

Abstract
Comparisons among evolutionarily related enzymes offer opportunities to reveal how structural differences produce different catalytic activities. Two structurally-related enzymes, E. coli alkaline phosphatase (AP) and X. axonopodis nucleotide pyrophosphatase/phosphodiesterase (NPP) have nearly identical binuclear Zn2+ catalytic centers, but show tremendous differential specificity for hydrolysis of phosphate monoesters or phosphate diesters. To determine if there are differences in Zn2+ coordination in the two enzymes that might contribute to catalytic specificity, we analyzed both x-ray absorption spectroscopic and x-ray crystallographic data. We report a 1.29 Å crystal structure of alkaline phosphatase with bound phosphate, allowing evaluation of interactions at the AP metal site with high resolution. To make systematic comparisons between AP and NPP, we measured zinc extended x-ray absorption fine structure (EXAFS) for AP and NPP in the free enzyme forms, with AMP and inorganic phosphate ground-state analogs, and with vanadate transition state analogs. These studies yielded average zinc-ligand distances in AP and NPP free-enzyme forms and ground-state analog forms that were identical within error, suggesting little difference in metal ion coordination among these forms. Upon binding of vanadate to both enzymes, small increases in average metal-ligand distances were observed, consistent with an increased coordination number. Slightly longer increases were observed in NPP relative to AP, which could arise from subtle rearrangements of the active site or differences in the geometry of the bound vanadyl species. Overall, the results suggest that the binuclear Zn2+ catalytic site remains very similar between AP and NPP during the course of a reaction cycle.
Keywords: x-ray absorption spectroscopy, crystal structure, nucleotide pyrophosphatase/phosphodiesterase, catalytic promiscuity, phosphoryl transfer
Introduction
Enzyme superfamilies are composed of evolutionarily and structurally related enzymes that catalyze different reactions yet share mechanistic features 1. These related enzymes often exhibit catalytic promiscuity, where members of a superfamily catalyze reactions of other superfamily members at a low level as a result of common mechanistic and structural features 1-3. This catalytic promiscuity presumably played a role in the evolution of new enzymatic activities 2,4 and currently offers the opportunity to make mechanistic comparisons to uncover how structural differences can lead to specialization and optimization of related active sites for different reactions. In addition to informing fundamental aspects of enzyme evolution and mechanism, this information may ultimately prove valuable in the development of new enzymes with practical uses.
The enzymes of the alkaline phosphatase superfamily catalyze several types of phosphoryl and sulfuryl transfer reactions, often with enormous rate accelerations 5,6. Escherichia coli alkaline phosphatase (AP), the most extensively studied member of the superfamily, catalyzes the hydrolysis of a broad range of phosphate monoester substrates with a two-metal-ion Zn2+ catalytic core 7-10 (Figure 1). Its acceleration of the second-order rate constant for hydrolysis of methyl phosphate is 1027-fold over the corresponding second-order reaction with water, one of the largest rate accelerations known 11-14. Members of another class of enzymes within the AP superfamily, the nucleotide pyrosphosphatases/phosphodiesterases, have the same two-metal-ion Zn2+ core as AP but preferentially hydrolyze phosphate diesters 15,16. The nucleotide pyrosphosphatase/phosphodiesterase from Xanthomonas axonopodis (NPP) has been recently characterized and found to have a nearly superimposable Zn2+ site with that in AP, and yet there is a differential specificity of 1015-fold for monoesters versus diesters between AP and NPP 11,15.
Figure 1.

(A) Alkaline phosphatase (AP) efficiently catalyzes phosphate monoester hydrolysis (top) and nucleotide pyrophosphatase/phosphodiesterase (NPP) efficiently catalyzes phosphate diester hydrolysis (bottom). The position of the leaving group is indicated by LG, and the second substituent of phosphodiesters is indicated by R. (B) and (C) Expected active site interactions in the transition state for phosphoryl transfer in AP and NPP, respectively.
The similarities and differences between AP and NPP outside of the Zn2+ site have provided insight into enzymatic features that allow the two enzymes to specialize toward monoester and diester hydrolysis (Figure 1). Two structural elements unique to AP have been investigated: an active-site arginine residue that can stabilize phosphate monoesters through dual hydrogen bonding interactions and the enzyme's third metal ion, Mg2+, the removal of which yielded a 105-fold loss in activity for monoesters with little effect on diesters 11,17-19. In NPP, interactions of substrate with a nucleotide-binding pocket facilitate reaction of specific diester substrates and could destabilize negative charge on monoesters 15,20.
Although the Zn2+ sites in the two enzymes appear similar and are expected to make analogous interactions in the transition states of the reaction (Figure 1), the Zn2+ sites themselves may also be involved in defining the catalytic specificity of the two enzymes by mediating effects of second-shell or more remote differences between the enzymes. It has been proposed, for example, that the distance between the metal ions might be used to predict whether a two-metal-ion enzyme catalyzes phosphate monoester or diester hydrolysis 21. This proposal is based on differences in the transition states of monoester and diester reactions in solution; monoesters proceed through looser transition states than diesters, with more extensive bond cleavage and minimal formation of a bond to the nucleophile 13,22-26. The Zn2+ sites in AP and NPP could also mediate catalytic specificity through other potential routes. The Zn2+ sites in the two enzymes could differ in Zn2+ coordination number or geometry in response to binding of ligands, they could differ in sensitivity of the coordination geometry to substrate charge or geometry, or the Zn2+ sites could present different charge distributions resulting from structural elements surrounding the Zn2+ site. In this paper, we utilize both x-ray crystallography and zinc x-ray absorption spectroscopy (XAS) to compare the Zn2+ sites in AP and NPP in the free enzyme forms, bound to substrate analogs phosphate and AMP, and bound to the vanadate transition state analog. These studies provide high-resolution experimental data as benchmarks for emerging QM/MM studies of AP and NPP 27-30 and they suggest some lengthening of average coordination distances upon binding of the vanadate transition state analog, but little overall difference in Zn2+ coordination between AP and NPP.
Results
1.29 Å Structure of E. coli Alkaline Phosphatase
The structure of wild-type alkaline phosphatase was determined with bound inorganic phosphate and was refined using data of up to 1.20 Å resolution with good signal to noise (I/σ > 3). Thus, although an effective resolution of 1.29 Å is reported here to reflect the range of data with >90% completeness (Table 1), the true resolution of the structure is actually somewhat better than 1.29 Å. The structure offers improvement over prior AP structures, which have had resolutions of ≥1.75 Å and, in the highest-resolution case, mixed metal occupancy of the Mg2+ site 31. Figure 2 shows electron density for the six Zn2+ ligands, the nucleophile, and noncovalently bound phosphate. Unambiguous density for the Zn2+ ions and phosphate was seen in all four active sites of the asymmetric unit. As was observed in prior AP structures with noncovalently bound phosphate 8,31, the Ser102 sidechain approaches closely to the noncovalent phosphate (∼1.8 Å O-O distance) in all four chains. Several possible alternative models to account for this apparently short contact were tested, but these models did not fully account for the observed electron density, suggesting that a close and likely repulsive contact between Ser102 and phosphate exists a significant fraction of the time. These and other comparisons to prior AP structures are included in the Supplemental Material. A repulsive interaction between Ser102 and phosphate could be a consequence of very favorable electrostatic interactions of phosphate and serine with the two zinc ions, as well as the protein fold that serves to position the zinc ions and Ser102. Such a repulsive interaction is also consistent with recent structural and binding results with wild-type and Ser102 mutants of AP 32 (Logan D. Andrews and D.H., manuscript in preparation), and similar short interaction distances are found in small molecule structures (see Supplemental Material).
Table 1.
