled receptor (GPCR) signaling pathway of CXCL12 and CXCL13 chemokines. This provides a prolonged stable interaction of B and T cells in the GC that induces high levels of activation-induced cytidine deaminase (AICDA) thereby enabling development of pathogenic autoantibody-producing B cells.
Introduction
Chemotaxis is essential not only to promote the influx of cells to a site of immune responses but also for orchestrating the motion of immune cells during lymphoid organogenesis (Bende et al., 2009; Fukuyama et al., 2006). For the most part, the analysis of the mechanisms by which chemotaxis shapes immune responses has focused on the expression and activity of the chemokines and their receptors. Approximately 50 chemokines have now been identified in humans (Schaerli & Moser, 2005) and dysregulation of the activity of some of these chemokines or their receptors has been associated with specific autoimmune and rheumatic diseases (Koch, 2005; Loetscher & Moser, 2002; Nanki et al., 2009; Patel & Haynes, 2001; Shadidi, 2004; Szekanecz et al., 2010).
The objective of this review, however, is to highlight the importance of a second level of regulation of migration in the shaping of immune response; namely, the regulation of chemokine receptor signaling by cytokines. This is illustrated by a summary of the recent insights into the regulation of B-cell chemotaxis by interferon-α (IFNα) and interleukin-17 (IL-17) through modification of the G protein-coupled receptor (GPCR) chemotactic responses and how localized elevations in the levels of these proinflammatory cytokines can act to promote germinal center (GC) function (Hsu et al., 2008; Wang et al., 2011; Wang et al., 2010). It is well established that the trafficking of B cells in the lymphoid organs and target tissues is a mechanism of regulation of B-cell activation and differentiation (Bende et al., 2009; Hart et al., 2011; Hoek et al., 2010). One unique aspect of B cell trafficking in the regulation of immune responses is that B cells can serve as an antigen delivery system that transports blood borne antigens into the follicular dendritic cell (FDC) network region of the spleen (Cinamon et al., 2008). This regulates the formation of the sites where high affinity antibody-forming B-cell differentiation occurs, i.e., the GCs. These migratory responses are extremely dynamic in that they involve ongoing shuttling of the B cells between the different anatomic sites and rely on rapid “stop-go” switches that regulate the duration of retention in the vicinity of specific cells vs. directed migration to other locations. Our analyses of GC formation in BXD2 autoimmune mice have revealed that IFNα acts locally in the marginal zone to promote the release of the antigen-transporting and highly costimulatory marginal zone precursor B cells by affecting the activity of S1P1, a GPCR receptor (Goetzl et al., 2004; Wang et al., 2011). In contrast, the elaboration of IL-17 by the CD4+ T cells in the follicles (Hsu et al., 2008) may promote the retention of marginal zone precursor B cells in the vicinity of the FDC network in the GC light zone. This effect is mediated by the IL-17-mediated upregulation of the intracellular regulators of G-protein signals (RGS) that modify the response of the receptors for CXCL12/CXCL13, which also are GPCR receptors (Shi et al., 2002). The net consequence is the development of spontaneous GCs, production of pathogenic autoantibodies and development of autoimmune disease.
Migration and Differentiation of B cells in Germinal Centers
GCs (Figure 1) are specialized structures that develop within the B-cell follicles of secondary lymphoid tissues, including lymph nodes, spleen, tonsils, and the Peyer's patches of mucosal-associated lymphoid tissues. Ectopic GCs can also be found in some patients with autoimmune diseases (Jonsson et al., 2007; Xu et al., 2009; Zuckerman et al., 2010). Initiation of the formation of GCs is characterized by polarization of secondary follicles into rapidly proliferating centroblasts in the T-cell proximal “dark” zone and slowly proliferating centrocytes in the T cell distal “light” zone (Figure 1). There is a high density of FDCs in the “light zone” of centrocytes (Figure 1). These FDCs have Fcγ and complement receptors that trap and display antigen as immune complexes (Barrington et al., 2002; Qin et al., 1998). It has been reported that the enzyme that is responsible for both the generation of diversity through hypermutation and class switching (activation-induced cytidine deaminase, AICDA) is expressed primarily by germinal cell B cells and that only a few AICDA+ cells can be identified among marginal zone B cells (Willenbrock et al., 2005). It is well established that the autoimmune spleen differs from normal spleen and normal human tonsil in that there is spontaneous development of GCs with increased T- and B-cell interactions (Melchers & Rolink, 2006).
Figure 1.
