

SUMMARY
Long intergenic noncoding RNAs (lincRNAs) are key regulators of chromatin state, yet the nature and sites of RNA-chromatin interaction are mostly unknown. Here we introduce Chromatin Isolation by RNA Purification (ChIRP), where tiling oligonucleotides retrieve specific lincRNAs with bound protein and DNA sequences, which are enumerated by deep sequencing. ChIRP-seq of three lincRNAs reveal that RNA occupancy sites in the genome are focal, sequence-specific, and numerous. Drosophila roX2 RNA occupies male X-linked gene bodies with increasing tendency toward the 3’ end, peaking at CES sites. Human telomerase RNA TERC occupies telomeres and Wnt pathway genes. HOTAIR lincRNA preferentially occupies a GA-rich DNA motif to nucleate broad domains of Polycomb occupancy and histone H3 lysine 27 trimethylation. HOTAIR occupancy occurs independently of EZH2, suggesting the order of RNA guidance of Polycomb occupancy. ChIRP-seq is generally applicable to illuminate the intersection of RNA and chromatin with newfound precision genome-wide.
INTRODUCTION
Long noncoding RNAs are key regulators of chromatin states for important biological processes such as dosage compensation, imprinting, and developmental gene expression (Kelley et al., 1999; Koziol and Rinn, 2010; Mercer et al., 2009; Pandey et al., 2008; Rinn et al., 2007; Wang et al., 2011; Zhao et al., 2008). The recent discovery of thousands of lincRNAs in association with specific chromatin modification complexes, such as Polycomb Repressive Complex 2 (PRC2) that mediates histone H3 lysine 27 trimethylation (H3K27me3), suggests broad roles for numerous lincRNAs in managing chromatin states in a gene-specific fashion (Khalil et al., 2009; Zhao et al., 2010). While some lincRNAs are thought to work in cis on neighboring genes, other lincRNAs work in trans to regulate distantly located genes. For instance, Drosophila ncRNAs roX1 and roX2 bind numerous regions on the X chromosome of male cells, and are critical for dosage compensation (Franke and Baker, 1999; Meller et al., 1997). However, the exact locations of their binding sites are not known at high resolution. Similarly, human lincRNA HOTAIR can affect PRC2 occupancy on hundreds of genes genome-wide (Gupta et al., 2010; Rinn et al., 2007; Tsai et al., 2010), but how specificity is achieved is unclear. LincRNAs can also serve as modular scaffolds to recruit the assembly of multiple protein complexes. The classic trans-acting RNA scaffold is the TERC RNA that serves as the template and scaffold for the telomerase complex (Zappulla and Cech, 2006); HOTAIR can also serve as a scaffold for PRC2 and a H3K4 demethylase complex (Tsai et al., 2010).
Prior studies mapping RNA occupancy at chromatin have revealed substantial insights (Carter et al., 2002; Nagano et al., 2008), but only at single gene locus at a time. The occupancy sites of most lincRNAs are not known, and the roles of lincRNAs in chromatin regulation have been mostly inferred from the indirect effects of lincRNA perturbation. Just as chromatin immunoprecipitation followed by microarray or deep sequencing (ChIP-chip or ChIP-seq, respectively) have greatly improved our understanding of protein-DNA interactions on a genomic scale, here we introduce a strategy to map long RNA occupancy genome-wide at high resolution, and apply this strategy to illuminate the mechanisms of RNA guided PRC2 localization.
RESULTS
Development and optimization of ChIRP
We developed a method termed ChIRP that allows unbiased high-throughput discovery of RNA-bound DNA and proteins (Fig. 1A). Briefly, cultured cells are crosslinked in vivo, and their chromatin extracted and homogenized. Biotinylated complementary oligonucleotides that tile the RNA of interest were hybridized to target RNAs, and isolated using magnetic streptavidin beads. Co-purified chromatin was eluted for protein, RNA, or DNA, which was then subject to downstream assays for identification and quantitation.
Figure 1. Chromatin isolation by RNA purification.

(A) Workflow of ChIRP. Chromatin is crosslinked to lincRNA:protein adducts in vivo. Biotinylated tiling probes are hybridized to target lncRNA, and chromatin complexes are purified using magnetic streptavidin beads, followed by stringent washes. We elute lncRNA bound DNA or proteins with a cocktail of Rnase A and H. A putative lincRNA binding sequence is schematized in orange. (B) Northern blot shows HOTAIR RNA is sheared into the size range of 100-500nt by sonication. (C) Design of antisense DNA tiling probes, grouped into “even” and “odd” sets based on their positions along the RNA. (D) Complementary DNA tiling oligonucleotides effectively retrieve ~95% of HOTAIR RNA from chromatin, as compared to ~10% by morpholino probes. Mean ± s.d. are shown.
A key feature of our approach is the use of tiling oligonucleotides, which we found to be critical for success. We initially attempted to capture a lncRNA with morpholino probes, which are high-affinity ribonucleotide analogues resistant to nuclease digestion. As lncRNAs are known to be highly structured (Tsai et al., 2010), we designed three morpholino probes against single-stranded portions of HOTAIR, as determined by prior high-throughput RNA secondary structure measurements (Kertesz et al., 2010), Fig. S1A). As a negative control we synthesized a morpholino probe that bore no sequence homology with any human RNA. We titrated a wide array of hybridization parameters, and consistently obtained best results under high ionic strength (results not shown), higher hybridization temperature (Fig. S1B) and moderately denaturing conditions (Fig. S1C). However, with the 3-probe approach we could retrieve at most ~10% of HOTAIR RNA (Fig. S1C). Importantly, we found that the HOTAIR transcript was sheared into the size range of ~100nt to 500nt during sonication, a step necessary for the solubilization of chromatin (Fig. 1B). We suspected that the 3-oligo approach was ineffective at recovering all fragments of long RNAs such as HOTAIR, and indeed qRT-PCR primers targeting distinct regions of HOTAIR reported drastically different efficiencies of recovery from the same pull down (~10 fold range, Fig. S1D). This raised a serious concern because functional domains of HOTAIR at its 5’ and 3’ ends (Tsai et al., 2010) could potentially be lost due to their distance away from arbitrarily chosen probes. Moreover, without prior knowledge of the DNA interacting domain within a lncRNA, it would be difficult to decide where to target a small number of oligonucleotide probes or insert a RNA aptamer to consistently retrieve a lncRNA of interest on chromatin.