X-ray crystallographic data collection and refinement statistics for AP.
| Wild-type AP | |
|---|---|
| Space Group | P21 |
| Unit Cell | |
| a | 66.0 Å |
| b | 98.5 Å |
| c | 152.1 Å |
| β | 94.6° |
| Wavelength | 1.0332 Å |
| Unique reflections | 501,190 |
| Resolution range | 50.0 Å – 1.20 Å |
| Effective resolution (completeness > 90%) a | 1.29 Å |
| Completeness | |
| 50.0 Å – 1.29 Å | 99% |
| 1.35 Å – 1.29 Å | 94.2% |
| 1.29 Å – 1.24 Å | 57.4% |
| 1.24 Å – 1.20 Å | 28.3% |
| I/σb | 17.9 (3.1) |
| Redundancyb | 3.4 (1.8) |
| Rmergeb,c | 0.06 (0.48) |
| Rfactor | 0.149 |
| Rfree | 0.169 |
| Number of residues | 1763 |
| Number of water molecules | 1878 |
| Mean B value | 12.2 Å2 |
| RMSD from standard geometry | |
| bond lengths | 0.013 Å |
| bond angles | 1.4° |
| Ramachandran plot statistics | |
| Most favored regions | 92.5% |
| Additional allowed regions | 7.5% |
| Others | 0% |
Reflections were included to 1.20 Å with high signal-to-noise (I/σ >3). However, because of low completeness of these highest-resolution shells, we indicate an effective resolution of the structure of 1.29 Å, corresponding to the resolution at which the completeness is > 90%.
Values in parentheses are for the highest resolution shell, 1.24 Å – 1.20 Å.
Rmerge = Σ| Iobs - Iave| / ΣIobs
Figure 2.

Electron density for E. coli alkaline phosphatase Zn2+ site and noncovalently bound inorganic phosphate. The active site of chain A is shown with 2Fo-Fc density contoured at 2.0 sigma.
X-ray Absorption Spectroscopy Studies of AP and NPP
XAS is a quantitative structural technique that probes the local environment of a metal ion at an atomic-level resolution. For XAS data collected at the metal K edge, the edge region of a spectrum is sensitive to the effective charge on the metal ion and the coordination geometry, while the extended x-ray absorption fine structure (EXAFS) region provides average metal-ligand bond distances and coordination numbers typically with ∼0.02 Å and 20-25% (± 1 coordination bond) accuracy, respectively. Zinc K-edge XAS has been used to investigate the structure of the active center in a number of zinc enzymes including carboxypeptidase A 33, HIV-2 integrase 34, protein farnesyltransferase 35, and alcohol dehydrogenase 36. A zinc EXAFS study for AP has been previously reported 37 but the protein contained only partially occupied or partially cobalt-substituted active sites. Here, zinc K-edge XAS has been measured to probe the local structure of the native binuclear Zn2+ site in AP and NPP, both unbound and in the presence of ground state and transition state analogs, with estimated Zn2+ occupancies of 90% or more.
Figure 3 compares the normalized zinc K-edge spectra for the free, phosphate-bound, and vanadate-bound forms of AP and NPP; for each sample, the data represent an average XAS signal of two zinc atoms at the active center. For all AP protein forms, the energy position of the rising edge is identical (Figure 3A), indicating that the average local charge on the zinc remains the same throughout the series, despite the addition of anionic ligands. At the same time, the main edge peak at 9667–9670 eV changes in shape and intensity from free to phosphate-bound to vanadate-bound AP. Although Zn K-edge features are sensitive to the three-dimensional structure of an absorbing zinc site, they typically cannot be directly attributed to specific structural properties 38. Thus, the observed spectral changes suggest differences in zinc coordination among the three AP forms but do not reveal the nature of the differences. Similarly to AP samples, the NPP samples display no differences in the energy position of the rising edge (Figure 3C). However, while the edge spectra for free and AMP-bound NPP are similar, the edge shape and intensity for vanadate-bound NPP differ considerably from those for free NPP, suggesting a significant change in the average zinc coordination environment upon vanadate binding to NPP.
Figure 3.

Zn K-edge XAS data for AP (left) and NPP (right) protein samples: free proteins (black), phosphoryl-group bound (red), vanadate-bound (blue). (a,c) Normalized Zn K-edge spectra; (b,d) Fourier transforms (nonphase shift-corrected) and the EXAFS data (inset). The plotted Fourier transforms were not corrected for the phase shift, i.e., the Fourier transform peaks corresponding to the Zn-ligand distance of R are shifted to lower distances by Δ = 0.4-0.5 Å.
The k3-weighted Zn EXAFS data to k of 12.5 Å-1 and corresponding non-phase-shift-corrected Fourier transforms (FT) for the AP and NPP series are illustrated in Figures 3b and 3d. For all samples, the Fourier transform, which is related to the radial distribution of atoms around an absorbing zinc atom, displays a single first shell peak at R + Δ = 1.5 Å and a series of weaker outer-shell peaks at R + Δ = 2.0–4.0 Å1. Based on the crystal structures of AP and NPP (see Methods), the first-shell peak represents the scattering of 4-5 oxygen and nitrogen atoms located at R = ∼2.0 Å from zinc, whereas the outer shell features arise from the single and multiple scattering of the carbon, nitrogen, and oxygen atoms of His, Asp and Ser/Thr residues at distances from R = ∼3-5 Å, and possibly of the phosphorus and vanadium atoms in the phosphate- and vanadate-bound protein derivatives. Although all AP and NPP protein forms contain an average of 1.5 histidine ligand per zinc, the observed outer-shell peak pattern does not match that of zinc proteins with predominantly histidine environments 34,39, suggesting static disorder of the histidine ligands or comparable contributions of other ligands to the outer-shell scattering. In addition, no obvious Zn···Zn scattering peak (which could be expected at R + Δ = 3.5–4 Å if Zn···Zn scattering was substantial) is visible in any of the Fourier transforms. Similarly to the observations from the free AP zinc K-edge data, the EXAFS and FT spectra for phosphate- and vanadate-bound AP are slightly perturbed relative to free AP (Figure 3b). There is a larger difference in the NPP series; the EXAFS and FT for vanadate-bound NPP exhibit larger differences from those for free and AMP-bound NPP than the differences seen with AP (Figure 3c & d). These differences were quantitatively evaluated by curve-fitting analysis of the EXAFS data in k space (see Methods).
The EXAFS fitting results for the first coordination shell of zinc for AP and NPP protein derivatives are summarized in Table 2, which lists average zinc-ligand distances R and bond variance values σ2 for fits with selected average zinc coordination numbers N. In all cases, the first coordination sphere was modeled well with a single shell of 4-5 oxygen and nitrogen ligands at a short distance from each zinc. The free and phosphate-bound forms of AP and NPP display essentially identical 1.97–1.98 Å average bond lengths, while vanadate-bound AP and NPP show longer 2.00 and 2.03 Å distances, respectively. For all samples, the one-shell fit was not improved by the addition of another oxygen scatterer at a short (1.80–1.85 Å) or long (2.15–2.30 Å) distance from zinc.
Table 2.
First-shell parameters for the final EXAFS fits for AP and NPP protein samples for selected coordination numbers N.a
| Zn-N/O | |||
|---|---|---|---|
| Sample | Nb | R (Å)c | σ2 × 103 (Å2)d |
| AP-free | 4 | 1.98 | 5.6 |
| AP-phosphate | 4 | 1.97 | 5.8 |
| AP-vanadate | 4.5 | 2.00 | 6.4 |
| NPP-free | 4 | 1.97 | 4.0 |
| NPP-AMP | 4 | 1.97 | 4.0 |
| NPP-vanadate | 5 | 2.03 | 7.8 |
Fitting parameters that were varied in each fit are denoted in italics.
Average zinc coordination number, fixed at the best-fit values determined by bond-valence sum analysis.
Average zinc-ligand distances. The estimated uncertainty in R is 0.02 Å.
Bond variance values.
The first-shell coordination numbers N for AP and NPP samples could not be derived from the EXAFS data alone. The fit error changed insignificantly when N was varied between 4.0 and 5.5 for free and phosphate-bound AP and NPP, and between 4.5 and 6.0 for the vanadate-bound forms (See Supplemental Material). The low precision of EXAFS coordination numbers (uncertainty of ±20-25%) is a well-known limitation of the EXAFS technique 38, which arises in part from the significant correlation between N and σ2 values in a fit, as well as from combining nonequivalent atoms into a single shell and assuming the distribution of their distances to be Gaussian. On the other hand, average EXAFS bond lengths are determined with a significantly better accuracy (±0.01-0.02 Å) and are much less correlated to either N or σ2 values. For example, the EXAFS-derived bond lengths for AP and NPP remain the same when the coordination number is varied from 4 to 6 in the fits (Supplemental Material). Because metal-ligand bond lengths themselves are related to the metal coordination number, it is possible to estimate the coordination number from EXAFS-derived distances using a bond-valence sum analysis.