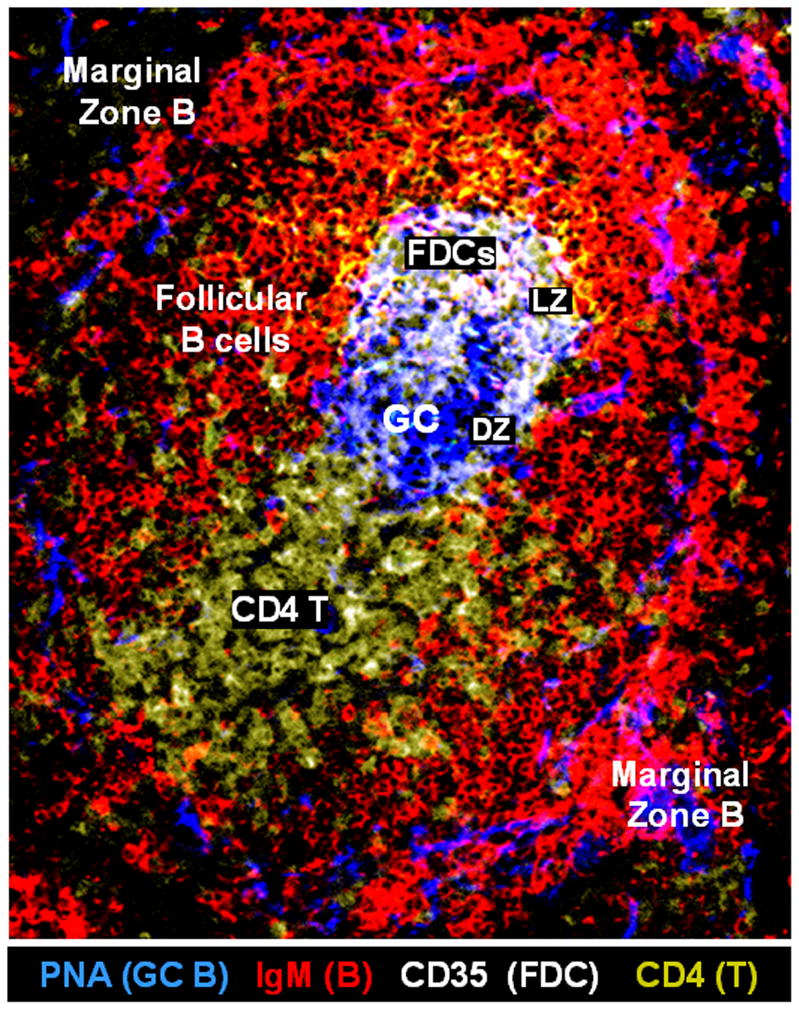
Anatomic structure of a spontaneous germinal center (GC) in the spleen of a representative BXD2 mouse. Confocal image of a representative BXD2 spleen section stained with anti-IgM (red), anti-CD35 (white), and anti-CD4 (yellow) antibodies, and peanut agglutinin (PNA) (blue). Anti-CD35 antibodies are used to stain follicular dendritic cells (FDCs) that express CD35 (DZ: dark zone; FDCs: follicular dendritic cells; GC: germinal center; LZ: light zone; PNA: peanut agglutinin).
The migration and sequential repositioning of the immune cells within the GCs is essential to their formation, stabilization and function. Specific migration events of importance include the migration and positioning of the FDCs, CD4-positive T cells, and B cells near the FDC network region at the outer margin of the GC light zone (Pereira et al., 2010). The delivery of antigens and immune complexes to, and captured by, the FDCs also has been shown to be of importance in the production of the antibody-producing B cells (Cinamon et al., 2008; Phan et al., 2009). It is well established that secretion of the chemokine CXCL13 by the FDCs plays an important role in the recruitment of both CXCR5+ B cells and CXCR5+ CD4 T cells into the FDC network region (Schaerli et al., 2000). The recruitment, localization, and retention of the immune cells in this region is of critical importance as this is where the interactions of antigen-bearing cells, highly costimulatory cells and CD4 T cells occur (Meyer-Hermann & Maini, 2005; Wu et al., 2008).
RGS and Regulation of an Immune Response
All of the CXC and CCR chemokines receptors are GPCRs (Moratz et al., 2004a). In a typical mammalian GPCR-regulated chemotaxis pathway, signaling begins with the binding of a chemoattractant to the extracellular surface of the GPCR. Subsequently, the receptor generates a transmembrane signal that enables its cytoplasmic region to replace guanosine triphosphate (GTP) for guanosine diphosphate (GDP). Binding of G protein alpha subunit (Gα) with GTP enables the dissociation from Gβγ subunits. Each active subunit, Gα:GTP and free Gβγ, independently initiates downstream signals (Siderovski & Willard, 2005; Sjogren et al., 2010). The activity of the heterotrimeric G protein can be modified by a family of proteins termed regulators of G protein signaling (RGS) (Lippert et al., 2003). These proteins increase the intrinsic GTPase activity of Gα (i.e., act as GTPase-activating proteins or GAPs) as well as interfere with the interaction of the activated Gα subunits with their down-stream effectors. As described below, this mechanism can play a role in desensitizing chemotaxis. More than 30 members of the RGS protein family have now been identified in humans (Siderovski & Willard, 2005).
Three different RGS proteins, RGS1, RGS13, and RGS16, have been implicated in the regulation of the B-cell migration response (Estes et al., 2004; Hsu et al., 2008; Moratz et al., 2004b; Shi et al., 2002). In normal mice, Rgs1 was found to be expressed exclusively in CD21+CD23+ B220+ follicular but not CD21hiCD23lowB220+ marginal zone or CD21dimCD23−B220+ transitional B cells (Moratz et al., 2004b). Analysis of B-cell responses in RGS1-deficient mice indicated an enhanced migration response of B cells to CXCL12 and CXCL13 (Moratz et al., 2004b) and enhanced GC formation in both the naïve and immunized Rgs1−/ − mice. These findings suggest that RGS1-induced desensitization of follicular B cells to the chemoattractants CXCL12 and CXCL13 is important for maintenance of B-cell homeostasis (Moratz et al., 2004b).