To develop a method that is applicable to any lncRNA without prior knowledge of its secondary structure or functional domains, we decided to target all parts of HOTAIR equally. We were inspired by the technique of single molecule RNA fluorescent in situ hybridization (Fusco et al., 2004; Raj et al., 2008), where dozens of short oligonucleotide probes that tile the length of an RNA generate highly specific signals. Thus, we designed 48 complementary DNA oligonucleotides that were 20mer each and tiled the entire length of HOTAIR across 2.2 kb (~50% tiled) (Fig. 1C). Sequences that have extensive complementarity to other sites in the genome or are repetitive are excluded (Methods). As a negative control, we designed a similar set of probes that targeted the LacZ mRNA, normally absent from human cells. With the tiling probes, we could pull down almost all HOTAIR RNA from chromatin (Fig. 1D), a substantial improvement over the 3-morpholinos approach. HOTAIR probes did not retrieve GAPDH nor did LacZ probes retrieve HOTAIR (Fig. 1D), demonstrating the specificity of the method. Furthermore, HOTAIR fragments were equally recovered (<2-fold difference between different qRT-PCR primers, Fig. S1E), further demonstrating the strength of a non-biased targeting method.
We next examined whether lncRNA-associated DNA and proteins could be co-purified. As a positive control, we examined the TERC RNA, which functions as the template and scaffold for the telomerase complex. In HeLa S3 cells transduced with human TERC and TERT, TERC RNA expression is ~4 fold over control and constitutively bound at telomeric ends of dividing chromosomes (Abreu et al., 2010). Using 19 probes complementary DNA against TERC RNA (84% tiled) or LacZ, ~90% of total TERC RNA was specifically retrieved (Fig. 2A), showing that the method is easily compatible with other lncRNAs. We gently eluted DNA off of beads using a combination of RNase A and RNase H so that only DNA retrieved via a RNA bridge, but not direct probe-DNA interaction, could be preferentially released. We evaluated various crosslinking strategies for fixing RNA:DNA:protein interactions. Consistent with classic electron micrographs showing RNA at chromatin using the thermo-stable crosslinker glutaraldehyde (Hopwood, 1972; Sabatini et al., 1962), we found that glutaraldehyde crosslinking consistently yielded the highest signal-to-noise ratio in comparison to ultraviolet light or formaldehyde crosslinking (Fig. 2B). TERC ChIRP specifically retrieved telomere DNA but not Alu repeats, while LacZ ChIRP retrieved neither (Fig. 2B). The telomere ChIRP signal could not have arisen due to direct probe-telomere interaction: The CCCTAA template region on TERC was avoided in probe design (for this reason), and no probe shared homology with telomeric sequences. Furthermore, the ChIRP signal was crosslinking-dependent, suggesting that it was specific to telomere-TERC interaction. As another positive control for the method, we found that TERC ChIRP specifically retrieved TCAB1, a subunit of the telomerase holoenzyme that facilitates telomerase trafficking (Venteicher et al., 2009; Zhong et al., 2011) (Fig. 2C). Thus, ChIRP is compatible with the simultaneous analysis of DNA and proteins associated with specific RNAs.
Figure 2. ChIRP enriches for TERC RNA and detects TERC-associated telomere DNA and TCAB1 protein.

(A) TERC-asDNA probes retrieve ~88% of cellular TERC RNA and undetectable GAPDH. LacZ-asDNA probes retrieve neither RNAs. Mean ± s.d. are shown. (B) Effect of different crosslinking agents on ChIRP-southern. After 1% glutaraldehyde crosslinking, TERC retrieval co-purifies telomeric repeats, but not Alu repeats. (C) TERC ChIRP retrieves TCAB1, a known telomerase holocomplex chaperone proteins. As a negative control tubulin was not detected.
One potential source of noise in ChIRP-seq is the precipitation of non-specific DNA fragments from off-target hybridization of the pool of oligonucleotide probes. In order to eliminate such artifacts, we devised the “split-probe” strategy, where we ranked all probes based on their relative positions along the target RNA, and split them into two pools such that all even probes were in one set and all odd probes in another. As the two sets of probes shared no overlapping sequences, the only target they have in common is the RNA of interest and its associated chromatin. Similar to using two independent polyclonal antibodies to obtain high confidence ChIP-seq signal, we performed two independent ChIRP-seq runs with “even” and “odd” probes separately, and focused our analysis exclusively on the overlap between their signals. Notably, “even” and “odd” probe sets enriched each of the target RNAs below with similar efficiency, yielding comparable ChIRP-seq libraries in terms of signal-to-noise ratio (Fig. S2).
ChIRP-seq elucidates roX2 binding sites on X chromosome
To assess the sensitivity and specificity of ChIRP when applied genome-wide, we need a biological system where the binding sites of a lincRNA are already known genome-wide, and we should show that ChIRP-seq selectively retrieves most of these sites. The Drosophila dosage compensation system is ideal for this purpose. In Drosophila, male cells up-regulate the expression of genes on their single X chromosome by two-fold; this dosage compensation requires a ribonucleoprotein complex containing the Male-Specific Lethal (MSL) proteins and two lncRNAs, roX1 and roX2 (Lucchesi et al., 2005). Staining of polytene chromosomes showed that roX and MSL co-localize exclusively on the male X chromosome but not on autosomes (Franke and Baker, 1999), and pioneering work by Kuroda and colleagues have defined the occupancy landscape of MSL proteins at high resolution (Alekseyenko et al., 2008).
