

Abstract
Individually, the three protein components of anthrax toxin are nontoxic, but when they assemble, they form active holotoxin complexes. The role of the protective antigen (PA) component of the toxin is to deliver the two other enzyme components, lethal factor (LF) and edema factor (EF), across the plasma membrane and into the cytoplasm of target cells. PA is produced as a proprotein, which must be proteolytically activated, and generally cell-surface activation is mediated by a furin-family protease. Activated PA can then assemble into one of two non-interconverting oligomers, a homoheptamer and homooctamer, which have unique properties. Herein we describe molecular determinants that influence the stoichiometry of PA in toxin complexes. By tethering PA Domain 4 to Domain 2 with two different length cross-links, we can control the relative proportions of PA heptamers and octamers. The longer cross-link favors octamer formation, whereas the shorter one favors formation of the heptamer. X-ray crystal structures of PA (up to 1.45 Å resolution), including these cross-linked PA constructs, reveal that a hinge-like movement of Domain 4 correlates with the relative preference for each oligomeric architecture. Furthermore, we report the conformation of the flexible loop containing the furin-cleavage site and show that for efficient processing, the furin site cannot be moved ~5 or 6 residues within the loop. We propose that there are different orientations of Domain 4 relative to the main body of PA that favor the formation of either the heptamer or the octamer.
Introduction
Anthrax toxin (Atx) is a virulence factor secreted by pathogenic strains of Bacillus anthracis. Atx consists of three nontoxic protein components,3-5 namely protective antigen (PA, 83-kDa), lethal factor (LF, 90-kDa), and edema factor (EF, 89-kDa). The PA component assembles into a ring-shaped oligomer capable of forming a membrane-spanning translocase channel, which delivers the two enzyme components, LF and EF, into the cytosol of a host cell. LF is a Zn2+-dependent protease,6-8 which cleaves host-cell mitogen-activated protein kinase kinases. PA, LF, and EF are individually nontoxic; however, LF plus PA creates lethal toxin (LT), which can alter normal cell function and may even cause death.9 EF is Ca2+- and calmodulin-dependent adenylyl cyclase.10-12 Similarly, PA plus EF produces edema toxin (ET), which induces tissue swelling and may also cause death.
The PA component contains four different domains (Domain 1-4), which are critical to various stages of toxin assembly and translocation. To form cytotoxic complexes, PA must first assemble with LF and/or EF. Two different types of assembly pathways have been described: a cell-surface pathway (for review, see ref. 1) and a plasma-based/extracellular pathway. On the surface of host cells, PA binds to one of two known Atx receptors: ANTXR1 (ref. 15) and ANTXR2 (ref. 16). The PA-ANTXR2 interaction is rather stable and dissociates with a half-life measured in days.19 The interaction involves Domain 2 and Domain 4 in PA, such that the latter domain coordinates the receptor’s Ca2+ or Mg2+ dependent metal ion adhesion site.17-21 ANTXR2-bound PA is subsequently cleaved by a furin-family protease yielding the proteolytically-activated or nicked form, called nPA. After a 20-kDa portion of nPA (PA20) dissociates, the remaining 63-kDa, receptor-bound portion (PA63) self-assembles into an ~2:1 mixture of heptameric (PA7) and octameric (PA8) ring-shaped oligomers. The complexes are endocytosed25 and transferred to an acidic compartment,26 where the lower pH conditions induce the PA oligomers to form transmembrane translocase channels.27-29 LF and EF translocate through the PA channel to enter the cytosol of the host cell.
Recent studies investigating the molecular mechanism of PA oligomerization have revealed that the assembly of the octameric PA oligomer is largely dependent on the degree to which even-numbered PA intermediates (PA2 and PA4) are populated. Co-assembly factors such as LF, EF, or dimeric ANTXR2 (dANTXR2) presumably favor the formation of these PA2 intermediates and, correspondingly, PA8 formation13 (Fig. 1a). Interestingly, when PA’s membrane insertion loop (MIL) is deleted (PAΔMIL), these mutant subunits can form 5-10 fold more octamer than heptamer—even in the absence of co-assembly factors. The structural explanation for the propensity of PAΔMIL to form the octamer is not well understood. Alignment of the PA subunits from the heptameric23 and octameric14 oligomer structures reveals that the conformation of Domain 4 (D4, residues 596-735) is quite flexible (Fig. 1b). Relative to the main body of the protein, PA D4 may be treated as a rigid body whose “pitch” may be described as a rotation around the “hinge” joining Domain 3 (D3, residues 458-595) and D4. While oligomerization may occur at a large contiguous surface containing residues from Domain 1 (D1, residues 1-258), Domain 2 (D2, residues 259-457) and D3, it is unknown how the orientation of D4 may modulate the oligomerization pathway and determine the stoichiometry of PA in the complex.
Figure 1. Anthrax toxin assembly is heterogeneous and requires proteolytic processing.

(a) Cartoon depiction of anthrax toxin assembly. PA domains are colored as follows: D1 (gold), D2 (cyan), D3 (violet), and D4 (green). Upon oligomerization, PA forms both PA7 and PA8 complexes. The oligomeric ratio of PA complexes (PA7:PA8) decreases from about 20:1 for unliganded PA assembly to 2:1 in the presence of ligand (LF, EF, or dANTXR2). The PAΔMIL construct can form complexes at a 2:1 oligomeric ratio; however, it does so in the absence of ligand. (b) Global Cα alignment of a PA subunit (chain G) from the PA7 crystal structure (PDB 1TZO)18 onto a PA subunit (chain G) from the PA8 crystal structure (PDB 3HVD).14 The ribbons are colored based on the RMS displacement of the Cα positions. Small, intermediate and large RMS displacements are colored blue, white and red, respectively. (c) (Above) The PA sequence containing the furin-cleavage site, where the scissile bond is indicated with an arrow. (Below) The furin-dependent cleavage reaction of the PA proprotein releases the 20-kDa fragment (PA20) and yields the 63-kD portion, called PA63. PA63 ultimately assembles into PA7 and PA8 oligomers.
Mammalian furin-type proteases are members of the prohormone convertase (PC) family.31-34 A close homolog of furin in yeast, kexin, functions analogously by cleaving prohormone proteins.35-37 Furin, the most well-characterized PC (EC 3.4.21.75),33 cleaves its substrates at a cationic, R-X-(R/K/X)-R↓, consensus site (where the scissile bond is indicated with an ↓).38 Furin is involved in diverse cellular processes, such as homeostasis and embryogenesis, and it is expressed in all tissues and cells as a secreted membrane N-linked-type glycoprotein that matures in the trans-Golgi network. Furin and its yeast homolog, kexin, each anchor to the cell membrane via a homologous single-pass transmembrane helix located on its carboxy terminus. As a membrane protein, furin can cycle between the cell surface and the endosomal compartment. Furin functions to proteolytically process many types of membrane-bound and soluble factors, including: growth factors, extracellular matrix proteases, viral proteins and bacterial toxins.
The role of furin and related PCs in viral and bacterial pathogenesis is widespread. Many bacterial and viral proproteins require proteolytic activation prior to maturation into either active enzymes or assembled virulence factors. Generally, but not always, furin-activated proteolysis permits these pathogenic proteins to subsequently fuse with or penetrate the host cell membrane. Included amongst these virulence factors are the bacterial toxins,40 anthrax,41 shiga,42 diphtheria,43 tetanus, botulinum, Clostridium septicum α-toxin,44 and aerolysin;45 the influenza A virus fusion protein, hemagglutinin;46 human immunodeficiency virus-1 envelope glycoprotein, gp160;47 flaviviruses, such as tick-borne encephalitis and West Nile;48 and filoviruses, such as Marburg and Ebola.49
In order for Atx to assemble, PA must first be cleaved by a furin-type protease. Cleavage occurs after the sequence, 164RKKR↓S, in a solvent-exposed loop within D1.41 Selective50 and nonselective inhibition of furin and furin-like proteases strongly impairs Atx function. Furthermore, substrate-mapping studies of many related furin-type proteases show that PA’s RKKR furin-recognition sequence (Fig. 1c) has maximized the motif for recognition by a broad number of these furin-type PC enzymes.38 While NMR techniques have elucidated the structure of a furin cleavage site in a 19-residue peptide from gp160,53 atomic-resolution structural information for furin cleavage sites in proviral and protoxin proteins have been challenging to obtain, because the sites are typically found on flexible loop structures. To gain an understanding on the structural basis of toxin assembly and furin activation we crystallized a variant of anthrax toxin PA and solved its structure to 1.45-Å resolution.