RGS13 is highly expressed by GC B cells (Shi et al., 2002) and has been implicated as the key RGS in the regulation of the GPCRs of CD4+ T cells and B cells in the GCs (Estes et al., 2004). The expression of Rgs13 has been localized to the GC regions of mouse spleens and Peyer's patches and to the thymus medulla by in situ hybridization with sense and anti-sense probes (Shi et al., 2002). RGS13-expressing cells have been identified in the light zone of the GC using an affinity-purified RGS13 antibody (Shi et al., 2002). The RGS13 was found to desensitize signaling through the chemokine receptors CXCR4 and CXCR5. Moreover, prolonged CD40 signaling up-regulated RGS13 expression in human tonsil B cells (Shi et al., 2002). Recently, it was noted that nuclear expression of RGS13 might affect cAMP signaling in that RGS13 can interact with the pCREB-CBP/p300 complex to reduce both pCREB binding to the CRE and its association with CBP/p300 independently of the G protein (Xie et al., 2008).
RGS16 has been implicated in the regulation of the migration response of T cells in response to CXCR4, CCR3, and CCR5, but not CCR2 or CCR7 (Lippert et al., 2003) and is expressed by both GC CD4 T and B cells including those that are in close proximity to FDCs (Estes et al., 2004). Notably, the expression of RGS16 is highly dependent upon local environmental factors and, in an in vitro coculture condition, the presence of FDCs can alter the migration response of both GC and non-GC T cells (Estes et al., 2004). Despite the strong anatomic association of RGS13 and RGS16 in GC T cells and B cells, it has not yet been shown if genetically disrupted expression of RGS13 or RGS16 affects the development of spontaneous or antigen-stimulated GCs.
S1P1-Regulation of Lymphocyte Egress
In contrast to CXCL12 and CXCL13, and RGS1, RGS13, and RGS16, which appear to regulate the influx and retention of B cells to follicular centers, the egress of B cells within and from a lymphoid organ is mediated by a separate set of GPCRs known as the sphingosine 1-phosphate (S1P) receptors (Pappu et al., 2007; Schwab & Cyster, 2007). There are five S1P receptors, S1P1-S1P5, with S1P1 being the most studied. The only known physiologic ligand of S1P1 is S1P (Lee et al., 1998), which is generated as a by-product of normal sphingolipid turn-over in many cells (Hannun et al., 2001; Spiegel & Milstien, 2003). There is an inverse relationship between the presence of S1P and its receptor S1P1 as the surface expression of S1P1 is very sensitive to the presence of S1P (Schwab et al., 2005). While surface expression of S1P1 on T cells in blood was undetectable, surface expression of S1P1 on T cells in spleen and lymph nodes was detectable, which is consistent with the lower concentrations of S1P inside the lymphoid organs than in the blood (Pappu et al., 2007; Schwab et al., 2005). The relatively high concentrations of S1P in the plasma are maintained by the red blood cells, which lack S1P-degrading enzymes (Ito et al., 2007). In contrast, the S1P-degrading S1P lyase inside the secondary lymphoid organs reduces the concentrations of S1P (Schwab et al., 2005). Thus, under normal physiologic conditions, the S1P/S1P1 system favors the egress of lymphocytes from the lymphoid organs. Interestingly, the S1P- and CXCL13-mediated migratory responses appear to counteract each other and the relative balance can determine the migration pattern of B cells. Thus, a stronger CXCL13 than S1P signal promotes the follicular-oriented migration of B cells whereas a stronger S1P than CXCL13 signal facilitates marginal zone-oriented migration (Sinha et al., 2009). Shuttling of B cells between the follicular and marginal zone compartments has been found to be regulated by the relative signaling strength of B cells that can be influenced by these two chemoattractive signals (Cinamon et al., 2008; Shiow et al., 2006).
Modulation of Chemokine Receptor Signaling by Inflammatory Cytokines in BXD2 Autoimmune Mice
The dynamics and orientation of B-cell migration have to be tightly regulated to ensure a homeostatic immune response and to maintain the well-orchestrated formation and function of the GCs. Although the immune system is dysregulated in autoimmune diseases, the pathology of these diseases requires the generation of autoantibody-producing B cells, presumably by functional GCs. This raises the issue of how the normal physiologic responses are perturbed in a manner that promotes autoantibody formation and GC formation rather than loss of function. To address this issue, we have used the BXD2 autoimmune mouse model. We have demonstrated previously that these mice spontaneously produce pathogenic autoantibodies that can induce and exacerbate glomerulonephritis and erosive arthritis (Hsu et al., 2006). Development of spontaneous GCs that harbor the production of highly mutated and pathogenic autoantibody-forming B cells is the major immune pathogenic feature in the spleens of BXD2 mice (Hsu et al., 2007; Hsu et al., 2006).
IFNα produced by plasmacytoid dendritic cells in the marginal zone promotes GC stimulation by marginal zone-precursor B cells in BXD2 mice
While investigating if spleen B cells from BXD2 mice are predisposed to differentiation into GC B cells, we observed that there is expansion of a population of B cells that can be characterized in terms of displaying the CD93loCD1dhiCD21hiIgMhiCD23hi phenotype and which are commonly known as marginal zone precursor B cells (Wang et al., 2010). Whether or not these CD93loCD1dhiCD21hiIgMhiCD23hi can only differentiate into CD93loCD1dhiCD21hiIgMhiCD23lo marginal zone B cells remains a matter of debate. CD21hi T2 B cells with a similar phenotype have been shown to be able to differentiate into either marginal zone or follicular B cells depending on environmental factors (Verma & Waldschmidt, 2007). It has not been shown if marginal zone precursor cells can also differentiate into GC B cells. For convenience, we use the term marginal zone precursor cells to describe this CD93loCD1dhiCD21hiIgMhiCD23hi population of B cells in this review.