We performed ChIRP-seq on endogenous roX2 in male S2 cells. Using the software MACS (Zhang et al., 2008), we identified 308 roX2 binding sites--all of them are on the X chromosome and none on autosomes (Fig. 3A, B, Table S1). Autosomes constitute ~80% of the Drosophila genome. Thus, ChIRP-seq is highly specific even on a genome-wide scale, and has a negligible false discovery rate (FDR ~0). The 308 rox2 binding sites recovered 89.3% of known Chromosomal Entry Sites (CES), which are high affinity binding sites of the roX-MSL complex that have been previously defined by genetic epistasis (Alekseyenko et al., 2008). This number compares favorably with ChIP-seq of single MSL components, whose top 309 peaks identified 91% of CES (Alekseyenko et al., 2008). roX2 ChIRP-seq profile is highly correlated with MSL ChIP-seq profile, and both show very strong peaks at CES (Fig. 3A). The roX2 and MSL occupancy profiles show a Pearson correlation of 0.77, which is in the range of correlation for biological replicates of a single MSL protein, or ChIP-seq of different MSL subunits in parallel (Fig. 3A, C; R= 0.65 - 0.94) (Alekseyenko et al., 2006). Remarkably, direct comparison of roX2 ChIRP-seq vs. MSL3 ChIP-seq shows that ChIRP-seq has better dynamic range and discrimination of X chromosome vs. autosomes than ChIP-seq (Fig. 3D). Aligning roX2 ChIRP signal across all bound genes, we discovered that the roX2 occupancy is enriched over gene bodies of X chromosome genes, and increases from 5’ to 3’ end of each gene. This pattern provides independent support for a recent notion that the roX-MSL complex acts by promoting transcriptional elongation rather than initiation (Larschan et al., 2011) (Fig. 3D). In addition, motif analysis of roX2 ChIRP-seq data revealed a very significantly enriched DNA motif that is nearly identical to the MSL motif (a sequence shown to function as CES when inserted into autosomes) (Alekseyenko et al., 2008) (Fig. 3E). These data demonstrate that ChIRP-seq is highly sensitive and specific, and retrieves biologically useful signal.
Figure 3. ChIRP-seq reveals roX2 binding sites on X chromosome.

(A) roX2 co-localizes with MSL complex and CES. (B) 308 roX2 binding sites are all on the X chromosome, indicating an FDR ~ 0. (C) roX2 ChIRP-seq is overall highly correlated to MSL3 ChIP-seq (R = 0.77 for log2 intensity > 10). (D) roX2 binds across X-linked gene bodies with bias towards the 3’ end, in a manner similar to MSL3 but with higher dynamic range. ChIRP-seq or ChIP-seq signal intensity for all bound genes on X or genes on chromosome 2L were averaged and to gene start and end annotations. (E) roX2 binding sites are strongly enriched for a sequence motif that is nearly identical to MSL3 motif.
Consistent with our hypothesis that the shared signal between even and odd probes will improve ChIRP-seq accuracy, we found that the shared signal between the two independent probe sets are highly correlated with that of MSL3 ChIP-seq while the unique signals in either probe sets alone were not (Fig. S2D). Based on these findings, we performed at least two ChIRP-seq experiments using independent sets of non-overlapping probes for each target RNAs, and we only accept binding sites that are concordant in both experiments (Methods). Only the shared signal between from two independent ChIRP-seq experiments can be considered meaningful signal; signal present in only even or odd experiments alone should not be interpreted. These data also suggest a lower bound of false discovery rate in larger genomes. The roX2 results showed no off-target peaks in all Drosophila autosomes, which is ~100 MB. Assuming a worst case scenario that the off-target effect is actually 1 peak per 100 MB and that this scales linearly on the human genome (3GB, 30x the fly autosomal genome), then one expects ~30 false positive peaks, a far smaller number than actually observed peaks in actual experiments below (estimated FDR<0.05 for each). Thus, our data indicate that off-target effects should be limited even in larger mammalian genomes.
ChIRP-seq reveals TERC occupancy sites genome-wide
We next performed ChIRP-seq of TERC in HeLa S3 cells transduced with TERT and TERC. TERC ChIRP-seq showed significant enrichment of telomeric DNA sequences (~9 fold) relative to input reads, whereas Alu repeats were not (Fig. 4A). In addition, we observed numerous specific TERC binding events throughout the genome with signal intensities comparable to conventional ChIP-seq. TERC binding sites were focal; most binding sites are “peaks” of <600 bp that do not spread beyond 1 kb (Fig. 4B), which is a pattern reminiscent of ChIP-seq peaks of transcription factors. Using the same analysis pipeline employed in roX2 analysis, we identified over 2198 TERC binding sites in the genome, which represents a large resource to study potential non-canonical functions of TERC RNA and telomerase (Table S2). It is known that TERT can bind to and co-activate Wnt target genes at chromatin (Park et al., 2009), and we hypothesized that TERC, as a component of the TERT complex, may also co-occupy some of the same genes. Unbiased analysis of the TERC-bound peaks revealed that one of the top three enriched Gene Ontology terms is Wnt receptor signaling pathway (p = 1.3 × 10-6), strongly supporting our initial hypothesis. We found that TERC occupied multiple Wnt genes directly, including Wnt11, which is transcriptionally induced by TERT overexpression in vivo (Choi et al., 2008). ChIRP-seq revealed a series of TERC binding peaks near the MYC gene, concordant with previously documented binding sites of TERT (Fig. 4C). Analysis of TERC-bound sequences identified an enriched cytosine rich sequence motif (Fig. 4D), suggesting that specific DNA motifs may be involved in TERC occupancy. ChIRP-seq of TERC in wild type HeLa cells identified largely similarly pattern of chromatin occupancy [r= 0.80; 1549 of 2198 peaks independently identified (70% overlap), p <10-20, hypergeometric distribution], indicating that endogenous TERC bind similar genomic sites (Fig. S3). These results bolster the concept of direct connections between chromosome replication and self-renewal pathways (Park et al., 2009). It will be of great interest to use ChIRP-seq to interrogate TERC binding in various biological systems where TERT has been shown to assume non-canonical roles.
Figure 4. TERC ChIRP-seq.