Results
Crystal structure of the PAΔMIL monomer
For our initial crystallographic studies, we chose a PA construct (PAΔMIL) in which the MIL in D2 (residues 303-324) was replaced with the Pro-Gly dipeptide (which generally favors the formation of a Type II turn). The highly dynamic MIL is absent from previous structures of the PA monomer; therefore, we assumed its absence would improve crystal quality. Furthermore, we had previously solved the structure of PA8 to 3.2-Å resolution14 and the PA8(LFN)4 complex to 3.1-Å resolution24 using the PAΔMIL construct. These PAΔMIL crystal forms diffracted to higher resolution than the PA7 crystal forms using the wild-type (WT) PA construct, which previously diffracted to 3.6 Å18 and 4.5 Å.23
The PAΔMIL construct yielded large, well-diffracting orthorhombic crystals belonging to the P212121 space group and diffracted X-rays to 1.45 Å. The structure was solved using molecular replacement; one PA protein was identified in the asymmetric unit. Interestingly, the unit cell dimensions and space group are identical to what Petosa et al. referred to as “Crystal form 1”. From their study, Crystal form 1 did not diffract as well as “Crystal form 2,” the basis for the PA structure with Protein Data Bank (PDB) accession code 1ACC.23 Coordinates for Crystal form 1 were not deposited in the PDB, however. Contiguous electron density was identified for residues 15-735 of PAΔMIL (Fig. 2a). Loop regions lacking in the 1ACC search model were built manually in the experimental electron density using standard methods.
Figure 2. The structure of PAΔMIL.

(a) (left) Ribbons depiction of PAΔMIL. D1 (gold), D2 (cyan), D3 (violet), D4 (green), and calcium ions (dark green). (right) Loop structures not present in 1ACC23 are indicated as follows: L99-102 (orange), L162-173 (red) containing the furin-cleavage site, L275-287 (blue), the loop containing 2α1 (navy blue), and 3α1 (purple). (b) Sample 2Fo-Fc electron density calculated at the end of refinement to 1.45 Å for the twin Ca2+-coordination site contoured at σ=2.5. (c) LSQ Cα alignment of residues 16-340 for PA monomer (green, 1ACC),23 ANTXR2-bound PA (blue, 1T6B)17 and PAΔMIL (yellow). ANTXR2 (coral) from IT6B is included for reference. (d) Relative D4 orientation for PA monomer, ANTXR2-bound PA, and PAΔMIL (colored as in panel c) LSQ Cα superposition for residues 25-595.
The resolution of our structure allowed us to refine all of PA’s atomic coordinates individually, and the atomic displacement parameters (ADP) were refined anisotropically. Numerous solvent molecules were resolved, and their ADP values were also refined anisotropically. Overall, the model is in good agreement with the electron density, with an R value of 19.4% and an Rfree value of 21.6% (Table 1). The resulting structure closely resembles previous PA monomer structures. D1 contains a jelly-roll motif with peripheral small helices and loops, including the furin-cleavage site-containing L162-174, as well as a twin-Ca2+ coordination site resembling the classical EF hand motif (Fig. 2b). D2 contains a Greek-key motif implicated in ultimately forming the 14- or 16-stranded β-barrel channel, through which LF and EF translocate. Additionally, a loop containing 2α1 was resolved, which has been implicated in binding to ANTXR2. D3 contains a mixed β sheet and associated helices, including 3α1, which was not present in 1ACC. D4 contains an immunoglobulin-like β-sandwich and is the most flexible of the PA domains.
Table 1. Data collection and refinement statistics.
| Data collection | PAΔMIL pH 8.5 | PAΔMIL C337-C664 | PA C337-C664-DCA | PAΔMIL pH 6.5 |
|---|---|---|---|---|
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 72.36, 93.39, 118.08 |
71.56, 94.60, 119.80 |
71.29, 93.69, 117.85 |
71.40, 93.93, 117.85 |
| α, β, γ(°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.1159 | 1.1159 | 1.1159 | 1.1159 |
| Resolution (Å) | 23.0-1.45 (1.49-1.45)a |
25.06-2.06 (2.09-2.06)b |
24.9-1.83 (1.87-1.83)c |
9.99-1.70 (1.73-1.70)d |
| Rmerge (%) | 9.6(74.8) | 11.4(86.8) | 12.0(75.7) | 8.5(85.5) |
| I / σI | 13.9(1.9) | 15.7(2.5) | 20.9(4.3) | 22.1(2.1) |
| Completeness (%) | 90.3(81.0) | 99.9(99.2) | 99.7(96.0) | 99.8(98.0) |
| Redundancy | 6.5(3.4) | 7.6(7.7) | 14.2(10.5) | 8.1(7.4) |
| Wilson B | 27.000 | 36.331 | 26.490 | 27.278 |
| Refinement | ||||
| Resolution (Å) | 23.0-1.45 | 23.86-2.06 | 24.9-1.83 | 9.99-1.70 |
| No. reflections | 126,263 | 50,702 | 70,160 | 87,171 |
| Rwork, Rfree (%) | 19.4/ 21.6 | 20.7 / 23.4 | 19.3 / 22.6 | 19.4 / 21.7 |
| No. atoms | ||||
| Protein | 5647 | 5589 | 5616 | 5590 |
| Ligand/ion | 17 | 2 | 13 | 2 |
| Water | 348 | 208 | 422 | 398 |
| Average ADP-values | ||||
| Protein | 21.7 | 47.8 | 36.6 | 40.3 |
| Ligand/ion | 29.5 | 31.0 | 33.4 | 27.0 |
| Water | 29.9 | 42.9 | 39.3 | 43.0 |
| RMS deviations | ||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.016 | 0.007 |
| Bond angles (°) | 1.151 | 1.041 | 1.087 | 1.053 |
| Ramachandran (%) | ||||
| Favored | 98.1 | 97.9 | 98.1 | 97.1 |
| Outliers | 0 | 0 | 0 | 0 |
| MolProbity score | 1.51 | 1.93 | 1.58 | 1.86 |
Values in parenthesis refer to data in the highest resolution shell (1.49-1.45 Å).
Values in parenthesis refer to data in the highest resolution shell (2.13-2.09 Å).
Values in parenthesis refer to data in the highest resolution shell (1.87-1.83 Å).
Values in parenthesis refer to data in the highest resolution shell (1.87-1.83 Å).
Ordered loop regions
We resolved continuous electron density for PAΔMIL from residues 15 to 735 at the carboxy-terminus. While acknowledging that the membrane insertion loop (residues 305-324) is missing by design, this is the most complete PA model to date. (N.B. the numbering convention of 1ACC is retained.) While our structure and the three reported monomeric structures, PA (PDB 1ACC),23 the ANTXR2-bound PA (PDB 1T6B),17 and the 2-fluoro-histidine substituted PA (PDB 3MHZ),54 align with close agreement with a root mean square (RMS) < 1, A number of surface-loop regions not modeled in 1ACC are present in the PAΔMIL structure. Residues 99-102 and 512-515 are not modeled in 1ACC; however, they are present in 1T6B, while the latter is present in 3MHZ. Residues 163 and 168-173, a region that includes the furin proteolytic-cleavage site, are not present in any deposited structure of PA, both monomeric and oligomeric. Interestingly, residues 159-161 are in a different conformation in 1ACC relative to our model, indicating that the loop containing the furin-cleavage site can adopt multiple conformations. Residues 162 and 164-167 are found in 3MHZ in approximately the same conformation as reported here. Also D2 residues 275-287 are present, modeled here as an extended loop whose atoms have ADP values comparable to the average protein atom. Residues 343-350, which are not present in 1ACC, are modeled in both 1T6B and 3MHZ.