Our observations of this population of B cells in autoimmune BXD2 mice suggest that the marginal zone precursor B cells are retained primarily in the follicle near the GC light zone whereas the marginal zone B cells are located in the marginal zone (Wang et al., 2010). Our results suggest that a key regulator of the differential distribution of these cells is the migration of the marginal zone precursor cells (Figure 2). As described above, location to the marginal zone requires expression of S1P1 that responds to the S1P in the marginal zone, which is in contact with sera. Upon activation by Toll-like receptor 7 (TLR7) or TLR9, the IFNα-producing plasmacytoid dendritic cells (pDCs) cluster in the marginal zone and in the outer T-cell area (Asselin-Paturel et al., 2005). IFNα acts directly on the type I IFN receptor (IFNRA) on marginal zone precursor B cells to upregulate CD69, which downregulates S1P1 thereby antagonizing the marginal zone-oriented migration of the marginal zone precursor B cells in the spleen of BXD2 mice (Wang et al., 2010). As it has been shown that signaling through the S1P receptor leads to upregulation of Rgs2, Rgs4 and Rgs16 mRNA in vascular smooth muscle cells (Hendriks-Balk et al., 2009; Hendriks-Balk et al., 2008), it is possible that in B cells, S1P-induced localization in the marginal zone B cells may be associated with upregulation of an RGS protein which has not yet been identified. Importantly, we have shown that type I IFN antagonizes the effects of S1P and leads to the predominant localization of marginal zone precursor B cells in the vicinity of the GC light zone in BXD2 mice (Wang et al., 2010).
Figure 2.
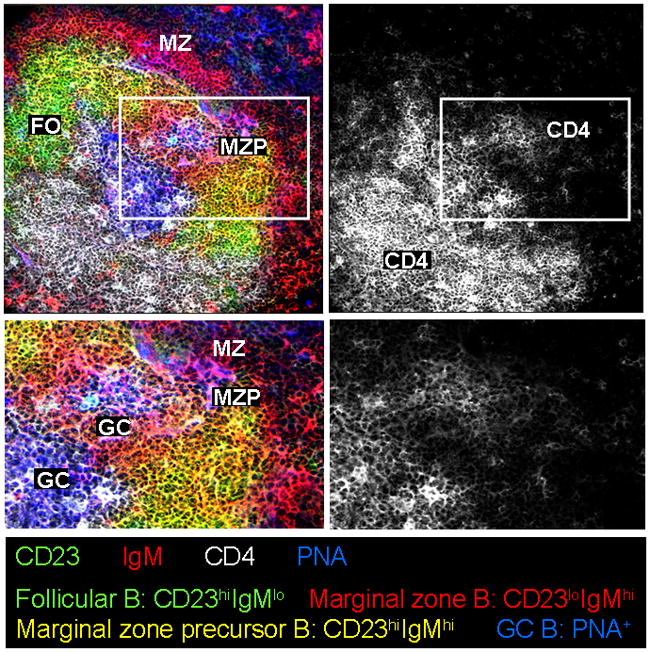
Marginal zone precursor B cells are near CD4 T cells in the vicinity of germinal center (GC) light zone end in the spleen of BXD2 mice. Confocal images of a representative BXD2 spleen section stained with anti-IgM (red), anti-CD23 (green), and anti-CD4 (white) antibodies, and peanut agglutinin (PNA) (blue). CD23hiIgMlo follicular (FO) B cells are green-colored; CD23loIgMhi marginal zone (MZ) B cells are red-colored; CD23hiIgMhi marginal zone precursor (MZP) B cells are yellow-colored; and PNA+ GC B cells are blue-colored. Bottom row displays higher magnifications of boxed areas in top row (FO: follicular B cells; GC: germinal center; MZ: marginal zone B cells; MZP: marginal zone precursor B cells; PNA: peanut agglutinin).
The above results do not address the issue as to whether the expansion of marginal zone precursor B cells in BXD2 mice has pathologic consequences. Cyster and colleagues have shown that antigen-transporting marginal zone B cells directly transport antigen into the naïve CD4 T-cell area (Cinamon et al., 2008). We have found that, in BXD2 mice, the marginal zone precursor B cells are potent antigen-transporting B cells and that the antigen-bearing marginal zone precursor B cells can migrate directly to the vicinity of the GC light zone (Wang et al., 2010). We also have found, in BXD2 mice, the marginal zone precursor B cells exhibit the highest expression of CD86 and that they provide a much stronger costimulatory signal for stimulation of CD4+ T cells than conventional marginal zone and follicular B cells (Wang et al., 2011). The upregulation of CD86 in marginal zone precursor B cells is highly associated with the presence of an increased number of type I IFN producing pDCs in marginal sinus in the spleens of BXD2 mice (Wang et al., 2011). Thus, it can be speculated that the relocation of the antigen-bearing CD86hi marginal zone precursor B cells into the GC light zone that is promoted by type I IFN is a necessary event in the formation of spontaneous GCs that are characteristic of autoimmune disease.