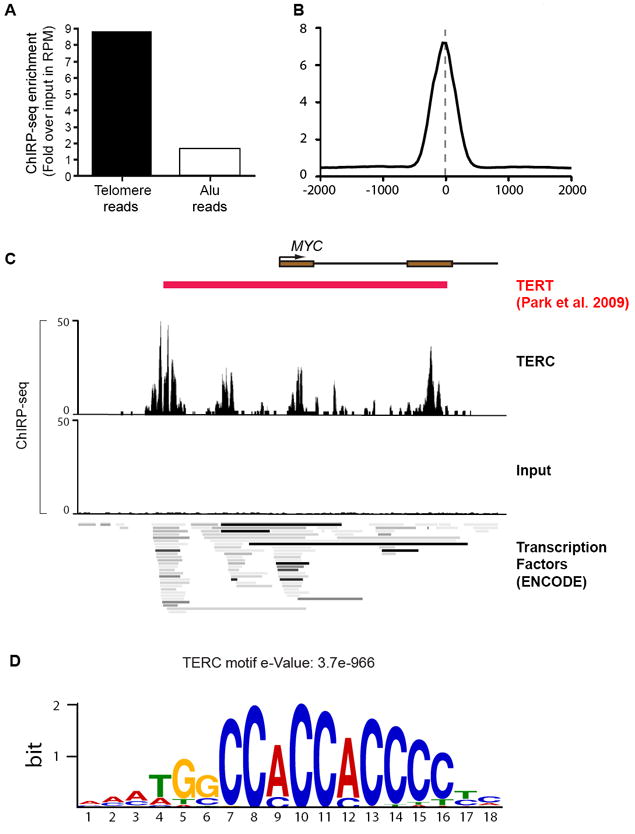
(A) Fold enrichment of reads from TERC-ChIRP-seq and Input sample that map to telomere and Alu sequences. (B) TERC peaks are focal. (C) TERC occupancy at the MYC promoter, overlaying regions of TERT occupancy (Park et. al., 2009) and regions of dense transcription factors occupancy identified by the ENCODE project (bottom). (D) A cytosine-rich motif enriched among TERC-binding sites (e-Value = 3.7e-966).
ChIRP-seq reveals HOTAIR nucleation of Polycomb domains
We next turned to discover the genomic binding sites of HOTAIR and their relationship with Polycomb occupancy. HOTAIR is a 2.2 kb lincRNA from the HOXC locus that binds the Polycomb Repressive Complex 2 (PRC2) and affects PRC2 occupancy to target genes throughout the genome. How HOTAIR guides PRC2 to target genes is not understood. Overexpression of HOTAIR alters the positional identity of cancer cells and promotes cancer metastasis (Gupta et al., 2010). We chose to map HOTAIR occupancy genome-wide by ChIRP-seq in MDA-MB-231 breast cancer cells expressing HOTAIR, which matches the HOTAIR level and phenotypic consequences in metastasis-prone human breast cancers (Gupta et al., 2010),
We identified 832 HOTAIR occupancy sites genome-wide, using the same analysis pipeline described above with two independent ChIRP-seq probe sets (Table S3). HOTAIR binding sites occur on multiple chromosomes and are enriched in genic regions, notably regions annotated as enhancers and introns (Fig. 5A). HOTAIR binding events are focal; typical HOTAIR peaks are no more than a few hundred base pairs, a pattern reminiscent of transcription factors. When overlaid with previous generated genomic-binding data of PRC2 subunits EZH2, SUZ12, and H3K27Me3 in the same cell type, we discovered a significant pattern of co-occupancy (Fig. 5B). Focal sites of HOTAIR occupancy are associated with more broad domains of PRC2 occupancy and H3K27me3, suggesting that HOTAIR may nucleate Polycomb domains. One prime example of this pattern is in the human HOXD locus, where HOTAIR is known to target PRC2 to silence multiple HOXD genes across 40 kilobases (Rinn et al., 2007). One of the high confidence HOTAIR ChIRP-seq peaks mapped to the intergenic region between HOXD3 and HOXD4, which corresponds to middle of a broad domain of H3K27me3 and SUZ12 occupancy loss upon HOTAIR depletion (Fig. 5C) (Rinn et al., 2007; Tsai et al., 2010). Endogenous HOTAIR in primary human fibroblasts also bound the same sites (four of four tested, including in HOXD), as indicated by ChIRP-qPCR (Fig. S4). HOTAIR occupancy sites are significantly enriched for genes that gain PRC2 occupancy in a HOTAIR-dependent manner in the same cell type, or become de-repressed when endogenous HOTAIR is depleted (Gupta et al., 2010; Tsai et al., 2010) (p = 2.4 × 10-5 and p = 8.57 × 10-3 respectively, hypergeometric distribution). Unbiased analyses of HOTAIR occupied genes revealed enrichment for genes involved in pattern specification processes (p= 8.7 × 10-7), consistent with prior data that HOTAIR enforces the epigenomic state of distal and posterior positional identity (Gupta et al., 2010). These results provide additional evidence that HOTAIR-chromatin interaction is associated with PRC2 relocalization and gene silencing. Despite these significant overlaps, it is clear that the correspondence between HOTAIR occupancy and downstream effects (PRC2 occupancy, gene silencing) does not map one-to-one, which may suggest additional layers of complexity.
Figure 5. HOTAIR ChIRP-seq suggests mechanisms of HOTAIR-recruitment of PRC2.

(A) HOTAIR binding sites are enriched in genic regions, notably enhancers and introns. (B) Metagene analysis of genomic regions aligned by 832 HOTAIR ChIRP peaks show focal HOTAIR peaks in association with broad domains PRC2 occupancy (evidenced by subunits EZH2 and Suz12) and H3K27Me3. (C) HOTAIR nucleates broad domains of PRC2 occupancy. A HOTAIR binding site between HOXD3 and HOXD4 lies in the center of a broad domain of Suz12 and H3K27Me3 occupancy that are both lost upon HOTAIR knock down (Tsai et. al., 2010, Rinn et. al., 2007). (D) GA-rich homopurine motif enriched in HOTAIR binding sites.