Receptor-binding loop-helix 2α1
In the published PA monomer structure (1ACC),23 residues 343-350 are not modeled. In the co-crystal structure of PA and ANTXR2 (1T6B)17 and the structure of the 2-fluoro-histidine substituted PA (3MHZ),54 these residues are well-ordered in a loop-helix motif (Fig. 2c). Presumably, this ordering is due to the strong van der Waals interaction of PA residue L320 with ANTXR2 L154, which contributes to the strong energetics of PA-receptor binding. This loop-helix is also well-ordered for PAΔMIL; the degree of order in this structure may reasonably be explained by the crystal contacts between 2α1 and 3α1. PAΔMIL and ANTXR2-bound PA from 1T6B align well and are in good agreement (RMS <0.3 Å). Alignment of the structures by Cα of residues 16-340 results in a slight shift of 2α1 in the PAΔMIL structure relative to 1T6B. This movement frees up potential steric clashes of PA 2α1 with the extracellular PA-binding domain of ANTXR2; therefore, the positioning of 2α1 in the structure presented here likely represents the biological conformation in the absence of receptor and agrees well with the 3MHZ structure.54
Changes in orientation of PA D4
In the structure of PA in complex with ANTXR2 (1T6B), D4 undergoes a rigid-body conformational change, resulting in a slight tilting of the domain relative to the unbound structure of PA (1ACC). This movement is actually required so that ANTXR2 may bind PA at two discontinuous contact points, i.e., at 2α1 in D2 and around D683 in D4. Interestingly, a backbone alignment of the PAΔMIL structure with the ANTXR2-bound PA structure from D1-D3 reveals that the orientation of D4 relative to the main body (D1-D3) does not significantly deviate. Even though both the PAΔMIL and 1ACC structures are not bound to receptor, the orientation of D4 in PAΔMIL more closely resembles that of the receptor-bound structure, 1T6B (Fig. 2d). Thus the orientation of D4 is the most significant structural change we can identify in the PAΔMIL structure.
Crystal structures of cross-linked PA monomers
In order to test the structural consequence of the orientation of D4 relative to the main body of PA, we engineered a pair of Cys mutations into the D2-D4 interface. We selected the residues, S337C and N664C, and incorporated the mutations into both the WT PA and PAΔMIL backgrounds. We then produced two different length cross-links. For the shorter-length cross-link, we allowed a disulfide bond to form between the two Cys residues, and we refer to this cross-link as C337-C664. For the longer-length cross-link, we used the small, thiol-specific cross-linker, 1,3-dichloroacetone (DCA),55 and we refer to this DCA link as C337-C664-DCA (Fig. 3a). We solved the crystal structures of both PAΔMIL C337-C664 and PA C337-C664-DCA (Table 1). We found that C337-C664 contains a disulfide bond (Fig. 3b), while PA C337-C664-DCA contains a dithioether cross-link consistent with the DCA modification (Fig. 3c). The two structures agree well with other PA monomer structures. We then performed a global, Cα alignment of PAΔMIL C337-C664 to PAΔMIL to assess whether the orientation of D4 was impacted by the cross-links. We find that domains D2 and D4 showed the largest displacement, resulting in maximum RMS displacements greater than 0.8 Å (Fig. 3d). This deviation increases with distance from the hinge between D3 and D4, indicating that the deviation is the result of a change in the hinge angle relating D4 with the main body of the protein. When we performed a similar RMS-displacement analysis, comparing PA C337-C664-DCA to PAΔMIL. We found that the backbone positions did not move appreciably (Fig. 3d). Finally, for the purpose of comparison, we aligned the Cα’s of PA from 1ACC to PAΔMIL to assess the RMS deviation contribution of the MIL. While surface loops and other protein periphery contributed to large RMS-displacements in excess of 11 Å, the backbone atoms comprising the major secondary structures in D1, D2 and D3 show relatively strong agreement. We found that most of the major deviations of backbone positions (> 1.2 Å) were localized in D4 (Fig. 3d).
Figure 3. Cysteine cross-linking of PA D2 and D4.

(a) Cartoon showing the location of the Cys-Cys cross-link introduced into PA, where domains are colored as in Fig. 1a. Two different cross-links were introduced via either a disulfide tether or the bifunctional cross-linking reagent, 1,3-dichloroactetone. Simulated-annealing 2Fo-Fc electron density map calculated at the end of refinement for (b) PAΔMIL C337-C664 and (c) PA C337-C664-DCA, contoured at σ=2.0 and σ=1.5, respectively, for which Cys337 and Cys664 and the bridging acetone cross-linker have been omitted. Sulfur and oxygen atoms are colored yellow and red, respectively. (d) Global Cα alignment of PAΔMIL to (left to right) PA from PDB accession code 1ACC, PAΔMIL acidic pH, PAΔMIL C337-C664 and (right) PA C337-C664-DCA and the corresponding RMS deviations colored from low (blue) to high (red).
Crystal structure of PAΔMIL at pH 6.5
During our analysis of structural changes in the relative orientation of D4, we realized that certain structures were obtained at slightly alkaline pH values of ~8.5 and other crystals were obtained at slightly acidic pH values of ~6.5. In order to rule out the possibility that the D4 movements we observe are simply due to differences in the pH of the crystallization condition, we crystallized PAΔMIL at pH 6.5. The structure was of similar quality to the others described thus far (Table 1). We then compared the structure of PAΔMIL at pH 6.5 to that obtained at pH 8.5 by computing the RMS displacement of the Cα backbone positions between the two different pH conditions. We found that the orientation of D4 is identical in the pH 8 and pH 6.5 crystal forms, and this change in pH does not dictate the relative orientation of D4 in PA (Fig. 3d).
The D2-D4 interface modulates the propensity of PA to form octameric oligomers
In order to study the contribution of PA’s D2-D4 interface to its oligomeric preference, we co-assembled native and cross-linked PA and PAΔMIL constructs with WT LFN and analyzed the assembly products by negative-stain electron microscopy (Fig. 4). We obtained ~1500-5000 images of ring-shaped particles in each sample; these images were processed using single-particle alignment, classification, and class-averaging image analysis. From these analyses, we determine the proportion of heptameric and octameric oligomers in each sample.
Figure 4. The interface of PA D2 and D4 controls oligomeric stoichiometry.
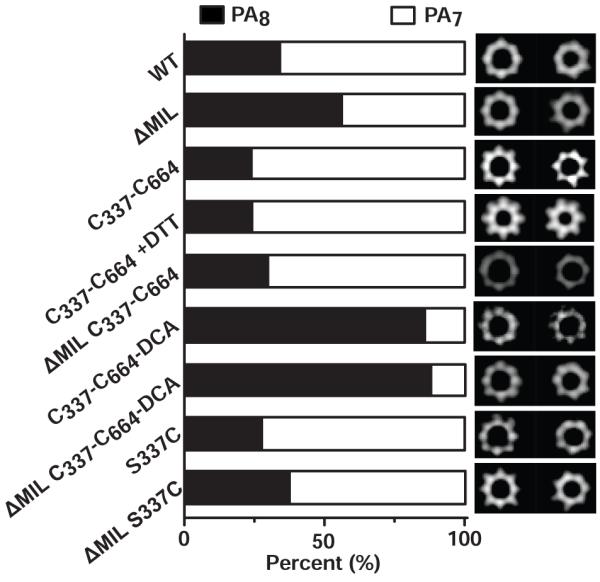
The percentages of PA7 (white) and PA8 (black) complexes are given following the assembly of various PA constructs in the presence of LFN. Representative class-average images for octamers (left) and heptamers (right) are shown at the right. The number (N) of particles analyzed are 2218, 3241, 2534, 2147, 2054, 1972, 1535, 1750 and 4845 for PA WT, PAΔMIL, PA C337-C664, PA C337-C664+DTT, PAΔMIL C337-C664, PA C337-C664-DCA, PAΔMIL C337-C664-DCA, PA S337C and PAΔMIL S337C, respectively. Note: “+DTT” indicates that 10 mM of DTT was included in the oligomerization reaction.