IL-17 induction of RGS13 and RGS16 via the canonical NF-κB pathway promotes development of pathologic autoreactive GCs in BXD2 mice
Our analyses of the BXD2 mice have established that IL-17, as well as IFNα, plays an important role in the development of autoimmune disease in BXD2 mice (Hsu et al., 2008). Because either blocking of IL-17R signaling by AdIL-17R:Fc or IL-17R deficiency led to dissipation of B cells in the follicles and a reduction of the spontaneous GC response in BXD2 mice, we showed that the IL-17 is important to stabilize B cells within the GC and in the vicinity of the GC (Hsu et al., 2008). We have established that IL-17 can direct the formation of the spontaneous GCs in these mice through a previously undescribed mechanism in which the IL-17 upregulates the expression of RGS13 and RGS16 in B cells thereby slowing the migration response of B cells stimulated by CXCL12 and CXCL13 (Hsu et al., 2008; Xie et al., 2010).
Cinamon et al. (2008) previously showed that antigen+ marginal zone B cells are constantly shuttling between the marginal and follicular zones. To fully exploit the costimulatory functions of the CD4+ T cells, however, it would be anticipated that the marginal zone precursor B cells should be retained in the immediate vicinity of CD4 T cells. Indeed confocal imaging analysis on the anatomic location of marginal zone precursor B cells in the spleen of BXD2 mice shows that the majority of these cells are in the GC light zone end (Figure 2). This is the region where high numbers of FDCs and CXCR5+ CD4 T cells distribute. Although the precise mechanisms that stabilize the marginal zone precursor B cells in this area have not been elucidated fully, our data suggest that IL-17-regulated upregulation of Rgs16 may play a role in promoting the retention of the marginal zone precursor B cells. Analysis of the expression of CXCR4 and CXCR5 in follicular, marginal zone, and marginal zone precursor B cells from BXD2 mice revealed that the follicular B cells expressed the highest levels of CXCR4 whereas the marginal zone precursor B cells expressed the highest levels of CXCR5. Interestingly, IL-17 stimulation of FACS-sorted subpopulations of B cells from BXD2 mice further revealed that Rgs16 was upregulated only in the marginal zone precursor B cells, but not in the follicular or marginal zone B cells (Wang et al, unpublished data). Interestingly, deficiency of either IL-17R or RGS16 suppressed the expansion of the marginal zone precursor B cells and their location in the GC light zone in the spleens of BXD2 mice (Wang et al., unpublished data). These results suggest that the migration arrest of the marginal zone precursor B cells at the GC light zone may be associated with the increased number of IL-17-producing T helper 17 cells (TH17) in these autoimmune mice, leading to the upregulation of Rgs16 and stabilization of marginal zone precursor B cells in this area.
We have recently identified the signaling pathway utilized by IL-17R to enhance the expression of RGS genes (Xie et al., 2010). IL-17 signaling is known to be mediated through NF-κB activator 1 (ACT1) and TNFR-associated factor 6 (TRAF6) (Chang et al., 2006; Li, 2008). The cytoplasmic tails of the IL-17R family have homology with the IL-1/TLR/IL-1R domain (referred to as the SEFIR). The SEFIR domain in IL-17RA is essential for IL-17R signaling (Chang et al., 2006; Maitra et al., 2007; Qian et al., 2007). ACT1, an activator of NF-κB, has been linked to the B-cell activation factor in the TNF family and CD40L signaling (Li, 2008; Novatchkova et al., 2003). ACT1 also contains a TRAF6-binding motif, and IL-17 activation of the NF-κB and MAPK pathways requires TRAF6 to induce the expression of IL-6 (Chang et al., 2006). We found that B cells from BXD2 mice express high levels of IL-17RA. IL-17 rapidly activates the canonical NF-kB signaling pathway in BXD2 B cells, and these cells exhibit higher basal and activated phosphorylated p65 than B6 or BXD2-Il17ra−/− B cells (Xie et al., 2010). We also showed that inhibition of p65 phosphorylation downregulates RGS16 expression and abrogated the IL-17 induced chemotactic arrest of B cells in response to CXCL12. Knock-down of Traf6 or Act1 in 70Z/3 pre-B cells led to decreased Rgs16 expression, indicating that both of these genes are involved in IL-17-mediated activation of NF-κB signaling in B cells (Xie et al., 2010). Thus, these findings identify the signaling pathway regulated by IL-17 that contributes to the development of spontaneous GCs in autoimmune BXD2 mice.
Conclusion
This review outlines the mechanisms by which proinflammatory cytokines can modify the immune responses in autoimmune mice leading to the development of GCs and the promotion of production of pathogenic antibody-producing B cells. In conclusion, we would like to emphasize the importance of the localized (paracrine) nature of the regulation of the migration responses by the cytokines. Both IL-17 and type I IFN have been implicated in the development of autoimmune diseases, including lupus, rheumatoid arthritis, psoriasis and multiple sclerosis, through human and animal studies (Crow et al., 2003; Doreau et al., 2009; Kirou et al., 2004; Koutouzov et al., 2006; Kyttaris et al., 2010). Our accumulated data indicate that both IL-17 and type I IFN play critical roles in the formation of GCs in BXD2 mice. However, this involvement of both IL-17 and type I IFN in autoimmunity seemed paradoxical given that the literature indicates that type I IFN can act to promote T helper 1 cell (TH1) responses and suppress TH17 cells (Hirohata et al., 2010; Moschen et al., 2008). Thus, the question arose as to how high levels of these two cytokines can coexist and how they can act together to promote autoantibody responses. Based on our studies on the effects of these cytokines on the migration behavior of marginal zone precursor B cells, we have formulated a model (Figure 3) that integrates the effects of IFNα- and IL-17 in the development of autoimmunity that is based on the differential regulation of the migration behavior of marginal zone precursor B cells by the localized production of high levels of these cytokines in discrete areas of the spleen. Our results suggest that type I IFN produced by plasmacytoid dendritic cells in the marginal sinus provides a signal for upregulation of CD69 (Wang et al., 2010). This downregulates S1P1 and the S1P-mediated egress response of B cells and facilitates marginal zone precursor B cells migration into the follicular area that is further facilitated by CXCL13 (Wang et al., 2010). CXCR5 is highly expressed on the marginal zone precursor B cells and CXCL13 produced by the FDCs can attract the marginal zone precursor B cell into an actively developing GC. IL-17 also acts to modulate the chemotactic responses. In this case, the IL-17 produced by TH17 cells localized in GC region can signal through the NF-kB pathway in B cells to upregulate RGS13 and RGS16 (Hsu et al., 2008; Xie et al., 2010). This upregulaton of RGS16 appears to be associated with the retention of marginal zone precursor B cells in the GC light zone. Thus, the production of IFNα and IL-17 at different sites promotes the herding of antigen-bearing and CD86hi marginal zone precursor B cells to the GC light zone and their retention at this site thereby stimulating the spontaneous GC response in BXD2 mice.