ChIRP-seq data enable potentially new mechanistic insights into RNA-chromatin interaction. Analysis of HOTAIR binding sites revealed enrichment of a GA-rich polypurine motif (e=3.8e-128, Fig. 5D), which we term the HOTAIR motif. Interestingly, Drosphila Polycomb Response Element (PRE) are known to bind GAGA protein (Horard et al., 2000), and recent studies of mammalian PREs also identified GA-repeats as a shared feature (Woo et al., 2010), (Sing et al., 2009), although other sequences are also required. In addition, the MSL/roX ribonucleoprotein complex responsible for dosage compensation in Drosophila also recognizes a GA-rich element on fly X chromosome (Alekseyenko et al., 2008), raising the intriguing possibility of similar mechanisms where lncRNAs could potentially serve as guides for chromatin-lncRNA complexes such as PRC2-HOTAIR and MSL-roX.
HOTAIR occupancy occurs independent of EZH2
HOTAIR may actively recruit PRC2 to it targets genes, or simply serve as a scaffolding molecule that gets passively transported along with PRC2. The observed pattern of focal HOTAIR occupancy in the midst of broader domains of PRC2 strongly suggests the former hypothesis. To formally distinguish between these two possibilities, we performed HOTAIR ChIRP-seq in isogenic cells depleted for EZH2 (Gupta et al., 2010), which directly binds HOTAIR (Kaneko et al., 2010). Notably, the pattern of HOTAIR occupancy was largely preserved upon EZH2 depletion (Fig. 6A), indicating that HOTAIR can bind chromatin without an intact PRC2. Independent ChIRP-qPCR validated the binding sites and confirmed the specificity of ChIRP-seq results in control and shEZH2 cells (Fig. 6B). Together, these results support the role HOTAIR lincRNA as an active recruiter of chromatin modifying complexes.
Figure 6. HOTAIR binds chromatin in a PRC2-independent manner.

(A) Heatmap of HOTAIR ChIRP-seq signal in peak regions. Each row is a 4 kB genomic window centered on a HOTAIR ChIRP peak in control cells; the peaks are aligned for the 832 HOTAIR bound sites (left panel). Red color intensity indicates the number of ChIRP-seq reads. The equivalent genomic windows in control and shEZH2 cells show that LacZ ChIRP retrieved no signal (right panel) while shEZH2 did not diminish or alter the profile of HOTAIR occupancy (middle panel). (B) ChIRP-qPCR validation of peaks from (A). TERC and GAPDH served as negative controls. Mean ± s.d. are shown.
DISCUSSION
Genomic maps of RNA-chromatin interaction by ChIRP-seq
Here we described ChIRP-seq, a method of mapping in vivo lincRNA binding sites genome-wide. The key parameters for success are the split pools of tiling oligonucleotide probes and glutaraldehyde crosslinking. The design of affinity-probes is straightforward given the RNA sequence and requires no prior knowledge of the RNA’s structure or functional domains. Our success with roX2, TERC, and HOTAIR-- three rather different RNAs in two species-- suggests that ChIRP-seq is likely generalizable to many lncRNAs. As with all experiments, care and proper controls are required to interpret the results. Different lincRNA may require titration of conditions, and judicious change of conditions, such as selection of different affinity probes or crosslinkers, may highlight different aspects of RNA-chromatin interactions. Like ChIP-seq, not all binding events are necessarily functional, and additional studies are required to ascertain the biological consequences of RNA occupancy on chromatin. Nonetheless, we foresee many interesting application of this technology for researchers of other chromatin-associated lncRNAs, which number now in the thousands (Khalil et al., 2009; Zhao et al., 2010). Just as ChIP-seq has opened the door for genome-wide explorations of DNA-protein interactions, ChIRP-seq studies of the “RNA interactome” may reveal many new avenues of biology.
Principles of lincRNA-chromatin interaction
ChIRP-seq has enabled the first genome-wide views of ncRNA occupancy on the human genome. Commonalities in the occupancy patterns of TERC and HOTAIR suggest several lessons for RNA-chromatin interactions. First, lincRNA binding sites are focal, specific, and numerous. In contrast to histone modifications which often broadly occupy certain genomic elements (e.g. promoters, enhancers, transcribed exons, or silent genes (Rando and Chang, 2009), the focal, interspersed, and gene-selective nature of lincRNA occupancy more resembles transcription factors. Even roX2, which binds across gene bodies of fly X-linked genes, shows focal peaks of high occupancy at CES sites. These results imply that certain lincRNAs may be “selector” elements that can access the genome in a highly discriminating fashion.
Second, lincRNAs access the genome through specific DNA sequences. Using ChIRP-seq, we show that genome-scale collections of RNA binding sites can be used to discover the enriched underlying DNA sequence motifs. These findings indicate the existence of an entirely new class of regulatory elements--lincRNA target sites--in the genome. For instance, we discovered a GA-rich homopurine motif for HOTAIR, a lincRNA known to recruit Polycomb. Importantly, mammalian Polycomb response elements are known to have a GAGA motif (Sing et al., 2009; Woo et al., 2010), but the cognate partner has been lacking. The HOTAIR motif also has similarities to the MSL binding motif in that both are GA-rich. But the HOTAIR motif is more degenerate than the MSL motif and does not strictly conserve a GAGA sequence. The discovery of specific RNA targeting motifs may start to unify at a mechanistic level many of the disparate phenomena that involve RNA-mediated chromatin states. The GA-rich HOTAIR motif may enable formation of RNA:DNA:DNA triplex (facilitated by homopurine runs and known mediate some lncRNA-chromatin interaction (Martianov et al., 2007; Schmitz et al., 2010), serve as the binding site of a protein that recruits HOTAIR, or indirectly configure a chromatin state that facilitates HOTAIR binding. Additional studies are required to evaluate these hypotheses, which are now possible due to ChIRP and knowledge of the candidate motif. HOTAIR also binds the LSD1-coREST-REST complex that can target DNA (Tsai et al., 2010), and multiple mechanisms may operate together to target lincRNAs.