Our data show that two separate processes control oligomeric preference. First, we find that when we co-assemble PAΔMIL with LFN, the resulting PA oligomer products are enriched with ~55% octamers (Fig. 4). Under identical solution conditions, when we co-assemble WT PA with LFN we find only ~35% of the resulting complexes are octameric.14 By further comparison, when WT and PAΔMIL assemble on their own, each produces ~2% and 24% octamer, respectively.14 Taken together these results imply two different mechanisms lead to octamer formation, and those mechanisms are additive. Second, the length of the cross-link between D4 and D2 changes the propensity of PA to form octameric complexes (Fig. 4). The shorter C337-C664 disulfide cross-link produces ~24% and ~30% octamer in the PA and PAΔMIL backgrounds, respectively, which is slightly less than non-cross-linked PA or PAΔMIL when co-assembled with LFN (Fig. 4). Interestingly, extending the length of the cross-link by 3 carbon atoms in the C337-C664-DCA construct resulted in complexes that were highly enriched with octamers (>85%) for both the WT PA and PAΔMIL backgrounds (Fig. 4). Control constructs in which a single Cys residue was introduced showed no preference for the formation of the octameric complex. We surmise that cross-linking D4 to D2 alters the degree of conformational restriction in D4. Therefore, the interface between D2 and D4 plays an important role in determining the oligomeric preference of PA.
Correlation of the conformation and D4 and oligomeric heterogeneity
Since D4 appears to have the largest global RMS deviation when comparing the various PA monomer structures (Fig. 3d), we hypothesize that its position may provide the structural basis for determining PA oligomeric stoichiometry. To correlate D4 conformations with oligomeric stoichiometry, we aligned the Cα atoms of D1-D3 residues for PA (derived from 1ACC), acidic PAΔMIL, PAΔMIL C337-C664, and PAΔMIL C337-C664-DCA to the corresponding residues from PAΔMIL. The center-of-mass (COM) for each construct’s D2 and D4 was determined, accounting for differences in sequence. We then defined the hinge angle (θ) for D4 movements. θ is defined by the center of the oligomer’s lumen (COL), the COM of D2, and the COM of D4. These centers were projected to a plane orthogonal to the central pore axis through the COL, and the COM of D2 is the angle vertex (Fig. 5a). We find that θ increases relative to PAΔMIL for the WT PA and PAΔMIL C337-C664, which gave rise to a lower fraction of octamers, increasing by 2.5 degrees and 0.8 degrees, respectively. In contrast, θ for PAΔMIL at pH 6.5 and PAΔMIL C337-C664-DCA, the constructs that form an excess of octamer, increases by only 0.15 and 0.3 degrees, respectively (Fig. 5b). Furthermore, the distance displaced (Δd) between the COM of D4, relative to PAΔMIL, changes by 1.4 Å and 0.5 Å for WT PA and PAΔMIL C337-C664, respectively, but it changes by only 0.2 Å for both PAΔMIL at pH 6.5 and PA C337-C664-DCA (Fig. 5b). Therefore, we conclude that there are two D4 conformations, termed Pro-PA7 and Pro-PA8, which differ in the linearity of D4 with respect to D2 and the oligomer lumen. These different conformations can give rise to lower and higher fractions of PA8, respectively (Fig. 5c).
Figure 5. Molecular basis for PA oligomeric stoichiometry.

(a) A model showing the angle, θ, which is used as a metric relating the observed movement of D4 relative to the amino-terminal domains of the PA63 moiety. θ is defined by the center of the oligomeric lumen (COL) and the centers of mass (COM) of D2 and D4. Also indicated in the model is the metric for the displacement of the D4 COM, called Δd. The PA7 coordinates, containing a model of the MIL, are from 1TZO.18 D2, D4, and the MIL of the adjacent PA subunit are colored cyan, green, and purple, respectively. (b) Deviations in θ (Δθ) (left) and Δd (right) are plotted against the percentage of PA8 oligomer produced (as shown in Fig. 4). The deviations are computed relative to the reference model from PAΔMIL: Δθ = θ(PAΔMIL) - θ(PAi), and Δd = d(PAΔMIL) - d(PAi), where PAi is the PA monomer measured. (c) A molecular mechanism for PA oligomerization, where the hinge-like movement of D4 can lead to the population of two different population of PA that favor the formation of PA7 and PA8, called Pro-PA7 (left) and Pro-PA8 (right), respectively. In Pro-PA8 D4 is rotated and translated relative to D4 for Pro-PA7. The Pro-PA8 conformation is sterically inhibited by the presence of the adjacent PA subunit’s MIL during assembly. Presumably, the intramolecular dithioacetone bridge linking D4 to D2 favors the Pro-PA8 conformation in both the presence and absence of a neighboring MIL.
The furin-cleavage site
The loop containing the furin-cleavage site is ordered in our PAΔMIL structure (Fig. 6a). The structure of the loop begins as a β hairpin with the preceding loop, as R164 hydrogen bonds back to S161. Notably, none of the residues make contact with residues in the PA63 domains, although the loop is in relatively close in proximity to the D2 loop containing F464. This result is expected given the fact that cleavage at the site functions to release PA20 from the rest of PA; and previous studies reported that the presence of the furin-site loop does not affect the dissociation kinetics of the PA20 fragment.57 Several residues in the furin-cleavage loop make contact with PA20. Most prominent is R164, whose guanidino group forms a salt bridge with E155, as well as ionic interactions with the ε-hydroxyl of Q152 and the backbone carbonyl of L153. The γ-hydroxyl of S160 forms a hydrogen bond with the amide nitrogen of K166, and the ε-amino group of K166 hydrogen bonds to the backbone of Q158. The side chain for R167, which is the P1 site residue for furin, is not well ordered; however, its backbone carbonyl forms a hydrogen bond with Q115. Finally, K117 enters a hydrogen-bond network with the carbonyl oxygens of T169 and P173 and loosely approaches one of the conformers for S168. We conclude that the furin-site loop is freely accessible to solvent, makes numerous contacts with PA20 residues, and yet forms few interactions with PA63.
Figure 6. Structural basis for furin-dependent cleavage of PA.

(a) Positioning of the furin-cleavage site loop relative to the rest of PA20. Electrostatic interactions are represented as dashed red lines; carbon (yellow), oxygen (red), and nitrogen (blue) atoms are indicated. Sample composite simulated-annealing electron density (gray mesh) calculated at 1.45 Å at the end of refinement for PA residues 164-168 is contoured at σ=1. (b) SDS-PAGE assay for furin processing of (top) PA83, PA+6, and PA−5 and (bottom) PA and ANTXR2-bound PA. Proteins and incubation times as well as molecular weights corresponding to PA and PA63 are indicated above and to the left, respectively. (c) LSQ all-atom alignment of PA residues 164-167 with the furin-inhibitor peptide from 1P8J.33 (d) Placement of the aligned (yellow) PA furin-cleavage site in the active site of furin (1P8J).33 Rotation around the peptide bond linking residues K166 and R167 (green) relieves steric clashes and positions the scissile bond in proximity to the catalytic S368 of furin.
We then asked whether protoxin processing by furin is dependent on the positioning of the site along the extended loop region. Using WT PA, we produced constructs in which the RKKR sequence was moved either six residues toward the N-terminus of PA (PA+6) or five residues toward to the C-terminus (PA−5). Samples were incubated with furin at various time points and analyzed by SDS-PAGE. While furin-dependent processing of WT PA is evident after 30 minutes and is essentially complete after 3 hours, virtually no processing was observed for either PA+6 or PA−5 (Fig. 6b). Therefore, we conclude that the positioning of the furin-cleavage site is important to proper protoxin processing, and all sites within the loop are not equally accessible to furin.