Figure 3.
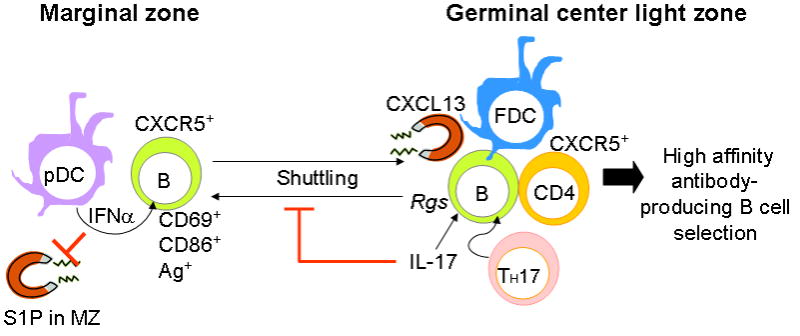
Migration of marginal zone precursor B cells regulated by type I interferon (IFN) and IL-17. This figure depicts our current view on the migration and migration arrest of CD86hi antigen-bearing marginal zone precursor B cells. Our results suggest that type I IFN produced by plasmacytoid dendritic cells (pDCs) in the marginal zone (MZ) provides a signal to upregulate CD69. This downregulates S1P1 and the S1P chemotactic response to favor the CXCL13-induced follicular entry of marginal zone precursor B cells. A second action in the germinal center (GC) results from production of CXCL13 by follicular dendritic cells (FDCs). CXCR5 is highly expressed on marginal zone precursors and CXCL13 from FDCs can attract these B cells into an active developing GC. A third action comes from IL-17 produced by T helper 17 cells (TH17) that were in the GC region. IL-17 signals through the NF-κB pathway in B cells to upregulate RGS. This locks the marginal zone precursor B cells into their position in the GC FDC region to interact with effector CXCR5+ CD4 T cells. Thus, the formation of spontaneous GCs in the spleens of BXD2 mice is highly associated with localized imbalance of cytokines that are normally invoked during immune responses. Such coupled effects of cytokines on different cells at different anatomic locations within a lymphoid organ may result in responses that are not predicted by the analysis of the effects the isolated cytokines on stationary cells (Ag: antigen; FDC: follicular dendritic cells; MZ: marginal zone; pDC: plasmacytoid dendritic cells; Rgs: regulators of G-protein signaling; S1P: sphingosine-1- phosphate; TH-17: T helper 17 cells)
Acknowledgments
This work was supported by the American Collegeof Rheumatology Research and Education Foundation for the Within OurReach: Finding a Cure for Rheumatoid Arthritis campaign, theAlliance for Lupus Research – Target Identification inLupus program, the Department of Veterans Affairs Merit ReviewGrant 1I01BX000600, Daiichi-Sankyo Co., Ltd., the National Institutesof Health Grants 1AI 071110 and ARRA 3RO1 AI71110-02S1(all to J.D.M.), the Lupus Research Institute Novel Research Project, and the Arthritis InvestigatorAward supported by the Arthritis Foundation (to H.-C.H).
Footnotes
Disclosure
The authors report no conflicts of interest.
Contributor Information
John D. Mountz, Professor of Medicine, Director of Rheumatic Diseases Core Center, Aging, Immunology, Rheumatology, Division of Clinical Immunology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA and Birmingham Veteran Administration Medical Center, Birmingham, AL 35233.
John H. Wang, Medical Student in the Med Scientist Training Program, Immunology, Pathology, Division of Clinical Immunology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294.
Shutao Xie, Post-doctoral Fellow, Immunology, Rheumatology, Division of Clinical Immunology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294.
Hui-Chen Hsu, Associate Professor of Medicine , Immunology, Rheumatology, Division of Clinical Immunology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294.