Third, comparison of lincRNA occupancy map with chromatin state maps can reveal the order and logic of the regulatory cascade. For instance, comparison of HOTAIR versus Polycomb occupancy suggested that HOTAIR nucleates Polycomb domains. Focal HOTAIR binding sites (<500 bp) occur in the midst of a broad domain of Polycomb that can extend in both directions for several kilobases. This pattern argues that HOTAIR does not simply bind to or stabilize pre-existing Polycomb, which would have predicted broad co-occupancy of the two. Rather, the maps suggested that the RNA may be a pioneering factor that recruits Polycomb, which then spreads out bilaterally. To directly test the order of occupancy, we depleted PRC2 subunit EZH2 and showed that HOTAIR can bind to target chromatin genome-wide. This result uncouples the formation of HOTAIR-PRC2 ribonucleoprotein complex (the RNA scaffold function) from RNA targeting to chromatin. Because EZH2 is the enzymatic subunit of PRC2, H3K27me3 is also presumably not required a priori for HOTAIR targeting. Thus, the information for target gene selectivity resides in the RNA, which then recruits Polycomb to chromatin. Prior efforts have identified sequence motifs associated with PRC2 occupancy as a function of HOTAIR (Tsai et al., 2010), which may facilitate the spreading of PRC2 occupancy. We previously showed that EZH2 depletion diminished the metastatic potential of HOTAIR-expression cancer cells (Gupta et al., 2010). The ChIRP-seq data indicate that it is the lack of PRC2, rather than the inactivation of HOTAIR function at chromatin, that is responsible for this epistatic interaction. Together, these experiments suggest that lincRNAs are surprisingly like sequence-specific transcription factors in dictating chromatin states, and again suggests the utility of ChIRP to generate mechanistic insights.
EXPERIMENTAL PROCEDURES
Cell Culture
SuperTelomerase, MDA-MB-231-HOTAIR, MDA-MB-231 HOTAIR-shEZH2 cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS (HyClone) and 1% Pen/Strep (Invitrogen).
Probe Design
Morpholino Probes against HOTAIR were designed on three open regions detected by PARS-seq (ref) by Gene-Tools LLC (HOTAIR Morpho-1: GAGCAGCTCAAGTCCCCTGCATCCA, HOTAIR Morpho-2: GCACCCGCTCAGGTTTTTCCAGCGT, HOTAIR Morpho-3: TACATAAACCTCTGTTCTGTGAGTGC, Mock Morpho: CCTCTTACCTCAGTTACAATTTATA). All probes were biotinylated at the 3’ end. Antisense DNA probes were designed against HOTAIR full-length sequence using online designer at www.singlemoleculefish.com. All probes were compared with the human genome using the BLAT tool and probes returning noticeable homology to non-HOTAIR targets were discarded (BLAT searches through a non-overlapping 11-mers index). 48 probes were generated and split into two sets based on their relative positions along HOTAIR sequence such as even-numbered and odd-numbered probes were separately pooled. A symmetrical set of probes against LacZ RNA was also generated as the mock control. All probes were biotinylated at the 3’ end with an 18-carbon spacer arm (Protein and Nucleic Acid Facility, Stanford University). 19 probes were generated against TERC RNA and 24 for roX2 by similar methods. Sequences of all probes are listed in Table S4. The absolute levels of the ncRNAs in this study are as follows in Ct values per 100 ng of total RNA: roX2 =16.6; TERC=18.4; HOTAIR= 22.95. Thus, the fly and mammalian experiments are roughly comparable, and the mammalian experiments in fact show that ChIRP is compatible with lower expressed ncRNAs.
Crosslinking and chromatin preparation
Cells were grown to log-phase in tissue culture plates and rinsed once with room temperature PBS. For UV crosslinking, the plates were irradiated in UV crosslinker (Stratagene) with lids off and PBS aspirated. UV strength was titrated from 240mJ to 960mJ. For chemical crosslinking, cells were fixed on plate with appropriate amounts of 1% formaldehyde or 1% glutaraldehyde in PBS for 10 minutes at room temperature. Crosslinking was then quenched with 0.125M glycine for 5 minutes. Cells were rinsed again with PBS, scraped into Falcon tubes, and pelleted at 800g for formaldehyde crosslinking and 2500g for glutaraldehyde crosslinking. Cell pellets were then snap frozen in liquid nitrogen and can be stored in -80C indefinitely.
To prepare chromatin, cell pellets were quickly thawed in 37C water bath and resuspended in Swelling Buffer (0.1M Tris pH7.0, 10mM KOAc, 15mM MgOAc. Before use, add 1% NP-40, 1mM DTT, 1mM PMSF, complete protease inhibitor (GE), and 0.1U/ul Superase-in (Ambion)) for 10’ on ice. Cell suspension was then dounced and pelleted at 2500g for 5’. Nuclei was further lysed in nuclear lysis buffer at 100mg/ml (50mM Tris 7.0, 10mM EDTA, 1% SDS, add DTT, PMSF, P.I., and Superase-in before use) on ice for 10’, and sonicated using Bioruptor (Diagenode) until most chromatin has solubilized and DNA is in the size range of 100-500bp. Chromatin can be snap frozen in liquid nitrogen and stored in -80C until use.
Hybridization and washing
Chromatin is diluted in 2 times volume of hybridization buffer (500mM NaCl, 1%SDS, 100mM Tris 7.0, 10mM EDTA, 15% Formamide, add DTT, PMSF, P.I, and Superase-in fresh). 100pmol probes were added to 3ml of diluted chromatin, which was mixed by end-to-end rotation at 37C for 4 hours. Streptavidin-magnetic C1 beads were washed three times in nuclear lysis buffer, blocked with 500ng/ul yeast total RNA and 1mg/ml BSA for 1 hour at room temperature, and washed three times again in nuclear lysis buffer before resuspended in its original volume. 100ul washed/blocked C1 beads were added per 100pmol of probes, and the whole reaction was mixed for another 30min at 37C. Beads:biotin-probes:RNA:chromatin adducts were captured by magnets (Invitrogen) and washed five times with 40x beads volume of wash buffer (2x SSC, 0.5% SDS, add DTT and PMSF fresh). After last wash buffer was removed carefully with P-10 pipette so that no trace volume was left behind. Beads are now poised for different elution protocols depending on downstream assays.