Bond-valence sum (BVS) analysis correlates the oxidation state of the metal to the sum of the metal-ligand bond strengths, or bond valences 40,41. The bond valence (BV) of a bond between two atoms is assumed to be a function of the bond length R:
| (1) |
where R0 values are empirically determined values tabulated for specific metal-ligand pairs and B is empirically determined and set to 0.37 Å 42. The sum of bond valences (BVS) for all bonds formed by an atom is expected to equal the valence of that atom, which for a metal cation is the same as the oxidation state, i.e., two for Zn2+. Studies of metal sites in a variety of metalloenzymes have indicated that calculated BVS values typically fall within ∼0.2 units from the metal oxidation state 40,41. Table 3 gives calculated BVS values for AP and NPP using EXAFS-determined bond lengths. These were evaluated for each of the possible coordination numbers for the two- Zn2+ system (4.0, 4.5, 5.0, 5.5, and 6.0) using R0 = 1.77 Å for Zn-N, 1.704 Å for Zn-O 43, and assuming 1.5 Zn-N bonds at an average site of AP and NPP. For comparison, the BVS values were also calculated for a number of crystallographically characterized 4-, 5-, and 6-coordinate small-molecule zinc complexes with mixed N/O first-shell ligation from the Cambridge Structural Database 44 (See Supplemental Material); only complexes with one or two nitrogen atoms in the first shell were considered, and an average Zn-O/N distance for each molecule was used to calculate its BVS. The results show that under these conditions the average BVS value for the entire set of 4-6 coordinate zinc is 2.08 ± 0.08 Å (Table 4). When this range is compared to the BVS values calculated for the EXAFS-derived distances in AP and NPP samples (Table 3), the observed distances of 1.97 and 1.98 Å suggest 4-coordinated complexes (BVS values of 2.09 and 2.04, respectively), while R = 2.00 Å is the closest to 4.5-coordination (BVS = 2.15). For R = 2.03 Å, the BVS values for both 4.5- (BVS = 1.99) and 5-coordination (BVS = 2.19) are close to the reference value and thus either model is compatible with the EXAFS data. Thus, based on average bond lengths from EXAFS, the BVS analysis yields a coordination number of 4.0 for free and phosphoryl-bound AP and NPP, 4.5 for AP-vanadate, and 4.5 or 5.0 for NPP-vanadate.
Table 3.
Zn bond-valence sum (BVS) values calculated using EXAFS Zn-O/N distances (R) of AP and NPP samples for a range of possible Zn coordination numbers (N).a
| Zn BVS | |||||
|---|---|---|---|---|---|
| N | |||||
| RZn-O/N(Å) | 4 | 4.5 | 5 | 5.5 | 6 |
| 1.97 | 2.09 | 2.34 | 2.58 | 2.82 | 3.07 |
| 1.98 | 2.04 | 2.27 | 2.51 | 2.75 | 2.98 |
| 2.00 | 1.93 | 2.15 | 2.38 | 2.60 | 2.83 |
| 2.03 | 1.78 | 1.99 | 2.19 | 2.40 | 2.61 |
Table 4.
Average Zn-L bond distances Rave and BVS values for small-molecule Zn complexes with mixed O/N ligation and total coordination number N. a
| NZn-O/N | Nobsb | Rave (Å) | BVS |
|---|---|---|---|
| 4 | 669 | 1.98 (0.02) | 2.06 (0.10) |
| 5 | 435 | 2.05 (0.02) | 2.09 (0.07) |
| 6 | 217 | 2.12 (0.02) | 2.08 (0.07) |
The CSD v5.29 was used to search for Zn(N1On) (n = 3, 4, 5) and Zn(N2Om) (m = 2, 3, 4) complexes. All searches required R-factor ≤ 0.065 and non-polymeric structures with no disorder and no unresolved errors. The average first-shell Zn-O/N distance Rave was calculated for each complex and then averaged over all complexes of the same total coordination number NZn-O/N. The BVS values were calculated for each molecule using its Rave; R0 and B values were the same as in Table 3. The BVS values were averaged over all molecules of the same NZn-O/N. The sample standard deviations are given in parentheses.
Number of crystallographically independent observations.
Discussion
X-ray crystallography and x-ray absorption spectroscopy offer complementary approaches to evaluating active-site interactions in metalloenzymes. Crystal structures provide a structural context and reveal identities of groups interacting with the metals, while XAS allows high-precision comparisons of metal coordination environments across a series of enzyme and ligand combinations. Here, we combine analyses of crystal structures and XAS data to systematically evaluate Zn2+ coordination in the AP and NPP active sites.
The 1.29 Å crystallographic structure reported herein provides a high-resolution picture of protein-metal ion interactions in AP. Several prior structures of AP have been reported, including wild-type 8,31,45-47 and mutant enzymes 11,19,48-58 and structures with alterative metal ions occupying the Zn2+ sites 59,60. The improved resolution of the current structure relative to prior structures of AP provides a more precise determination of the coordination geometry around the Zn2+ ions, with average coordinate errors of ∼0.04 Å, as compared to an estimated ≥∼0.2 Å for prior structures 8,31,45-47. Figure 4 shows a schematic of the Zn2+ interactions with the average and standard deviations from the four chains. The two Zn2+ ions are coordinated by three His and three Asp sidechains, with additional close contacts to the Ser102 nucleophile and inorganic phosphate bound in the active site.
Figure 4.

Schematic of Zn2+ interactions in 1.29 Å structure of E. coli alkaline phosphatases with phosphate bound. For each interaction, distances in Ångströms correspond to the averages and standard deviations from the four chains of the asymmetric unit.
Comparison of the Zn2+ coordination distances to those from other available AP and NPP crystal structures (Figure 5 and Supplemental Figure S6) illustrates that variation in structural resolution complicates the analysis. The available structures include free enzymes as well as enzymes bound to substrate analogs and vanadate transition state analogs. These different forms of the enzymes span wide ranges of resolution, and thus differences in metal coordination distances may arise from differences between AP and NPP, differences upon binding substrate or transition state analogs, differences in protonation states, and/or differences in resolution. The highest-resolution structure of AP reported here has bound phosphate, while the highest-resolution NPP structure available (2GSO, 1.45 Å) has bound vanadate, preventing direct comparison even among the highest-resolution structures.
Figure 5.

Comparison of Zn-sidechain and Zn-Zn distances among the available structures of AP (blue) and NPP (red). Zinc contacts with His 412 (AP) and His363 (NPP) are given as an example, and comparisons for all other zinc-ligand interactions are provided in the Supplemental Material. Wild-type AP structures are shown in closed squares and mutants are shown in open squares. Both free enzymes and enzymes bound to substrate and transition state analogs are included in the plots. Each point represents a single active site within the asymmetric unit of the corresponding crystal structure. The dashed line indicates average interaction distances from zinc-nitrogen interactions from small-molecule crystal structures 63,66.
Plotting Zn-ligand distances for AP and NPP as a function of resolution (Figure 5 and Supplemental Figure S6) illustrates that much of the variation in Zn-ligand distances occurs at lower resolutions and among AP mutants; variations among different bound and unbound enzyme forms may also contribute but cannot be distinguished from variations in resolution. The Zn-ligand distances in the 1.29 Å structure reported here are very similar to average Zn-N and Zn-O distances observed in high-resolution structures of small molecules (dashed lines in Figure 5 and Supplemental Figure S6), consistent with prior observations that protein-metal interactions tend to converge to near-canonical values with higher resolution 61-64. An exception is Asp327 (Asp210 in NPP), which forms a semi-bidentate interaction with Zn2+ and thus has longer interaction distances 63. The Zn-Zn distance shows a similar degree of variation among lower-resolution structures. The average Zn-Zn distances in all structures (4.18 ± 0.18 Å) and all wild-type structures (4.15 ± 0.12 Å) closely resemble the average Zn-Zn distance in the highest resolution AP structure of 4.12 ± 0.01 Å.