References
- Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O'garra A, Vicari A, Trinchieri G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201(7):1157–1167. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrington RA, Pozdnyakova O, Zafari MR, Benjamin CD, Carroll MC. B lymphocyte memory: role of stromal cell complement and FcgammaRIIB receptors. J Exp Med. 2002;196(9):1189–1199. doi: 10.1084/jem.20021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bende RJ, Van Maldegem F, Van Noesel CJ. Chronic inflammatory disease, lymphoid tissue neogenesis and extranodal marginal zone B-cell lymphomas. Haematologica. 2009;94(8):1109–1123. doi: 10.3324/haematol.2009.005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281(47):35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9(1):54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36(8):481–490. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, Fabien N, Cochat P, Pouteil-Noble C, Trolliet P, Durieu I, Tebib J, Kassai B, Ansieau S, Puisieux A, Eliaou JF, Bonnefoy-Berard N. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10(7):778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- Estes JD, Thacker TC, Hampton DL, Kell SA, Keele BF, Palenske EA, Druey KM, Burton GF. Follicular dendritic cell regulation of CXCR4–mediated germinal center CD4 T cell migration. J Immunol. 2004;173(10):6169–6178. doi: 10.4049/jimmunol.173.10.6169. [DOI] [PubMed] [Google Scholar]
- Fukuyama S, Nagatake T, Kim DY, Takamura K, Park EJ, Kaisho T, Tanaka N, Kurono Y, Kiyono H. Cutting edge: uniqueness of lymphoid chemokine requirement for the initiation and maturation of nasopharynx-associated lymphoid tissue organogenesis. J Immunol. 2006;177(7):4276–4280. doi: 10.4049/jimmunol.177.7.4276. [DOI] [PubMed] [Google Scholar]
- Goetzl EJ, Wang W, Mcgiffert C, Huang MC, Graler MH. Sphingosine 1-phosphate and its G protein-coupled receptors constitute a multifunctional immunoregulatory system. J Cell Biochem. 2004;92(6):1104–1114. doi: 10.1002/jcb.20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Luberto C, Argraves KM. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 2001;40(16):4893–4903. doi: 10.1021/bi002836k. [DOI] [PubMed] [Google Scholar]
- Hart GT, Wang X, Hogquist KA, Jameson SC. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A. 2011;108(2):716–721. doi: 10.1073/pnas.1013168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks-Balk MC, Hajji N, Van Loenen PB, Michel MC, Peters SL, Alewijnse AE. Sphingosine-1-phosphate regulates RGS2 and RGS16 mRNA expression in vascular smooth muscle cells. Eur J Pharmacol. 2009;606(1–3):25–31. doi: 10.1016/j.ejphar.2009.01.018. [DOI] [PubMed] [Google Scholar]
- Hendriks-Balk MC, Van Loenen PB, Hajji N, Michel MC, Peters SL, Alewijnse AE. S1P receptor signalling and RGS proteins; expression and function in vascular smooth muscle cells and transfected CHO cells. Eur J Pharmacol. 2008;600(1–3):1–9. doi: 10.1016/j.ejphar.2008.09.041. [DOI] [PubMed] [Google Scholar]
- Hirohata S, Shibuya H, Tejima S. Suppressive influences of IFN-alpha on IL-17 expression in human CD4+ T cells. Clin Immunol. 2010;134(3):340–344. doi: 10.1016/j.clim.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Hoek KL, Gordy LE, Collins PL, Parekh VV, Aune TM, Joyce S, Thomas JW, Van Kaer L, Sebzda E. Follicular B cell trafficking within the spleen actively restricts humoral immune responses. Immunity. 2010;33(2):254–265. doi: 10.1016/j.immuni.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Wu Y, Yang P, Wu Q, Job G, Chen J, Wang J, Accavitti-Loper MA, Grizzle WE, Carter RH, Mountz JD. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178(8):5357–5365. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Zhou T, Kim H, Barnes S, Yang P, Wu Q, Zhou J, Freeman BA, Luo M, Mountz JD. Production of a novel class of polyreactive pathogenic autoantibodies in BXD2 mice causes glomerulonephritis and arthritis. Arthritis Rheum. 2006;54(1):343–355. doi: 10.1002/art.21550. [DOI] [PubMed] [Google Scholar]
- Ito K, Anada Y, Tani M, Ikeda M, Sano T, Kihara A, Igarashi Y. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem Biophys Res Commun. 2007;357(1):212–217. doi: 10.1016/j.bbrc.2007.03.123. [DOI] [PubMed] [Google Scholar]
- Jonsson MV, Skarstein K, Jonsson R, Brun JG. Serological implications of germinal center-like structures in primary Sjogren's syndrome. J Rheumatol. 2007;34(10):2044–2049. [PubMed] [Google Scholar]
- Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, Ly N, Woodward RN, Fry KE, Lau AY, Prentice JG, Wohlgemuth JG, Crow MK. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004;50(12):3958–3967. doi: 10.1002/art.20798. [DOI] [PubMed] [Google Scholar]
- Koch AE. Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 2005;52(3):710–721. doi: 10.1002/art.20932. [DOI] [PubMed] [Google Scholar]
- Koutouzov S, Mathian A, Dalloul A. Type-I interferons and systemic lupus erythematosus. Autoimmun Rev. 2006;5(8):554–562. doi: 10.1016/j.autrev.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184(9):4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279(5356):1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine. 2008;41(2):105–113. doi: 10.1016/j.cyto.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Lippert E, Yowe DL, Gonzalo JA, Justice JP, Webster JM, Fedyk ER, Hodge M, Miller C, Gutierrez-Ramos JC, Borrego F, Keane-Myers A, Druey KM. Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol. 2003;171(3):1542–1555. doi: 10.4049/jimmunol.171.3.1542. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Moser B. Homing chemokines in rheumatoid arthritis. Arthritis Res. 2002;4(4):233–236. doi: 10.1186/ar412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, Gaffen SL. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci U S A. 2007;104(18):7506–7511. doi: 10.1073/pnas.0611589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchers F, Rolink AR. B cell tolerance -- how to make it and how to break it. Curr Top Microbiol Immunol. 2006;305:1–23. doi: 10.1007/3-540-29714-6_1. [DOI] [PubMed] [Google Scholar]