Furin-dependent PA processing is not ANTXR2-dependent
We were intrigued by the degree to which the furin-cleavage site was ordered in these crystal structures. Is ordering of the ANTXR2 binding site linked to the ordering of the furin-cleavage site in PA? We suspected that crystal packing contacts resulted in the stabilization of 3α1, which is a structure implicated in ANTXR2-binding. Also it is known that anthrax toxin assembly is coordinated on cell surfaces by means of cholesterol-rich microdomains.25 Could receptor binding alter the dynamics of the furin-cleavage site to either stimulate or inhibit cleavage to allow PA to assemble within microdomains? We tested this hypothesis by monitoring furin-dependent processing of both PA and PA in complex with ANTXR2 using SDS-PAGE (Fig. 6b). We used both monomeric and dimeric ANTXR2 (dANTXR2) constructs to also test whether clustering of PA monomers affected the rate of furin processing (data from dANTXR2 not shown). The disappearance of the full-length PA band, and corresponding appearance of the PA63 band occurs at similar rates, that is, either when PA is free of receptor or it is complexed with dimeric or monomer versions of ANTXR2. Therefore, we conclude that the binding of PA to ANTXR2 has no effect on the rate of furin processing.
Discussion
Elucidating the thermodynamic mechanisms governing the formation of specific macromolecular architectures in the cell has remained a significant barrier to understanding the assembly and function of molecular machinery. Macromolecular assembly is a ubiquitous cellular process central to normal physiology as well as the mechanisms of microbial pathogenesis. Viruses and bacterial virulence factors, including anthrax toxin, must properly assemble for function. Because each PA subunit contains four folded domains, a number of functions including host-receptor recognition, oligomerization, channel formation, enzyme subunit binding, and enzyme activity may be attributed to each individual domain and/or inter-domain cross-talk. The fact that anthrax toxin assembles heterogeneously is a testament to the inherent complexity and flexibility involving the interactions of these multidomain subunits during assembly. Heterogenous assembly mechanisms can lead to new functions for the macromolecular machine, such as altered complex stability, dynamics and function. Furthermore, the complexity of the intra-domain and inter-domain interactions allow for allosteric regulators to bind and alter either the assembly or the function of the complex.
Due to the primacy of virus and virulence-factor assembly to pathogenesis, small-molecule inhibitors that disrupt or alter assembly may seem at first glance to be reasonable drug-development strategies; however, these types of therapeutics have lagged considerably relative to those that target metabolic pathways and traditional enzyme activities. The lack of assembly-targeted drugs is, in part, due to the difficulty of assembly assays compared to enzyme assays or binary-component binding reactions. But more likely, the interfacial surface areas that define the thermodynamic interactions between subunits in these molecular machines are too extensive to impair or inhibit with a small-molecule drug by means of direct interference. Thus, the most-likely path to developing small molecules that inhibit virulence factor assembly would likely involve the triggering of a cryptic and/or native site that allosterically controls assembly. The allosteric-modulation strategy enables the development of a more practically-sized molecule to modulate the assembly pathway. It is in this spirit that we investigate the molecular mechanism of anthrax toxin assembly.
Protoxin maturation by furin
Since many viruses and toxins are secreted as proproteins, they must be proteolytically processed or activated prior to assembly. This mechanism ensures that these virulence factors can localize properly and infect targeted cells. In our structure of PAΔMIL, we modeled residues 162-173, which contain the site for furin-dependent cleavage and subsequent activation of PA (Fig. 6a). Furin has been implicated as the protease responsible for the proprotein processing of a myriad of disease pathogens.40-49 The crystal structure of furin, in complex with an inhibitor peptide, has been solved; thus the structural basis for furin specificity has been reported.33 Aligning the PA sequence containing the furin-cleavage motif (S163-R-K-K-R) to the dec-R-K-V-R-cmk inhibitor (Fig. 6c), where the amino and carboxy termini of the inhibitor peptide were modified with the decyl (dec) and carboxymethyl ketone (cmk) groups, respectively, shows the backbone atoms are in close agreement, with the exception of the P1 site, R167. In order to align well with the inhibitor, the peptide bond between K166 and R167 must be rotated. We find that residues remain well within the β-strand region of the Ramachandran plot upon rotation.58 Given that R167 is freely accessible to solvent and its movement is not restricted in our structure of PAΔMIL, we expect that this movement is not unreasonable. Successful rotation of R167 positions the scissile bond site in proximity to the catalytic Ser and His residues of furin and allows the residues carboxy to 167 to exit the furin catalytic site (Fig. 6d). Furthermore, we show that moving the furin-cleavage site either upstream or downstream results in a loss of protoxin processing in vitro (Fig. 6b). We propose that steric clashes with PA20 residues accounts for the specific requirement of the furin-cleavage site positioning on L158-173. The atomic model of PA presented here provides a starting point for understanding the structural basis for furin-dependent cleavage of PA. Furthermore, being a protease-sensitive site, the ordering of the furin loop in the PAΔMIL structure suggested that there was some level of cross talk between the furin loop, D2, and D4. We tested this hypothesis and found that receptor binding across D2 and D4 does not alter the rate of furin processing. Furin processing and PA assembly on cell surfaces, which appears to be coordinated with cholesterol-rich microdomains,25 may be controlled through other means.
Furin is a critical housekeeping enzyme involved in proprotein and prohormone maturation. Protoxins have taken advantage of the ubiquitous convertase, using it to activate virulence factors and infect a plethora of cell types. While furin-specific inhibitors have emerged as attractive therapeutics for preventing protoxin activation, such treatments may also obstruct normal cell processes that rely on furin processing. A recent study found that the efficacy of furin targets transforming growth factor beta (TGFβ1) and matrix metalloprotein (MT1-MMP) were not affected by furin inhibition, and it was proposed that the inherent redundancy of cell-surface convertases might compensate for the loss in furin activity.50 However, other furin targets have not been studied, and a broader picture of the side effects associated with inhibiting furin activity remains unclear. Here, we report for the first time the atomic structure of a furin-cleavage site, which may lead to the development of drugs that are specifically tailored to inhibiting the furin-protoxin interaction, rather than furin itself. Indeed, one such inhibitor that binds to the viral envelope protein gp160 in a region proximal to the furin-cleavage site has been implicated as a possible treatment for HIV-1.59 In this manner, the inhibitor interacts exclusively with the target antigen, preventing furin processing; thus, potential side effects arising from furin inhibition activity would be minimized. This avenue may be worth pursuing with anthrax toxin, as furin-loop accessibility can affect the rate of processing (Fig. 6b) and drug binding may lead to altered conformational dynamics.
Molecular determinants of PA’s heterogeneous oligomerization mechanism
Another goal of this study is to obtain a molecular-level understanding of anthrax toxin assembly. How does the PA subunit assemble into a heterogeneous mixture of two different oligomeric architectures? From this effort we expect to draw broader conclusions about macromolecular assembly paradigms and to inspire novel therapies for anthrax disease. Previous studies indicate that PA subunits form mixtures of PA7 and PA8 complexes, yielding of a PA8 content of ~5% to 30%. The PA8 complex in particular is interesting because it is more pH-stable and thermostable than the more abundant PA7 complex.13 PA8 complexes can survive in the plasma fraction of bovine blood with an ~30-minute half life, while the PA7 complexes precipitate and inactivate in a few minutes.13 Our present model is that allosteric control of the level of PA8 complexes during pathogenesis may alter the level of toxicity achieved by the toxin. An alternative, non-mutually-exclusive model is that the heterogeneity allows for more efficient assembly on cell surfaces, preventing situations where the chance occurrence of odd- or even-numbered subcomplex stoichiometries would impede proper assembly if a strict oligomeric architecture were prerequisite for assembly.14 In either case, it is likely that some level of control is present in the system, and this control determines the ultimate stoichiometry of the oligomeric mixture produced.