To make direct comparisons between AP and NPP in the same states with high precision, we used x-ray absorption spectroscopy. We first considered whether there were differences in Zn2+ coordination in the free-enzyme forms of AP and NPP. As one example of a possible difference, a prior proposed model holds that the geometry of the Zn2+ sites in the resting states of the enzyme, and specifically the Zn-Zn distance, determines the specificity of the enzyme for monoester or diester reactions 21. Crystal structures suggested that this model did not hold for AP and NPP 15.2 However, as mentioned above, direct comparison of crystal structures is complicated by both differences in resolution and differences among bound and unbound states of the enzyme, and other differences in metal coordination beyond Zn-Zn distance could play roles in defining the specificity of the two enzymes. Thus, comparison of free enzyme EXAFS provides an alternative route to evaluate intrinsic differences between the AP and NPP coordination spheres, although the ∼4 Å Zn-Zn separation is not detectable in the absence of bridging ligands. The EXAFS data indicated nearly identical average Zn-ligand distances for AP and NPP of 1.97-1.98 Å, and the EXAFS and BVS analysis suggested coordination numbers of 4.0 for both enzymes. These values suggest, most simply, that there are no significant intrinsic differences in the Zn2+ coordination environment between AP and NPP. The strong overall similarity between AP and NPP EXAFS spectra for AP and NPP (Supplemental Figure S5) suggests that marked differences in the Zn2+ environment between the two enzymes are unlikely in the free enzyme.
Differences between AP and NPP could also arise in response to binding of substrates and transition states to the bimetallo site. QM/MM computational studies of the Zn2+ sites in AP and NPP have predicted changes in geometry of the Zn2+ sites during the process of the reaction pathway 27-30, in one case reaching the remarkably and unexpectedly long Zn-Zn distance of 7 Å in the transition state, as compared to ∼4.1 Å in crystal structures 29. While the EXAFS data cannot determine Zn-Zn distances of this length, these changes in geometry of the metal sites would likely result in additional changes to the first-shell coordination numbers, distances, and geometries, particularly since the bimetallo site is anchored to the structural core of the two enzymes, which shows low B-factors in all crystal structures and no evidence of significant deformability. Conversely, differences in Zn2+ coordination between AP and NPP could arise independently of Zn-Zn distances. To evaluate whether differences in Zn2+ coordination could be identified in AP and NPP in response to binding of ground-state substrate analogs, we measured EXAFS in the two enzymes in response to binding of inorganic phosphate and AMP, respectively.
AP binds inorganic phosphate with Kd = 1 μM at pH 8 19. NPP does not bind phosphate tightly, so we used the phosphate monoester AMP for comparison. AMP binds NPP with a Kd of 260 μM at pH 8, with the adenosine group occupying a binding pocket on the enzyme that is unique to NPP 15. The binding modes of the phosphoryl group in the substrate analogs differ between the two enzymes, with the phosphate group making two Zn2+ contacts in AP and one contact in NPP (Figure 6A). Prior studies have shown that formally the dianionic form of phosphate (HPO42-) binds to AP and the monoanionic form of AMP binds to NPP (R-HPO41-) 15,65. However, recent vibrational spectroscopy studies strongly suggest that upon binding to AP, the phosphate dianion proton is transferred to Ser102, such that the stable bound form has protonated SerOH and phosphate trianion (PO43-) (Figure 6C) 32. An analogous transfer would seem likely in NPP, but this possibility has not been tested. Based on typical Zn-O distances in small molecule structures, differences in average coordination distances due to alternative protonation states of the Ser/Thr alkoxide nucleophile may be on the order of 0.01-0.02 Å for the binuclear Zn2+ sites and may not be detectable in the EXAFS experiments 66.
Figure 6.
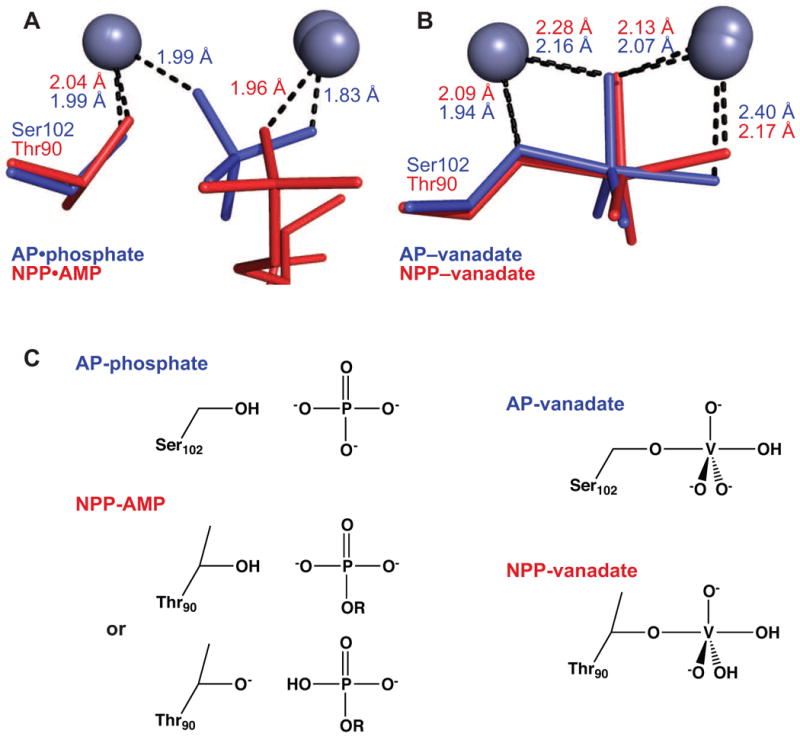
Interactions of AP and NPP with ground-state and transition-state analogs. The indicated distances are averages over all monomers in the asymmetric unit. (A) Overlay of structures of AP bound to inorganic phosphate (reported herein, 1.29 Å) and NPP bound to AMP (2GSU, 2.0 Å). (B) Overlay of vanadate-bound structures of AP (1B8J, 1.9 Å) and NPP (2GSO, 1.45 Å). (C) Schematic of expected protonation states in AP and NPP upon binding phosphate, AMP, and vanadate. No experimental evidence addresses the position of the proton in the NPP•AMP complex, but the proton may transfer to Thr90, analogous to a model based on isotope-edited IR studies in AP 32.
The EXAFS data for these bound species indicate no measurable change in Zn-ligand coordination numbers or distances relative to the free enzyme forms (Table 2). In AP, the average Zn-ligand distance remained at 1.97 Å upon phosphate binding, with coordination number 4.0. This average Zn-ligand distance from EXAFS (1.97 ± 0.02 Å) is within error of that in the x-ray structure (2.02 ± 0.04 Å). Moreover, the narrow distribution of the first-shell Zn-ligand distances in the x-ray structure (1.97–2.10 Å for eight bonds out of nine) is consistent with the low value of the bond variance σ2 of 0.0058 Å2 observed in EXAFS. In NPP, the average Zn-ligand distance remained at 1.97 Å upon binding of AMP with coordination number 4.0. These results suggest, most simply, that no significant changes in Zn coordination geometry occur upon binding of the ground state analogs for both AP and NPP. Despite the binding of the phosphate groups, the different binding modes in the two enzymes, and possibly differences in protonation states of the phosphate groups in AP and NPP, the spectra, the average distances, and the estimated coordination numbers are nearly identical. We next considered the binding of transition state analogs to AP and NPP.
Vanadate can adopt both tetrahedral and trigonal bipyramidal forms, and thus vanadate esters have been used in numerous systems to mimic the trigonal bipyramidal transition states of phosphoryl transfer reactions 67,68. Vanadate binds AP and NPP with Kd values of 10 μM and 50 μM, respectively, at pH 8 9,15. Crystal structures of AP and NPP show trigonal bipyramidal vanadate esters bound in the active sites in similar orientations that are consistent with stabilization of the nucleophile and leaving group oxygens by the Zn2+ ions (Figure 6B) 15,45. Differences in protonation state between AP- and NPP-bound forms of vanadate are expected, as discussed below.
In contrast to the data for substrate analogs, the EXAFS data for the transition state analog vanadate indicated longer Zn-ligand distances (2.00 for AP and 2.03 Å for NPP) and BVS analysis suggested higher coordination numbers in both AP and NPP (4.5 for AP and 4.5-5.0 for NPP) (Tables 2 and 3). These observations are consistent with an additional coordinating interaction with oxygen in the position of the leaving group oxygen in the transition state, as suggested from crystal structures of AP and NPP with the bound pentavalent vanadyl group (Figures 1 and 6). A strong stabilizing interaction with the leaving group oxygen is expected to be a crucial feature of catalysis in phosphoryl transfer reactions of both phosphate monoesters and diesters. In transition states for phosphate monoesters and diesters, the P-O bond to the leaving group is substantially or partially broken, resulting in a development of negative charge, and stabilization of this charge could contribute several orders of magnitude to rate acceleration 13. While the vanadate transition state analog does not have the same distribution of charge as these transition states, its trigonal bipyramidal geometry is expected to mimic the interactions of the transition state with the Zn2+ ions 15,45.