- Meyer-Hermann ME, Maini PK. Cutting edge: back to "one-way" germinal centers. J Immunol. 2005;174(5):2489–2493. doi: 10.4049/jimmunol.174.5.2489. [DOI] [PubMed] [Google Scholar]
- Moratz C, Harrison K, Kehrl JH. Role of RGS proteins in regulating the migration of B lymphocytes. Arch Immunol Ther Exp (Warsz) 2004a;52(1):27–35. [PubMed] [Google Scholar]
- Moratz C, Hayman JR, Gu H, Kehrl JH. Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1−/− mice. Mol Cell Biol. 2004b;24(13):5767–5775. doi: 10.1128/MCB.24.13.5767-5775.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschen AR, Geiger S, Krehan I, Kaser A, Tilg H. Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology. 2008;213(9–10):779–787. doi: 10.1016/j.imbio.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Nanki T, Takada K, Komano Y, Morio T, Kanegane H, Nakajima A, Lipsky PE, Miyasaka N. Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2009;11(5):R149. doi: 10.1186/ar2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28(5):226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316(5822):295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- Patel DD, Haynes BF. Leukocyte homing to synovium. Curr Dir Autoimmun. 2001;3:133–167. doi: 10.1159/000060517. [DOI] [PubMed] [Google Scholar]
- Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol. 2010;22(6):413–419. doi: 10.1093/intimm/dxq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol. 2009;10(7):786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8(3):247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Qin D, Wu J, Carroll MC, Burton GF, Szakal AK, Tew JG. Evidence for an important interaction between a complement-derived CD21 ligand on follicular dendritic cells and CD21 on B cells in the initiation of IgG responses. J Immunol. 1998;161(9):4549–4554. [PubMed] [Google Scholar]
- Schaerli P, Moser B. Chemokines: control of primary and memory T-cell traffic. Immunol Res. 2005;31(1):57–74. doi: 10.1385/IR:31:1:57. [DOI] [PubMed] [Google Scholar]
- Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192(11):1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8(12):1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- Shadidi KR. New drug targets in rheumatoid arthritis: focus on chemokines. BioDrugs. 2004;18(3):181–187. doi: 10.2165/00063030-200418030-00004. [DOI] [PubMed] [Google Scholar]
- Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol. 2002;169(5):2507–2515. doi: 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440(7083):540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1(2):51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RK, Park C, Hwang IY, Davis MD, Kehrl JH. B lymphocytes exit lymph nodes through cortical lymphatic sinusoids by a mechanism independent of sphingosine-1-phosphate-mediated chemotaxis. Immunity. 2009;30(3):434–446. doi: 10.1016/j.immuni.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren B, Blazer LL, Neubig RR. Regulators of G protein signaling proteins as targets for drug discovery. Prog Mol Biol Transl Sci. 2010;91:81–119. doi: 10.1016/S1877-1173(10)91004-1. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci (Schol Ed) 2010;2:153–167. doi: 10.2741/s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Waldschmidt TJ. Characterization of splenic CD21hi T2 B cells. Immunol Res. 2007;39(1–3):240–248. doi: 10.1007/s12026-007-0072-5. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu Q, Yang P, Li H, Li J, Mountz JD, Hsu HC. Type I IFN-dependent CD86high marginal zone-precursor B cells are potent T-cell costimulators. Arthritis Rheum. 2011 doi: 10.1002/art.30231. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Li J, Wu Q, Yang P, Pawar RD, Xie S, Timares L, Raman C, Chaplin DD, Lu L, Mountz JD, Hsu HC. Marginal zone precursor B cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J Immunol. 2010;184(1):442–451. doi: 10.4049/jimmunol.0900870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbrock K, Jungnickel B, Hansmann ML, Kuppers R. Human splenic marginal zone B cells lack expression of activation-induced cytidine deaminase. Eur J Immunol. 2005;35(10):3002–3007. doi: 10.1002/eji.200535134. [DOI] [PubMed] [Google Scholar]
- Wu Y, Sukumar S, El Shikh ME, Best AM, Szakal AK, Tew JG. Immune complex-bearing follicular dendritic cells deliver a late antigenic signal that promotes somatic hypermutation. J Immunol. 2008;180(1):281–290. doi: 10.4049/jimmunol.180.1.281. [DOI] [PubMed] [Google Scholar]
- Xie S, Li J, Wang JH, Wu Q, Yang P, Hsu HC, Smythies LE, Mountz JD. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol. 2010;184(5):2289–2296. doi: 10.4049/jimmunol.0903133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Geiger TR, Johnson EN, Nyborg JK, Druey KM. RGS13 acts as a nuclear repressor of CREB. Mol Cell. 2008;31(5):660–670. doi: 10.1016/j.molcel.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hsu HC, Chen J, Grizzle WE, Chatham WW, Stockard CR, Wu Q, Yang PA, Holers VM, Mountz JD. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70(3):309–316. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman NS, Howard WA, Bismuth J, Gibson K, Edelman H, Berrih-Aknin S, Dunn-Walters D, Mehr R. Ectopic GC in the thymus of myasthenia gravis patients show characteristics of normal GC. Eur J Immunol. 2010;40(4):1150–1161. doi: 10.1002/eji.200939914. [DOI] [PubMed] [Google Scholar]