ChIRP RNA elution
For reversible crosslinking (formaldehyde), beads was resuspended in 10x original volume of RNA elution buffer (Tris 7.0, 1% SDS) and boiled for 15min, followed by trizol:chloroform extraction and RNeasy mini column purification. For non-reversible crosslinking (UV and glutaraldehyde), beads were resuspended in 10x original volume of RNA pK buffer (100mM NaCl, 10mM Tris 7.0, 1mM EDTA, 0.5% SDS) and 0.2U/ul Proteinase K (Invitrogen). pK treatment was carried out at 65C for 45’, followed by boiling for 15’, and trizol:chloroform extraction. Eluted RNA was subject to quantitative reverse-transcription PCR (QRTPCR) for the detection of enriched transcripts.
ChIRP Protein Elution and Dot Blot
Beads were resuspended in 3x original volume of DNase buffer (100mM NaCl and 0.1% NP-40), and protein was eluted with a cocktail of 100ug/ml RNase A (Sigma-Aldrich) and 0.1U/ul RNase H (Epicenter), and 100U/ml DNase I (Invitrogen) at 37C for 30’. Protein eluent was supplemented with 0.2 volume of 5x laemmeli buffer (without bromophenol blue or glycerol), boiled for 5’, and dot blotted to nitrocellulose membrane with Bio-Dot apparatus (Biorad). Membrane was then blotted against TCAB1 and tubulin antibodies (gifts from Artandi lab) per normal Western protocol.
ChIRP DNA Elution
Beads were resuspended in 3x original volume DNA elution buffer (50mM NaHCO3, 1%SDS, 200mM NaCl), and DNA was eluted with a cocktail of 100ug/ml RNase A (Sigma-Aldrich) and 0.1U/ul RNase H (Epicenter). RNase elution was carried out twice at 37C with end-to-end rotation and eluent from both steps was combined. For formaldehyde crosslinking, chromatin was reverse-crosslinked at 65C overnight. For non-reversible crosslinking, eluted chromatin was pK treated with 0.2U/ul pK at 65C for 45’. In either case, DNA was then extracted with equal volume of phenol:chloroform:isoamyl (Invitrogen) and precipitated with ethanol at -80C overnight. Eluted DNA was subject to QPCR, Dot Blots, or high-throughput sequencing.
DNA Dot Blot
DNA was denatured in 0.1 volume of denaturing solution (4M NaOH, 100mM EDTA) at 95C for 5’, and then chilled on ice for 5’. Equal volume of chilled 2M NH4OAC was added to neutralize DNA on ice, which is then dot blotted onto nitrocellulose membrane using a Bio-Dot apparatus. Membrane was immediately crosslinked at 120mJ in Stratalinker, and pre-hybridized in Rapid-Hyb (GE) at 42C for 30’. Telomere and Alu repeats were detected using end-labeled radioactive Southern probes CCCTAACCCTAACCCTAACCCTAACCCTAA and GTGATCCGCCCGCCTCGGCCTCCCAAAGTG respectively.
Deep Sequencing, Peak Calling, Motif and GO Term Analysis
High-throughput sequencing libraries were constructed from ChIRPed DNA according the ChIP-seq protocol as described(Johnson et al., 2007), and sequenced on Genome Analyzer IIx (Illumina), with read length of 36bp. Raw reads were uniquely mapped to reference genome (hg18 assembly for HOTAIR, TERC, LacZ and EZH2 ChIRP-seq samples, and dm3 for roX2) using Bowtie (Langmead et al., 2009).
ChIRP-seq workflow consists of three steps.
- Find concordance: from the two independent ChIRP-seq experiments, we generate a consensus track, taking the lower value of the two at each coordinate. Thus, any aberrant signal in only one of the two experiments is removed. For each sample, reads from even and odd lanes were aligned separately, and per-base coverage was normalized as if there were 10M mappable reads. For each base pair of the genome, true coverage of this base in this sample was defined as the minimum coverage of the even lane and odd lane.
Genome wide signal consists of a combine lane, based on which, a SAM file was generated for peak calling. Find peaks: Peaks of each sample were called using MACS against its corresponding input with p-value cutoff 1e-5 (Zhang et al., 2008).
Filter peaks: For each MACS peak, we filter for peaks that share the same shape in the raw data from the two independent experiments. Only peaks with substantial correlation of the raw data profile, and high coverage across the peak are accepted. For each MACS predicted peak, a window size of +/-2kbp around peak summit or peak width, whichever is smaller, is selected. Within this window, an average coverage of the combine lane and a Pearson correlation between the normalized per-base coverage of the even lane and odd lane were calculated. MACS predicted peaks were further filtered based on peak length, fold enriched against input lane, average coverage, and Pearson correlation to obtain a list of true peaks. For HOTAIR ChIRP-seq sample, thresholds of average coverage>1.5, Pearson correlation>0.3, and fold enrichment against input>2 were applied to filter MACS predicted peaks and obtained 832 true peaks. Same thresholds were used to obtain 2198 true TERC peaks. For roX2 ChIRP-seq, similar parameters were used with the additional cut-off of peak length >2300bp, based on the fact that roX2/MSL complexes cover entire genes. 308 true peaks were obtained.