The average Zn-ligand distances in AP and NPP vanadate-bound forms are similar and within experimental uncertainty (Table 2), suggesting that the two enzymes have very similar zinc coordination in the vanadate-bound complexes. Nevertheless, the longer average coordination distance of 2.03 Å in NPP relative to 2.00 Å in AP (with uncertainties of 0.02 Å) and the different appearance of the NPP-vanadate EXAFS spectrum relative to all other spectra (Figure 3) suggest that there are small differences in the Zn2+ sites of the two enzymes upon binding of vanadate.
Whereas the phosphate-bound forms of AP and NPP showed identical average distances by EXAFS but relatively different crystallographic binding modes (Figure 6A), the vanadate-bound forms of AP and NPP had different average EXAFS distances but overall similar binding modes in crystal structures15,45 (Figure 6B). Nevertheless, the vanadate-bound forms of AP and NPP show some variation in contact distances to Zn2+. Vanadate can adopt varied apical O-V-O angles, as seen in Figure 6B, and, further, the charge distribution and geometry of trigonal bipyramidal vanadate esters are not identical to those in phosphoryl transfer transition states67-70. The EXAFS differences between AP and NPP in the vanadate-bound forms could reflect mechanistically important distinctions between the two enzymes in the transition state or they could result from imperfections of vanadate as a transition state analog and in its ability to respond to its local environment.
Differences in zinc-vanadate interactions between AP and NPP could arise because vanadate lacks the ester substituent present in the native diester substrates of NPP. Interactions of the nucleotide-binding pocket of NPP with the second ester substituents of phosphate diesters can serve to position the phosphoryl group in the transition states of these reactions (Figure 1). Because the vanadate transition state analog lacks this group, the NPP active site may not be suited to optimal positioning of vanadate and, further, the relatively hydrophobic surface surrounding the nucleotide-binding pocket could destabilize a vanadate oxygen bound in that site. Indeed, mutational studies suggest that the hydrophobic side chains at this site reduce activity with phosphate monoesters (H.W.-K. and D.H., unpublished data). Thus, vanadate may be positioned differently in the NPP Zn2+ site relative to the AP active site, as AP can position the phosphoryl group in monoesters through direct interactions with the unsubstituted equatorial oxygens (Figure 1). Vanadate is also likely to have different protonation states in the two enzymes that could result in differences in Zn2+ coordination. pH-binding profiles indicate that AP formally binds primarily the dianion form of vanadate (HVO42-), whereas NPP binds primarily the monoanion form (H2VO41-) (See Supplemental Material) (Figure 6C). Similarly, pH-rate profiles suggest that AP reacts preferentially with dianionic phosphate monoesters, whereas the promiscuous reactivity of NPP toward monoesters preferentially occurs with monoanionic phosphate monoesters 15,65.
Overall, the EXAFS data for the free enzyme and enzyme with bound substrate and transition state analogs suggest that despite differences in protonation states and binding modes, the Zn2+ coordination in AP and NPP remains very similar throughout the reaction cycle. The structure of the Zn2+ site in each of these states is likely to resemble that in the 1.29 Å AP structure, the highest-resolution structure to date of an AP-superfamily two-metal enzyme. Although the structure and EXAFS data reported here were collected at cryogenic temperatures, prior AP structures determined at room temperature8,31 show very similar atomic positions and active-site temperature factors, further suggesting that the degree of variation in active-site structure is small.
The similarity of the EXAFS results for AP and NPP suggest, most simply, that the metal sites of AP and NPP act very similarly as catalytic centers in the two enzymes and that active site elements beyond the zinc sites, rather than differences in the structure of the bimetallo site, are responsible for specialization for monoester or diester hydrolysis. Small differences between AP and NPP may be expressed upon binding of the vanadate transition-state analog, but it remains to be determined whether these directly reflect differences in the transition states or indirectly reflect differences in the properties of the overall active sites. The systematic evaluation of AP and NPP variants that lack key distinguishing structural features offers the potential to further probe how structural differences define enzymatic specialization.
Materials and Methods
AP Structure Determination
Protein for structure determination was produced under control of the native AP promoter as previously described 71,72, including osmotic shock to collect the periplasmic fraction, a heat treatment step, and ion exchange and gel filtration chromatography steps. Wild-type AP was crystallized by the sitting drop method at 4 °C in the dark in 20% PEG 4000, 0.2 M HEPES, pH 8.0, 1 mM ZnCl2, and 10 μM MgCl2. A 30 mg/mL protein solution was used. The crystal used for the 1.3 Å structure reported here had been grown over ∼6 months under these conditions and was subsequently cryo-protected in 40% PEG 4000. These conditions yielded crystals of space group P21, with two dimers per asymmetric unit. Data were collected at Stanford Synchrotron Radiation Lightsource (SSRL) beamline 9-2 at 100 K and were processed with DENZO and SCALEPACK 73. Five percent of the data were set aside for calculation of Rfree. Molecular replacement was performed with Phaser 74 using wild-type AP structure 1ALK 8 as a search model. Cycles of refinement with PHENIX 75 and Refmac 76 and model building with Coot 77 were performed. Noncovalently bound phosphate was modeled in the active site of AP, as in prior structures 1ALK 8 and 1ED8 31. Although no additional phosphate was added during crystallization, phosphate copurifies with wild-type AP and is also found as a contaminant of commercial PEG solutions 11,31,32. The occupancy of non-covalently bound phosphate refined to 69-76% in the four chains of the asymmetric unit. Additional refinement tests were performed to evaluate alternative possible configurations of the active site (See Supplemental Material). In chain C only, difference density consistent with an alternate conformation of residues 403-408 was observed. However, the alternate conformation was not unambiguously assigned and modeling did not reduce Rfree. Accordingly, an alternative conformation was not included in the final model. Final refinement with anisotropic temperature factors was performed with Refmac 76.
Sample Preparation for X-ray Absorption Spectroscopy
Wild-type AP used for XAS was produced from a maltose binding protein fusion construct as previously described 11,17. This procedure yields the same full-length protein with identical activity to AP purified using the native promoter and signal peptide 11,17,71. Following elution from the final Hi-Trap Q Sepharose HP column in the reported procedure 11, peak fractions were pooled, buffer exchanged into 10 mM NaMOPS, pH 8.0, 50 mM NaCl, and concentrated to 2 mM by centrifugal filtration (Amicon Ultra, 10 kDa cutoff). Phosphate-containing AP samples were produced by diluting two-fold by addition of a solution of 70% glycerol and Na2HPO4 was added to a final concentration of 1 mM.
To remove phosphate from AP to produce the free AP and AP-vanadate samples, enzyme was diluted into 20 mL of 1 M Tris•HCl, pH 8.0, to a final concentration of 20 μM. This solution was dialyzed against 2 L of 1 M Tris•HCl, pH 9.0, at room temperature overnight with Spectra-Por 7 dialysis tubing, 10 kDa cutoff. The dialysate was changed and this procedure was repeated over four days. Following the final dialysis, protein was removed from dialysis, concentrated, and buffer exchanged into 10 mM sodium MOPS, pH 8.0, 50 mM NaCl by centrifugal filtration (Amicon Ultra, 10 kDa cutoff). Removal of phosphate from the enzyme samples was confirmed by either atomic emission spectroscopy or a malachite green assay 78. For the free AP sample, the enzyme was concentrated to 1 mM in the buffer solution and then diluted 2-fold by addition of 70% glycerol. For the AP-vanadate samples, the enzyme was concentrated to 1 mM in the buffer solution, and then diluted two-fold by addition of 70% glycerol. Sodium orthovanadate was added to a final concentration of 1 mM.
NPP was expressed and purified as previously described from a maltose binding protein fusion 15. Following elution from the final Source S ion exchange column as in the reported procedure, peak fractions were pooled and buffer exchanged into 20 mM sodium MES, pH 6.0, 100 mM NaCl. This lower pH was used for NPP to permit higher enzyme concentration 20; NPP exhibits a flat pH-rate profile within this region 15. EXAFS spectra of AP at pH 6.0 and NPP at pH 8.0 were within error of those reported here. The protein was concentrated to 1-2 mM in the buffer by centrifugal filtration (Amicon Ultra, 10kDa cutoff). The samples were then diluted 2-fold by addition of 70% glycerol, with AMP or Na3VO4 included when indicated for a final concentration of 1 mM.