In previous work, we demonstrated that two separate phenomena produce relatively heterogeneous PA oligomer populations: (i) Addition of protein assembly co-factors that facilitate the formation of dimeric PA intermediates can drive PA8 formation up to as high as ~30%; these proteins include the dANTXR2, LF, and EF. (ii) The PAΔMIL construct tends to form ~30% PA8 complexes in the absence of dimerization-enhancing protein co-factors.14 Mechanism (i) appears to have a straightforward intermolecular explanation, where dimerization favors the formation of the less prevalent even-numbered PA8. However, mechanism (ii), albeit an artificial one, is not clear. Does the presence of the MIL trigger an allosteric site that prevents octamer formation? Or does the MIL simply block octamerization by a steric mechanism, where it would interfere with D4 from an adjacent PA in the oligomer? In this report, we explored the mechanism (ii), which appears allosteric in nature. These results describe for the first time that a complex network of interactions exist within the PA monomer, which can coordinate oligomeric architecture. Interestingly, when PAΔMIL is co-assembled with LFN, PA8 complexes are enriched >50% (Fig. 4), or 24% more than when PAΔMIL is assembled in isolation.14 This result indicates that the mechanisms (i) and (ii) are in fact additive. Furthermore, we produced a PA construct whose D4 is loosely tethered to the main body of the protein via a dithioacetone cross-link. Independent of the presence of the MIL, this construct yields a highly enriched PA8 stoichiometry—in excess of 85% (Fig. 4). Therefore, we find that removing the MIL and loosely tethering D4 to D2 have similar effects on PA8 enrichment, albeit the latter is a noticeably stronger effect. On the other hand, tightly cross-linking D4 and D2 with a disulfide bond appears to inhibit the formation of the PA8 complex relative to WT (Fig. 4). Thus the conformation of individual PA subunits influences the outcome of an assembly reaction.
Based upon our structural observations that D4 can occupy a Pro-PA7 and Pro-PA8 conformation, we propose that these two conformations predispose the PA subunit to form either respective oligomeric architecture. The structure of PAΔMIL and various cross-linked variants reveal that the major conformational change in these monomers surrounds the hinge-like movement of D4 (Fig. 5b-c). D4 can adopt two different conformations, Pro-PA7 and Pro-PA8, which affect PA oligomer stoichiometry. Interestingly, the two conformations can be populated with different length cross-links tethering D4 to D2. The Pro-PA7 conformation is favored with the shorter disulfide bond cross-link, while the Pro-PA8 conformation is favored by the 3-atom extension inherent to the dithioacetone cross-link. We suspect that restricting the conformational space of the D2-D4 pitch angle with the dithioacetone cross-link essentially lowers the kinetic barrier for PA to adopt the Pro-PA8 conformation. Furthermore, the propensity of a particular PA monomer to adopt either D4 conformation is not limited to the presence of the MIL, as the cross-link phenomenon is consistent with both WT PA and PAΔMIL backgrounds (Fig. 4). Thus steric interference by the MIL may not be the only factor influencing the oligomeric architecture, since its presence can be overridden by the D2-D4 dithioacetone cross-link (Fig. 4). The Pro-PA8 conformation populated by the dithioacetone cross-link, therefore, also controls the PA oligomerization interface in a way that favors the PA8 species over the PA7 species. In conclusion, we propose that the flexibility and relative orientation of D4 holds an important key in deciding the outcome of PA’s heterogeneous oligomerization pathway. We suspect that other heterogeneous oligomeric systems may also have similar mechanisms that control product stoichiometry. Future work should address how small-molecule, drug-like molecules can influence virulence factor assembly, stoichiometry, and function.
Materials and Methods
Protein expression and purification
An expression plasmid encoding the PA deletion mutant, PAΔMIL, was produced by removing residues 305-324 while simultaneously introducing the mutations V303P and H304G.14 The resulting PA construct replaces the membrane insertion loop (residues 303-324), leaving in its place a type-II turn; residue numbering is consistent with 1ACC.23 Site-directed mutations of PA and PAΔMIL were produced using the QuikChange procedure (Agilent Technologies, Santa Clara, CA). PA and subsequent PA mutants were over-expressed in the periplasm of Escherichia coli BL21(DE3) and purified as PA monomers over Q-sepharose anion exchange (GE Biosciences), as described.14 PA constructs used for crystallography were further purified over S200 gel filtration chromatography (GE Biosciences) in buffer A (20 mM Tris-Cl, 150 mM NaCl pH 8), and concentrated to 25 mg/mL.
For PA S337C N664C and PAΔMIL S337C N664C mutants, monomers were split into two fractions prior to storage at −80°C. The first fraction was allowed to form a disulfide bond under oxidizing conditions while 10 mM dithiothriotol (DTT, Gold Biotechnology, St. Louis, MO) was added to the second fraction. DCA-modified constructs were produced following a prior method.55 Cys-containing PA proteins were immobilized on Q-sepharose anion exchange using N2-purged buffers. ~5 mg/mL fractions of DTT-free S337C N664C protein were incubated with 1 mM 1,3-dichloroacetone (Sigma-Aldrich, St. Louis, MO) on ice for 30 min, and the subsequent labeled protein was purified by S200 gel filtration in buffer A.
LFN (LF residues 1-263) was over-expressed from a pET15b construct60 in the cytoplasm of E. coli BL21(DE3) and purified as described.14 The hexahistidine tags were removed by incubation with 0.5 units of bovine α-thrombin (Enzyme Research, South Bend, IN) per mg of LFN for 30 minutes at room temperature in buffer A supplemented with 2.5 mM CaCl2 and 1 M dextrose.
Dimeric ANTXR2, corresponding to the soluble domain residues 40-217, was over-expressed from a pGEX vector (GE Healthcare) as a glutathione-S-transferase fusion protein in the cytoplasm of E. coli and purified on a glutathione-sepharose affinity column (GE Healthcare), as described.19 Monomeric ANTXR2 was produced by transferring residues 40-217 from pGEX into pET15b using the NdeI and BamHI restriction sites, yielding a N-terminal His-tag construct. Site-directed mutagenesis yielded the mutant C175A. The protein was over-expressed in E. coli and purified by Ni2+-affinity chromatography, as described.17
The PA+6 and PA−5 mutants were made using a three-step, gene-synthesis procedure, in a manner similar to a previous procedure.24 Overlapping oligonucleotides encoding the desired sequences were synthesized (Elim Biopharmaceuticals, Inc., Hayward, CA) and amplified by two rounds of polymerase chain reaction (PCR). In Round I, 20 nM of nested oligonucleotides with consistent annealing temperatures of ~55 °C were amplified in a standard PCR reaction. In Round II, 1 μL of the PCR product made in Round I was amplified with the two outermost PCR primers (1 μM each) to make the synthetic double-stranded DNA fragment. These synthetic DNA fragments were ligated via a 5′ Hind III site and 3′ Kpn I site into the PA reading frame from a pET22b vector containing an in-frame, silent Kpn I restriction site in PA at V175.
Protein crystallization
Initial crystallization trials were carried out by screening ~400 sparse-matrix61 conditions using a Mosquito nanoliter robot (TTP Labtech, Cambridge, MA) to set 200 nL drops by the hanging-drop vapor-diffusion method62 in 96-well format. Crystal conditions were further investigated using manual 24-well format trays using a 1:1 ratio of 1 or 2 μL drops of protein and reservoir solution. For PAΔMIL pH 8.5 and PA C337-C664-DCA the reservoir solution was composed of 15-25% (w/v) polyethylene glycol monomethyl ether (PEG ME) with an average molecular weight of 2000 Da, 100 mM Tris-Cl, 200 mM trimethylamine N-oxide, pH 8.3-8.7. These proteins formed trapezoidal prism-shaped crystals overnight, maturing to dimensions 300-800 μm. Crystals were harvested in an antifreeze solution containing a 1:1 mixture of 50% (v/v) PEG ME with an average molecular weight of 550 Da, and reservoir solution supplemented with 150 mM NaCl. PAΔMIL pH 6.5 and PAΔMIL C337-C664 reservoir solution contained 29-32% (v/v) pentaerythritol ethoxylate (15/4 EO/OH), 50 mM bis-tris Cl, 50 mM ammonium sulfate, pH 6.5-6.7. These proteins formed coffin-shaped crystals within 24 hours, maturing to dimensions 200-400 μm, and the crystals were harvested directly out of the drop. All crystal forms were flash-frozen by rapidly plunging the crystal into liquid N2.