Sequences of top 500 true peaks (ranked by fold enrichment) within +/-200bp around peak summits were extracted and motifs analysis against these 500 peaks was performed using MEME (Bailey and Elkan, 1994). Only motifs of the highest significance were reported. Enriched gene sets were obtained through GREAT (McLean et al., 2010) on all 2198 TERC true peaks and all 832 HOTAIR true peaks. Gene Ontology of both gene sets were performed using DAVID (Huang da et al., 2009; Wishart et al., 2009).
roX2 ChIRP-seq Analysis
roX2 peaks and motif were obtained in a way described above, within 308 predicted true peaks, none was in autosomes, resulted a false discovery rate (FDR) = 0. Normalized signal of both the combine lane of Rox2 ChIRP-seq and MSL3-TAP ChIP-seq was obtained in a similar way described in HOTAIR ChIRP-seq analysis. Only regions where normalized signal is >=10 were counted in calculating the Pearson correlation between Rox2 and MSL3-TAP samples. Genes who overlaps >=1bp with windows +/-2kbp of true Rox2 peak summits were included in the average diagram. In total, 1087 RefSeq transcripts were included in chrX average diagram, and 4260 RefSeq transcripts were included in that of chr2L. Distance on the diagram was scaled with gene length, so that the diagram shows signal in a region from 50% gene length upstream to 50% gene length downstream.
TERC ChIRP-seq Analysis
Reads from “TERC ChIRP” sample and “Input” sample were compared against telomere sequence (CCCTAAx5) and Alu sequence (GTGATCCGCCCGCCTCGGCCTCCCAAAGTG). Complete matches were tallied and divided by total number of reads in that sample to give Reads per Million (RPM). RPMs from TERC enriched sample were divided with those from the Input sample to give “Fold Enrichment.” We note that the odd probes yields better enrichment of telomere than the even probes. Because the genome-wide TERC binding sites require by definition comparable pull down by both sets of probes, this result raise the possibility that TERC interacts with telomeres vs other genomic binding site via different mechanisms.
HOTAIR ChIRP-seq Analysis
Normalized signal within 10kb upstream and downstream of the summits of true HOTAIR peaks were extracted with a smooth window size of 50bp. Within each 50bp, the normalized HOTAIR ChIRP signal is calculated via:
Suz12, Ezh2 and H3K27Me3 ChIP-chip data were generated previous by Gupta et. al., 2010, Tsai et. al., 2010, and Rinn et. al., 2007. ChIP-chip signal of Suz12, Ezh2 and H3K27Me3 of 10kb upstream and downstream of HOTAIR peak summits were also extracted in a similar way.
Supplementary Material
Highlights.
ChIRP-seq maps the binding sites of specific RNAs on chromatin genome-wide.
RNA-genome interactions are numerous, focal, and sequence-specific.
Telomerase RNA TERC binds telomeres and Wnt pathway genes.
HOTAIR lincRNA nucleates broader domains of Polycomb and H3K27me3 occupancy.
Acknowledgments
We thank T. Hung, MC. Tsai, O. Manor, E. Segal, M. Kuroda, T. Swigut, and I. Shestopalov for discussions. Supported by the Agency of Science, Technology and Research of Singapore (C.C., F.L.Z.), NIH R01-CA118750 and R01-HG004361 (H.Y.C.), and California Institute for Regenerative Medicine (H.Y.C.). H.Y.C. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
Accession Number Deep sequencing data in this study are available for download from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) (accession ID: GSE31332).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, Terns MP. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30:2971–2982. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 2006;20:848–857. doi: 10.1101/gad.1400206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nature genetics. 2002;32:623–626. doi: 10.1038/ng1051. [DOI] [PubMed] [Google Scholar]
- Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, Cheung P, Jun S, Artandi MK, Shah N, et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008;4:e10. doi: 10.1371/journal.pgen.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, Baker BS. The rox1 and rox2 RNAs are essential components of the compensasome, which mediates dosage compensation in Drosophila. Mol Cell. 1999;4:117–122. doi: 10.1016/s1097-2765(00)80193-8. [DOI] [PubMed] [Google Scholar]
- Fusco D, Bertrand E, Singer RH. Imaging of single mRNAs in the cytoplasm of living cells. Prog Mol Subcell Biol. 2004;35:135–150. doi: 10.1007/978-3-540-74266-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood D. Theoretical and practical aspects of glutaraldehyde fixation. Histochem J. 1972;4:267–303. doi: 10.1007/BF01005005. [DOI] [PubMed] [Google Scholar]
- Horard B, Tatout C, Poux S, Pirrotta V. Structure of a polycomb response element and in vitro binding of polycomb group complexes containing GAGA factor. Molecular and cellular biology. 2000;20:3187–3197. doi: 10.1128/mcb.20.9.3187-3197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes & development. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 1999;98:513–522. doi: 10.1016/s0092-8674(00)81979-0. [DOI] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol MJ, Rinn JL. RNA traffic control of chromatin complexes. Curr Opin Genet Dev. 2010;20:142–148. doi: 10.1016/j.gde.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature. 2011;471:115–118. doi: 10.1038/nature09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi JC, Kelly WG, Panning B. Chromatin remodeling in dosage compensation. Annu Rev Genet. 2005;39:615–651. doi: 10.1146/annurev.genet.39.073003.094210. [DOI] [PubMed] [Google Scholar]
- Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445:666–670. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell. 1997;88:445–457. doi: 10.1016/s0092-8674(00)81885-1. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460:66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–271. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DD, Barrnett RJ, Bensch KG. New Means of Fixation for Electron Microscopy and Histochemistry. Anat Rec. 1962;142:274. [Google Scholar]
- Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes & development. 2010;24:2264–2269. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing A, Pannell D, Karaiskakis A, Sturgeon K, Djabali M, Ellis J, Lipshitz HD, Cordes SP. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell. 2009;138:885–897. doi: 10.1016/j.cell.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323:644–648. doi: 10.1126/science.1165357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, et al. HMDB: a knowledgebase for the human metabolome. Nucleic acids research. 2009;37:D603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell. 2010;140:99–110. doi: 10.1016/j.cell.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulla DC, Cech TR. RNA as a flexible scaffold for proteins: yeast telomerase and beyond. Cold Spring Harb Symp Quant Biol. 2006;71:217–224. doi: 10.1101/sqb.2006.71.011. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes & development. 2011;25:11–16. doi: 10.1101/gad.2006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.