The conditions used for AP and NPP XAS measurements were selected to optimize precision for AP and NPP comparisons. Although the conditions used for EXAFS and structure determination could not be exactly matched, the differences in conditions are expected to have minimal effect. While structure determination and prior kinetic assays have included excess Zn2+ and Mg2+ in the buffers to ensure full metal occupancy, free metals had to be minimized for EXAFS, so the metal content of AP and NPP samples for EXAFS was tested directly by atomic absorption. The pH values of mock solutions for EXAFS were verified by a pH meter, and the same pH values were used for structure determination. While the EXAFS samples contained 35% glycerol and the AP crystals were grown with 20% PEG, these additives are not expected to alter the structures, and prior structures of AP determined with different precipitants superimpose closely.
X-ray Absorption Spectroscopy
For each protein sample, ∼100 μL of solution was transferred into a Lucite XAS cell with 37 μm Kapton tape windows and frozen in liquid nitrogen. The XAS data were recorded at SSRL on focused 16-pole wiggler beam line 9-3, with the ring operating at 3 GeV, 80– 100 mA. A Si(220) double-crystal monochromator was used for energy selection at the Zn K edge, and a Rh-coated mirror upstream of the monochromator was used for harmonic rejection and collimation. The samples were maintained at 10 K during data collection by using an Oxford Instruments CF1208 continuous-flow liquid-helium cryostat. Data were measured in fluorescence mode as Zn Kα fluorescence by using a Canberra (Meriden, CT) 30-element solid-state Ge array detector. The internal energy calibration was performed by simultaneous measurement of the absorption of Zn foil placed between two ionization chambers filled with nitrogen located after the sample. The first inflection point of the foil was assigned to 9660.70 eV. No photodegradation was observed for any of the samples. The averaged data included 14 scans for free AP, 11 scans for AP-phosphate, 17 scans for AP-vanadate, 11 scans for free NPP, 9 scans for NPP-AMP, and 12 scans for NPP-vanadate.
The averaged data were normalized with the program XFIT 79 by first subtracting a polynomial background absorbance that was fit to the pre-edge region and extended over the post-edge, followed by fitting a three-region polynomial spline of orders 2, 3, and 3 over the post-edge region. The data were normalized to an edge jump of 1.0 between the background and spline curves at 9675 eV. Theoretical EXAFS signals χ(k) were calculated using FEFF (version 7.02) 80 and fit to the nonfiltered data by EXAFSPAK (G.N. George, SSRL). The input structure for FEFF was revised according to the fit results obtained. The experimental energy threshold, E0, i.e. the point at which the photoelectron wavevector k equals zero, was initially chosen as 9675 eV and was varied in each fit using a common value ΔE0 for every component in the fit. The scale factor, S02, was set to 1.0. The structural parameters that were varied during the refinements included the bond distance (R) and the bond variance (σ2) for each shell of atoms. Atom types and coordination numbers (N) were systematically varied during the course of the analysis, but were not allowed to vary within a given fit. Data were fit over the k range of 2-12.5 Å-1. Fourier transforms of the EXAFS data and fits were calculated with EXAFSPAK.
Because the XAS signal is an average of that from all zinc atoms in a sample, modeling the structure of a binuclear zinc site in AP and NPP enzymes for the XAS analysis required consideration of the average coordination environment of the two structurally nonequivalent zinc atoms. Based on the available crystal structures of AP and NPP, the active site can be represented as the set of possible models I-VI shown in Figure 7. Model I is based on the structures of ligand-free AP and NPP (pdb codes 1ED9 and 2GSN, respectively). Model II is observed for NPP-AMP (2GSU), where the phosphate group of AMP is bound to Zn1 but not to Zn2. Models III and IV include a phosphate group bridging Zn1 and Zn2 and an additional Ser (AP) or Thr (NPP) ligand to Zn2 in IV (seen in structures 1ED8, 1ALK, and the AP structure reported herein). Model V is seen with vanadate-bound AP (1B8J), where one oxygen of vanadate bridges the zinc atoms, and there is an additional oxygen contact from the Ser residue to Zn2. Model VI is suggested by the structure of vanadate-bound NPP (1GSO) with an additional vanadate oxygen ligand to Zn1. After averaging the ligand environment over the two zinc atoms, average zinc coordination can be represented as ZnN1.5O(N-1.5), where total coordination number N varies between 4 and 5 depending on the model.
Figure 7.

Plausible coordination models used for EXAFS fits of the binuclear Zn2+ site in AP and NPP. These models were constructed based on the available crystal structures for AP and NPP.
Initial fits to the EXAFS data showed that first-shell Zn-N and Zn-O scattering could not be resolved into separate shells for any of the samples. To model a combined N/O first shell, the single scattering parameters for Zn-N and Zn-O bonds were calculated for Zn-N(His) and Zn-O(Asp) at bond distances of 2.00 and 1.97 Å, respectively, and then included in a fit with the coordination number of 1.5 for Zn-N and (N-1.5) for Zn-O, where N is an average coordination number tested in the fit. The bond distances R for Zn-O and Zn-N paths were restricted to the same value, as were the bond variances σ2. Thus, the combined O/N shell contributed two variable parameters to each fit.
For His ligands, single and multiple scattering contributions by the outer four atoms of the imidazole rings were included for all fits (1.5 His per zinc atom). The imidazole ring was assumed to have a rigid geometry, and the outer-shell Zn-N and Zn-C distances were linked to the first shell Zn-N distance. Theoretical phases and amplitudes were calculated with FEFF for a first shell Zn-N(His) distance of 2.00 Å. The σ2 values for single and multiple-scattering paths of an imidazole ring were calculated using a parameterized expression for σ2 as a function of temperature and first-shell Zn-N distance 81, and were fixed during the fit. A low degree of static disorder for the three His ligands was assumed, as suggested by relatively low first-shell σ2 values observed in the fits. Because the nearest C/N atoms of the imidazole are well separated spatially from the first-shell atoms (∼3.0 Å vs. ∼2.0 Å), the parameters for these two shells are relatively independent in a fit. For all samples, the freely refined first-shell bond variance value σ2 remains the same regardless of whether the outer-shell imidazole scattering is included or not, and whether the included imidazole scattering components are constrained or not.
Supplementary Material
Acknowledgments
We thank members of the Herschlag lab for helpful comments on the manuscript, Dr. Jun-yong Choe for assistance with diffraction data collection, Dr. Stefan Steinbacher for participating in the initial crystallographic refinement, Dr. Axel T. Brunger for the use of facilities for crystallographic refinement, and Jesse G. Zalatan for sharing unpublished NPP binding data. This work was supported by grants from the NIH to D.H. (GM64798), D.C.R., (GM045162) and K.O.H. (RR001209). J.K.L. was supported by an NIH postdoctoral fellowship (F32 GM080865). T.D.F. was supported by the Universitywide AIDS Research Program of the University of California (F03-ST-216). Facilities used for x-ray crystallography in the laboratory of Axel T. Brunger were supported by Howard Hughes Medical Institute. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the NIH, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The publication was partially supported by Grant Number 5 P41 RR001209 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Abbreviations
- XAS
x-ray absorption spectroscopy
- EXAFS
extended x-ray absorption fine structure
- AP
Escherichia coli alkaline phosphatase
- NPP
Xanthomonas axonopodis nucleotide pyrosphosphatase/phosphodiesterase
- BVS
bond valence sum
- PDB
protein data bank
- CSD
Cambridge structural database
Footnotes
Where Δ is approximately -0.4 to -0.5 Å, and arises because the Fourier transform data were not phase-shifted.
Note, for example, that in Figure 5 the NPP Zn-Zn distances on average actually appear slightly longer than those in AP, opposite of the proposed model.