X-ray diffraction data collection, solution and refinement
X-ray diffraction data were collected at a wavelength of 1.1159 Å (11111 eV) and a temperature of 100 K at the Lawrence Berkeley National Lab’s Advanced Light Source, Beamline 8.3.1 using a Quantum 315r CCD area detector (ADSC, Poway, CA).63 Crystals containing PAΔMIL pH 8.5 diffracted X-rays to 1.45 Å in the orthorhombic space group P212121 with unit cell dimensions of a = 72.36 Å, b = 93.39, c = 118.08, and angles, α, β, and γ, of 90°. Crystals containing PA C337-C664-DCA diffracted X-rays to 1.83 Å in the orthorhombic space group P212121 with unit cell dimensions of a = 71.29 Å, b = 93.69, c = 117.85, and angles, α, β, and γ, of 90°. For both samples, a second data set was collected from the same crystal with a shorter exposure time to obtain the overloaded, low angle reflections. Crystals containing PAΔMIL C337-C664 diffracted X-rays to 2.09 Å in the orthorhombic space group P212121 with unit cell dimensions of a = 71.56 Å, b = 94.60, c = 119.80, and angles, α, β, and γ, of 90°. Crystals containing PAΔMIL pH 6.5 diffracted X-rays to 1.70 Å in the orthorhombic space group P212121 with unit cell dimensions of a = 71.40 Å, b = 93.93, c = 117.85, and angles, α, β, and γ, of 90°. The diffraction data were indexed, scaled, and merged in HKL2000.64 Molecular replacement for PAΔMIL pH 8.5 was carried out in PHASER,65 using a loop-stripped model of PA (1ACC)23 as the search model. Missing loops with significant (σ > 2.5) Fo-Fc difference density were built manually in COOT,66 and the electron density was corroborated with composite simulated-annealing omit maps generated in PHENIX.67 The coordinates and ADP values for all atoms were refined individually and anisotropically, respectively, in PHENIX, followed by further rounds of model building and solvent picking in COOT. Some of the alternate side-chain conformations were identified using RINGER,68 and occupancy refinement was carried out in PHENIX. Structure solution and refinement for PA C337-C664-DCA and PAΔMIL pH 6.5 was carried out similarly, accept that PAΔMIL was used as the search model, and ADP values were refined isotropically. A similar strategy was also employed for PAΔMIL C337-C664 except that atomic coordinates were refined both individually and with rigid body groups, and ADP values were refined isotropically. Two molecules of 2-methoxyethanol were identified in the density for PAΔMIL pH 8.5 and PA C337-C664-DCA, as well as an acetone molecule linking Cys337 and Cys664 for PA C337-C664-DCA. The oxygen atom on the ketone was not resolved; therefore, its occupancy was set to 0. The final model geometry for all structures was validated using MOLPROBITY69 and PROCHECK70. Least-squares quotient (LSQ) Cα alignment of protein backbones was carried out in COOT.66 RMS deviations of protein backbone were computed with COMPAR in the CCP4 program suite.71 All molecular graphics were generated using CHIMERA.72
Furin proteolysis of PA
WT PA and PA mutants (1 mg/mL) were incubated with 10 units of furin (New England Biolabs, Ipswitch, MA) per mg of PA in buffer A supplemented with 1 mM CaCl2 at room temperature, as described.73 For the ANTXR2-PA proteolysis experiments, monomeric ANTXR2 C175A or dimeric GST-linked ANTXR2 was incubated with PA in a 4:1 molar ratio in buffer A supplemented with 1 mM CaCl2 and 1 mM MgCl2 for 10 minutes at room temperature. The co-complex was then processed with 20 units of furin per mg of PA. Samples were separated on a 10% SDS-PAGE gel and stained with Coomassie Brilliant Blue (Bio-RAD Laboratories, Hercules, CA), and the Pageruler protein ladder (Fermentas Life Sciences, Glen Burnie, MD) was used to determine the molecular weights of the products.
LFN-driven assembly of nPA constructs
PA constructs (1 mg/mL) were nicked with trypsin (Sigma-Aldrich) at a ratio of 1:1000 (w/w) for 15 minutes at room temperature and then treated with soybean tryspin inhibitor (Worthington Biochemical, Lakewood, NJ) at 1:100 (w/w). The resulting nPA mixture was co-assembled with LFN in a 1:1 molar ratio for 1 hour at room temperature. Samples were purified over S200 gel filtration in buffer A.
Electron microscopy
Grid preparation, data collection, and image processing were performed as described.14 Briefly, each PA oligomer was diluted to 100 nM (with respect to PA monomer concentration) in buffer A supplemented with 0.001% (w/v) n-dodecyl-β-D-maltopyranoside (Affymetrix, Maumee, OH). PA complexes containing Cys mutations were diluted in buffer containing 5 mM DTT to improve image quality. 400 mesh copper grids were successively covered by continuous carbon film via a formvar support. 4 μl of PA sample was applied to the grid for 1 minute, washed in 3 successive drops of water, and then stained with 2% uranyl acetate (Sigma-Aldrich). Negative-stain EM images were recorded with a Tecnai 12 (FEI Company, Hillsboro, OR) operated at 120 kV at 49,000× magnification. Images were taken using a CCD camera at a 2.13-Å/pixel specimen scale. Particle images were selected for each data set using automatic or manual particle picking using boxer in EMAN.74 Reference-free processing was computed using SPIDER.75 Images were subjected to three successive cycles of multi-reference alignment, multivariate statistical analysis, and classification, such that the last classification was done using only the lowest order eigenvectors.78 Following the last classification, we separated the images by size and by the heptameric and octameric oligomerization states. Also we used a second method of image processing, whereby crystal-structure-reference images were made from two-dimensional projections of low resolution density maps generated from the crystal structures of the PA heptamer18 and octamer14 in SPIDER.75 Crystal-structure-referenced images were aligned and classified using the lowest order eigenvectors. Final class-average images were manually inspected as being heptameric, octameric, or unclassifiable “junk” (<5% of the total particles). The number of particles per identifiable classification was used to determine the percentages of heptamers and octamers. Approximately 1500-5000 total particles were analyzed per sample. Either classification procedure produces similar results for the analysis of the composition of PA oligomer samples, agreeing with other biophysical measures of oligomeric heterogeneity.
Acknowledgements
G.K.F. and B.A.K. conceived and designed all experiments. G.K.F. produced the cross-linked PA constructs and oligomeric complexes; crystallized, solved, and refined the X-ray crystal structures; and performed the furin proteolysis experiments. A.F.K., G.K.F. & I.I.T. collected and processed the electron microscopy data. K.L.T. produced the furin-site mutations in PA. G.K.F. and B.A.K. wrote the manuscript. This work was supported by an NIH research grant R01-AI077703 (to B.A.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers. Coordinates and structure factors for PAΔMIL pH 8.5, PAΔMIL pH 6.5, PAΔMIL C337-C664, and PA83 C337-C664-DCA have been deposited in the PDB with accession codes 3TEW, 3TEX, 3TEY, and 3TEZ, respectively.