Accession Number: The crystallographic coordinates for E. coli AP have been deposited in the PDB with the accession code 3TG0.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: Mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien PJ, Herschlag D. Catalytic promiscuity and the evolution of new enzymatic activities. Chem Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 3.Khersonsky O, Tawfik DS. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 4.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 5.Galperin MY, Bairoch A, Koonin EV. A superfamily of metalloenzymes unifies phosphopentomutase and cofactor-independent phosphoglycerate mutase with alkaline phosphatases and sulfatases. Protein Sci. 1998;7:1829–1835. doi: 10.1002/pro.5560070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galperin MY, Jedrzejas MJ. Conserved core structure and active site residues in alkaline phosphatase superfamily enzymes. Proteins. 2001;45:318–324. doi: 10.1002/prot.1152. [DOI] [PubMed] [Google Scholar]
- 7.Coleman JE. Structure and mechanism of alkaline phosphatase. Annu Rev Biophys Biomol Struct. 1992;21:441–483. doi: 10.1146/annurev.bb.21.060192.002301. [DOI] [PubMed] [Google Scholar]
- 8.Kim EE, Wyckoff HW. Reaction mechanism of alkaline phosphatase based on crystal structures. Two-metal ion catalysis. J Mol Biol. 1991;218:449–464. doi: 10.1016/0022-2836(91)90724-k. [DOI] [PubMed] [Google Scholar]
- 9.Reid TW, Wilson IB. E. coli alkaline phosphatase. In: Boyer PD, editor. In The Enzymes. Academic Press; New York: 1971. pp. 373–415. [Google Scholar]
- 10.Strater N, Lipscomb WN, Klabunde T, Krebs B. Two-metal ion catalysis in enzymatic acyl- and phosphoryl-transfer reactions. Angew Chem Int Ed Engl. 1996;35:2024–2055. [Google Scholar]
- 11.Zalatan JG, Fenn TD, Herschlag D. Comparative Enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J Mol Biol. 2008;384:1174–1189. doi: 10.1016/j.jmb.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lad C, Williams NH, Wolfenden R. The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases. Proc Natl Acad Sci USA. 2003;100:5607–5610. doi: 10.1073/pnas.0631607100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lassila JK, Zalatan JG, Herschlag D. Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu Rev Biochem. 2011;80:669–702. doi: 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 15.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: Implications for mechanism and evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 16.Gijsbers R, Ceulemans H, Stalmans W, Bollen M. Structural and catalytic similarities between nucleotide pyrophosphatases/phosphodiesterases and alkaline phosphatases. J Biol Chem. 2001;276:1361–1368. doi: 10.1074/jbc.M007552200. [DOI] [PubMed] [Google Scholar]
- 17.Nikolic-Hughes I, O'Brien PJ, Herschlag D. Alkaline phosphatase catalysis is ultrasensitive to charge sequestered between the active site zinc ions. J Am Chem Soc. 2005;127:9314–9315. doi: 10.1021/ja051603j. [DOI] [PubMed] [Google Scholar]
- 18.O'Brien PJ, Herschlag D. Does the active site arginine change the nature of the transition state for alkaline phosphatase-catalyzed phosphoryl transfer? J Am Chem Soc. 1999;121:11022–11023. [Google Scholar]
- 19.O'Brien PJ, Lassila JK, Fenn TD, Zalatan JG, Herschlag D. Arginine coordination in enzymatic phosphoryl transfer: Evaluation of the effect of Arg166 mutations in Escherichia coli alkaline phosphatase. Biochemistry. 2008;47:7663–7672. doi: 10.1021/bi800545n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lassila JK, Herschlag D. Promiscuous sulfatase activity and thio-effects in a phosphodiesterase of the alkaline phosphatase superfamily. Biochemistry. 2008;47:12853–12859. doi: 10.1021/bi801488c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mildvan AS. Mechanisms of signaling and related enzymes. Proteins. 1997;29:401–416. [PubMed] [Google Scholar]
- 22.Cassano AG, Anderson VE, Harris ME. Understanding the transition states of phosphodiester bond cleavage: Insights from heavy atom isotope effects. Biopolymers. 2004;73:110–129. doi: 10.1002/bip.10517. [DOI] [PubMed] [Google Scholar]
- 23.Hengge AC, Onyido I. Physical organic perspectives on phospho group transfer from phosphates and phosphinates. Curr Org Chem. 2005;9:61–74. [Google Scholar]
- 24.Hengge AC. Mechanistic studies on enzyme-catalyzed phosphoryl transfer. Adv Phys Org Chem. 2005;40:49–108. [Google Scholar]
- 25.Cleland WW, Hengge AC. Enzymatic mechanisms of phosphate and sulfate transfer. Chem Rev. 2006;106:3252–3278. doi: 10.1021/cr050287o. [DOI] [PubMed] [Google Scholar]
- 26.Thatcher GRJ, Kluger R. Mechanism and catalysis of nucleophilic substitution in phosphate esters. Adv Phys Org Chem. 1989;25:99–265. [Google Scholar]
- 27.Huo G, Cui Q. QM/MM analysis suggests that alkaline phosphatase (AP) and nucleotide pyrophosphatase/phosphodiesterase slightly tighten the transition state for phosphate diester hydrolysis relative to solution: implications for catalytic promiscuity in the AP superfamily. doi: 10.1021/ja205226d. Manuscript Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez-Canut V, Marti S, Bertran J, Moliner V, Tunon I. Theoretical modeling of the reaction mechanism of phosphate monoester hydrolysis in alkaline phosphatase. J Phys Chem B. 2009;113:7816–7824. doi: 10.1021/jp901444g. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Canut V, Roca M, Bertran J, Moliner V, Tunon I. Theoretical study of phosphodiester hydrolysis in nucleotide pyrophosphatase/phosphodiesterase. Environmental effects on the reaction mechanism. J Am Chem Soc. 2010;132:6955–6963. doi: 10.1021/ja908391v. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Canut V, Roca M, Bertran J, Moliner V, Tunon I. Promiscuity in alkaline phosphatase superfamily: Unraveling evolution through molecular simulations. J Am Chem Soc. 2011;133:12050–12062. doi: 10.1021/ja2017575. [DOI] [PubMed] [Google Scholar]
- 31.Stec B, Holtz KM, Kantrowitz ER. A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J Mol Biol. 2000;299:1303–1311. doi: 10.1006/jmbi.2000.3799. [DOI] [PubMed] [Google Scholar]
- 32.Andrews LD, Deng H, Herschlag D. Paradoxical ligand binding properties of alkaline phosphatase resolved by isotope-edited FTIR. J Am Chem Soc. 2011;133:11621–11631. doi: 10.1021/ja203370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Auld DS. Structure of binary and ternary complexes of zinc and cobalt carboxypeptidase A as determined by x-ray absorption fine structure. Biochemistry. 1995;34:16306–16312. doi: 10.1021/bi00050a010. [DOI] [PubMed] [Google Scholar]
- 34.Feiters MC, Eijkelenboom APAM, Nolting HF, Krebs B, van den Ent FMI, Plasterk RHA, Kaptein R, Boelens R. X-ray absorption spectroscopic studies of zinc in the N-terminal domain of HIV-2 integrase and model compounds. J Synch Radiat. 2003;10:86–95. doi: 10.1107/s0909049502016205. [DOI] [PubMed] [Google Scholar]
- 35.Tobin DA, Pickett JS, Hartman HL, Fierke CA, Penner-Hahn JE. Structural characterization of the zinc site in protein farnesyltransferase. J Am Chem Soc. 2003;125:8862–9969. doi: 10.1021/ja035927o. [DOI] [PubMed] [Google Scholar]
- 36.Kleifeld O, Frenkel A, Martin JML, Sagi I. Active site electronic structure and dynamics during metalloenzyme catalysis. Nat Struct Biol. 2003;10:98–103. doi: 10.1038/nsb889. [DOI] [PubMed] [Google Scholar]
- 37.Mangani S, Carloni P, Viezzoli MS, Coleman JE. EXAFS Study of the Active Site of Alkaline Phosphatase from Escherichia Coli. Inorganica Chim Acta. 1992;191:161–165. [Google Scholar]
- 38.Penner-Hahn JE. Characterization of “spectroscopically quiet” metals in biology. Coord Chem Rev. 2005;249:161–177. [Google Scholar]
- 39.Vogel A, Schilling O, Niecke M, Bettmer J, Meyer-Klaucke W. ElaC encodes a novel binuclear zinc phosphodiesterase. J Biol Chem. 2002;277:29078–29085. doi: 10.1074/jbc.M112047200. [DOI] [PubMed] [Google Scholar]
- 40.Thorp HH. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. Inorg Chem. 1992;31:1585–1588. doi: 10.1021/ic9804420. [DOI] [PubMed] [Google Scholar]
- 41.Liu W, Thorp HH. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distances and other enzymes. Inorg Chem. 1993;32:4102–4105. [Google Scholar]
- 42.Brown ID, Altermatt D. Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Cryst. 1985;B41:244–247. [Google Scholar]
- 43.Brese NE, O'Keeffe M. Bond-valence parameters for solids. Acta Cryst. 1991;B47:192–197. [Google Scholar]
- 44.Allen FH. The cambridge structural database: a quarter of a million crystal structures and rising. Acta Cryst. 2002;B58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 45.Holtz KM, Stec B, Kantrowitz ER. A model of the transition state in the alkaline phosphatase reaction. J Biol Chem. 1999;274:8351–8354. doi: 10.1074/jbc.274.13.8351. [DOI] [PubMed] [Google Scholar]
- 46.Sowadski JM, Handschumacher MD, KrishnaMurthy HM, Foster BA, Wyckoff HW. Refined structure of alkaline phosphatase from Escherichia coli at 2.8 Å resolution. J Mol Biol. 1985;186:417–433. doi: 10.1016/0022-2836(85)90115-9. [DOI] [PubMed] [Google Scholar]
- 47.Holtz KM, Stec B, Myers JK, Antonelli SM, Widlanski TS, Kantrowitz ER. Alternate modes of binding in two crystal structures of alkaline phosphatase-inhibitor complexes. Protein Sci. 2000;9:907–915. doi: 10.1110/ps.9.5.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy JE, Stec B, Ma L, Kantrowitz ER. Trapping and visualization of a covalent enzyme-phosphate intermediate. Nat Struct Biol. 1997;4:618–622. doi: 10.1038/nsb0897-618. [DOI] [PubMed] [Google Scholar]
- 49.Le Du MH, Lamoure C, Muller BH, Bulgakov OV, Lajeunesse E, Menez A, Boulain JC. Artificial evolution of an enzyme active site: Structural studies of three highly active mutants of Escherichia coli alkaline phosphatase. J Mol Biol. 2002;316:941–953. doi: 10.1006/jmbi.2001.5384. [DOI] [PubMed] [Google Scholar]
- 50.Dealwis CG, Brennan C, Christianson K, Mandecki W, Abad-Zapatero C. Crystallographic analysis of reversible metal binding observed in a mutant (Asp153Gly) of Escherichia coli alkaline phosphatase. Biochemistry. 1995;34:13967–13973. doi: 10.1021/bi00043a001. [DOI] [PubMed] [Google Scholar]
- 51.Dealwis CG, Chen L, Brennan C, Mandecki W, Abad-Zapatero C. 3-D structure of the D153G mutant of Escherichia coli alkaline phosphatase: an enzyme with weaker magnesium binding and increased catalytic activity. Protein Eng. 1995;8:865–871. doi: 10.1093/protein/8.9.865. [DOI] [PubMed] [Google Scholar]
- 52.Stec B, Hehir MJ, Brennan C, Nolte M, Kantrowitz ER. Kinetic and X-ray structural studies of three mutant E-coli alkaline phosphatases: Insights into the catalytic mechanism without the nucleophile Ser102. J Mol Biol. 1998;277:647–662. doi: 10.1006/jmbi.1998.1635. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Kantrowitz ER. Trapping the tetrahedral intermediate in the alkaline phosphatase reaction by substitution of the active site serine with threonine. Protein Sci. 2006;15:2395–2401. doi: 10.1110/ps.062351506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma L, Tibbitts TT, Kantrowitz ER. Escherichia Coli AlkalinePhosphatase - X-Ray Structural Studies of a Mutant Enzyme (His-412-Asn) at One of the Catalytically Important Zinc-Binding Sites. Protein Sci. 1995;4:1498–1506. doi: 10.1002/pro.5560040807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma L, Kantrowitz ER. Kinetic and x-ray structural studies of a mutant Escherichia coli alkaline phosphatase (His412Gln) at one of the zinc binding sites. Biochemistry. 1996;35:2394–2402. doi: 10.1021/bi9523421. [DOI] [PubMed] [Google Scholar]
- 56.Li W, Bi L, Wang W, Li Y, Zhou Y, Wei H, Jiang T, Bai L, Chen Y, Zhang Z, Yuan X, Xiao J, Zhang XE. Development of a universal phosphorylated peptide-binding protein for simultaneous assay of kinases. Biosensors and Bioelectronics. 2009;24:2871–2877. doi: 10.1016/j.bios.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 57.Tibbitts TT, Murphy JE, Kantrowitz ER. Kinetic and structural consequences of replacing the aspartate bridge by asparagine in the catalytic metal triad of Escherichia coli alkaline phosphatase. J Mol Biol. 1996;257:700–715. doi: 10.1006/jmbi.1996.0195. [DOI] [PubMed] [Google Scholar]
- 58.Murphy JE, Tibbitts TT, Kantrowitz ER. Mutations at positions 153 and 328 in Escherichia coli alkaline phosphatase provide insight towards the structure and function of mammalian and yeast alkaline phosphatases. J Mol Biol. 1995;253:604–617. doi: 10.1006/jmbi.1995.0576. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Stieglitz KA, Kantrowitz ER. Metal specificity is correlated with two crucial active site residues in Escherichia coli alkaline phosphatase. Biochemistry. 2005;44:8378–8386. doi: 10.1021/bi050155p. [DOI] [PubMed] [Google Scholar]
- 60.Sowadski JM, Foster BA, Wyckoff HW. Structure of alkaline phosphatase with zinc/magnesium cobalt or cadmium in the functional sites. J Mol Biol. 1981;150:245–272. doi: 10.1016/0022-2836(81)90451-4. [DOI] [PubMed] [Google Scholar]
- 61.Glusker JP. Structural aspects of metal liganding to functional groups in proteins. Adv Prot Chem. 1991;42:1–76. doi: 10.1016/s0065-3233(08)60534-3. [DOI] [PubMed] [Google Scholar]
- 62.Vedani A, Huhta DW. A new force field for modeling metalloproteins. J Am Chem Soc. 1990;112:4759–4767. [Google Scholar]
- 63.Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Cryst. 2006;D62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- 64.Hsin K, Sheng Y, Harding MM, Taylor P, Walkinshaw MD. MESPEUS: a database of the geometry of metal sites in proteins. J Appl Cryst. 2008;41:963–968. [Google Scholar]
- 65.O'Brien PJ, Herschlag D. Alkaline phosphatase revisited: Hydrolysis of alkyl phosphates. Biochemistry. 2002;41:3207–3225. doi: 10.1021/bi012166y. [DOI] [PubMed] [Google Scholar]
- 66.Kuppuraj G, Dudev M, Lim C. Factors governing metal-ligand distances and coordination geometries of metal complexes. J Phys Chem B. 2009;113:2952–2960. doi: 10.1021/jp807972e. [DOI] [PubMed] [Google Scholar]
- 67.Davies DR, Hol WGJ. The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett. 2004;577:315–321. doi: 10.1016/j.febslet.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 68.Crans DC, Smee JJ, Gaidamauskas E, Yang L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem Rev. 2004;104:849–902. doi: 10.1021/cr020607t. [DOI] [PubMed] [Google Scholar]
- 69.Messmore JM, Raines RT. Pentavalent organo-vanadates as transition state analogues for phosphoryl transfer reactions. J Am Chem Soc. 2000;122:9911–9916. doi: 10.1021/ja0021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deng H, Callender R, Huang Z, Zhang ZY. Is the PTPase-vanadate complex a true transition state analogue? Biochemistry. 2002;41:5865–72. doi: 10.1021/bi016097z. [DOI] [PubMed] [Google Scholar]
- 71.O'Brien PJ, Herschlag D. Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry. 2001;40:5691–5699. doi: 10.1021/bi0028892. [DOI] [PubMed] [Google Scholar]
- 72.O'Brien PJ, Herschlag D. Sulfatase activity of E. coli alkaline phosphatase demonstrates a functional link to arylsulfatases, an evolutionarily related enzyme family. J Am Chem Soc. 1998;120:12369–12370. [Google Scholar]
- 73.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 74.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 75.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 76.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 77.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 78.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An Improved Assay for Nanomole Amounts of Inorganic Phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 79.Ellis PJ, Freeman HC. XFIT- An interactive EXAFS analysis program. J Synch Radiat. 1995;2:190–195. doi: 10.1107/S0909049595006789. [DOI] [PubMed] [Google Scholar]
- 80.Ankudinov AL, Rehr JJ. Relativistic calculations of spin-dependent x-ray-absorption spectra. Phys Rev B. 1997;56:R1712–R1715. [Google Scholar]
- 81.Dimakis N, Bunker G. XAFS Debye-Waller factors for Zn metalloproteins. Phys Rev B. 2004;70:195114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.