References
- 1.Young JA, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007;76:243–65. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 2.Thoren KL, Krantz BA. The unfolding story of anthrax toxin translocation. Mol. Microbiol. 2011;80:588–95. doi: 10.1111/j.1365-2958.2011.07614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith H, Keppie J. Observations on experimental anthrax: demonstration of a specific lethal factor produced in vivo by Bacillus anthracis. Nature. 1954;173:689. doi: 10.1038/173869a0. [DOI] [PubMed] [Google Scholar]
- 4.Stanley JL, Smith H. Purification of factor I and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961;26:49–66. doi: 10.1099/00221287-26-1-49. [DOI] [PubMed] [Google Scholar]
- 5.Beall FA, Taylor MJ, Thorne CB. Rapid lethal effects of a third factor of anthrax toxin. J. Bacteriol. 1962;83:1274–80. doi: 10.1128/jb.83.6.1274-1280.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duesbery NS, Vande Woude GF. Anthrax lethal factor causes proteolytic inactivation of mitogen-activated protein kinase kinase. J. Appl. Microbiol. 1999;87:289–93. doi: 10.1046/j.1365-2672.1999.00892.x. [DOI] [PubMed] [Google Scholar]
- 7.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–7. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 8.Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, Hanna P, Liddington RC. Crystal structure of the anthrax lethal factor. Nature. 2001;414:229–33. doi: 10.1038/n35101998. [DOI] [PubMed] [Google Scholar]
- 9.Pezard C, Berche P, Mock M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991;59:3472–7. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl Acad. Sci. U.S.A. 1982;79:3162–6. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leppla SH. Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eucaryotic cells. Adv. Cyclic Nucl. Prot. 1984;17:189–98. [PubMed] [Google Scholar]
- 12.Drum CL, Yan SZ, Bard J, Shen YQ, Lu D, Soelaiman S, Grabarek Z, Bohm A, Tang WJ. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 2002;415:396–402. doi: 10.1038/415396a. [DOI] [PubMed] [Google Scholar]
- 13.Kintzer AF, Sterling HJ, Tang II, Abdul-Gader A, Miles AJ, Wallace BA, Williams ER, Krantz BA. Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J. Mol. Biol. 2010;399:741–58. doi: 10.1016/j.jmb.2010.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–9. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 16.Scobie HM, Rainey GJA, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl Acad. Sci. U.S.A. 2003;100:5170–4. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–8. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 18.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13147–51. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J. Biol. Chem. 2004;279:23349–56. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 20.Lacy DB, Wigelsworth DJ, Scobie HM, Young JA, Collier RJ. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc. Natl Acad. Sci. U.S.A. 2004;101:6367–72. doi: 10.1073/pnas.0401506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scobie HM, Wigelsworth DJ, Marlett JM, Thomas D, Rainey GJ, Lacy DB, Manchester M, Collier RJ, Young JA. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLoS Pathog. 2006;2:e111. doi: 10.1371/journal.ppat.0020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994;269:20607–12. [PubMed] [Google Scholar]
- 23.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–8. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 24.Feld GK, Thoren KL, Kintzer AF, Sterling HJ, Tang II, Greenberg SG, Williams ER, Krantz BA. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nature Struct. Mol. Biol. 2010;17:1383–90. doi: 10.1038/nsmb.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003;160:321–8. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986;261:7123–6. [PubMed] [Google Scholar]
- 27.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–41. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 28.Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP, Collier RJ, Fisher MT. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nature Struct. Mol. Biol. 2008;15:754–60. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl Acad. Sci. U.S.A. 1989;86:2209–13. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mogridge J, Mourez M, Collier RJ. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol. 2001;183:2111–6. doi: 10.1128/JB.183.6.2111-2116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steiner DF. The proprotein convertases. Curr. Opin. Chem. Biol. 1998;2:31–9. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997;327(Pt 3):625–35. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nature Struct. Biol. 2003;10:520–6. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- 34.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nature Rev. Mol. Cell Biol. 2002;3:753–66. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holyoak T, Wilson MA, Fenn TD, Kettner CA, Petsko GA, Fuller RS, Ringe D. 2.4 A resolution crystal structure of the prototypical hormone-processing protease Kex2 in complex with an Ala-Lys-Arg boronic acid inhibitor. Biochemistry. 2003;42:6709–18. doi: 10.1021/bi034434t. [DOI] [PubMed] [Google Scholar]
- 36.Fuller RS, Brake A, Thorner J. Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc. Natl Acad. Sci. U.S.A. 1989;86:1434–8. doi: 10.1073/pnas.86.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuller RS, Brake AJ, Thorner J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science. 1989;246:482–6. doi: 10.1126/science.2683070. [DOI] [PubMed] [Google Scholar]
- 38.Remacle AG, Shiryaev SA, Oh ES, Cieplak P, Srinivasan A, Wei G, Liddington RC, Ratnikov BI, Parent A, Desjardins R, Day R, Smith JW, Lebl M, Strongin AY. Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J. Biol. Chem. 2008;283:20897–906. doi: 10.1074/jbc.M803762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molloy SS, Thomas L, VanSlyke JK, Stenberg PE, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon VM, Leppla SH. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infect. Immun. 1994;62:333–40. doi: 10.1128/iai.62.2.333-340.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 1992;267:16396–402. [PubMed] [Google Scholar]
- 42.Garred O, van Deurs B, Sandvig K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995;270:10817–21. doi: 10.1074/jbc.270.18.10817. [DOI] [PubMed] [Google Scholar]
- 43.Tsuneoka M, Nakayama K, Hatsuzawa K, Komada M, Kitamura N, Mekada E. Evidence for involvement of furin in cleavage and activation of diphtheria toxin. J. Biol. Chem. 1993;268:26461–5. [PubMed] [Google Scholar]
- 44.Gordon VM, Benz R, Fujii K, Leppla SH, Tweten RK. Clostridium septicum alpha-toxin is proteolytically activated by furin. Infect. Immun. 1997;65:4130–4. doi: 10.1128/iai.65.10.4130-4134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abrami L, Fivaz M, Decroly E, Seidah NG, Jean F, Thomas G, Leppla SH, Buckley JT, van der Goot FG. The pore-forming toxin proaerolysin is activated by furin. J. Biol. Chem. 1998;273:32656–61. doi: 10.1074/jbc.273.49.32656. [DOI] [PubMed] [Google Scholar]
- 46.Stieneke-Grober A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992;11:2407–14. doi: 10.1002/j.1460-2075.1992.tb05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360:358–61. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 48.Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997;71:8475–81. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volchkov VE, Feldmann H, Volchkova VA, Klenk HD. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl Acad. Sci. U.S.A. 1998;95:5762–7. doi: 10.1073/pnas.95.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Remacle AG, Gawlik K, Golubkov VS, Cadwell GW, Liddington RC, Cieplak P, Millis SZ, Desjardins R, Routhier S, Yuan XW, Neugebauer WA, Day R, Strongin AY. Selective and potent furin inhibitors protect cells from anthrax without significant toxicity. Int. J. Biochem. Cell. B. 2010;42:987–95. doi: 10.1016/j.biocel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarac MS, Peinado JR, Leppla SH, Lindberg I. Protection against anthrax toxemia by hexa-D-arginine in vitro and in vivo. Infect. Immun. 2004;72:602–5. doi: 10.1128/IAI.72.1.602-605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komiyama T, Swanson JA, Fuller RS. Protection from anthrax toxin-mediated killing of macrophages by the combined effects of furin inhibitors and chloroquine. Antimicrob. Agents Ch. 2005;49:3875–82. doi: 10.1128/AAC.49.9.3875-3882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oliva R, Leone M, Falcigno L, D’Auria G, Dettin M, Scarinci C, Di Bello C, Paolillo L. Structural investigation of the HIV-1 envelope glycoprotein gp160 cleavage site. Chemistry. 2002;8:1467–73. doi: 10.1002/1521-3765(20020315)8:6<1467::aid-chem1467>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 54.Wimalasena DS, Janowiak BE, Lovell S, Miyagi M, Sun J, Zhou H, Hajduch J, Pooput C, Kirk KL, Battaile KP, Bann JG. Evidence that histidine protonation of receptor-bound anthrax protective antigen is a trigger for pore formation. Biochemistry. 2010;49:6973–83. doi: 10.1021/bi100647z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yin L, Krantz B, Russell NS, Deshpande S, Wilkinson KD. Nonhydrolyzable diubiquitin analogues are inhibitors of ubiquitin conjugation and deconjugation. Biochemistry. 2000;39:10001–10. doi: 10.1021/bi0007019. [DOI] [PubMed] [Google Scholar]
- 56.Kintzer AF, Sterling HJ, Tang II, Williams ER, Krantz BA. Anthrax toxin receptor drives protective antigen oligomerization and stabilizes the heptameric and octameric oligomer by a similar mechanism. PLoS ONE. 2010;5:e13888. doi: 10.1371/journal.pone.0013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christensen KA, Krantz BA, Melnyk RA, Collier RJ. Interaction of the 20 kDa and 63 kDa fragments of anthrax protective antigen: kinetics and thermodynamics. Biochemistry. 2005;44:1047–53. doi: 10.1021/bi047791s. [DOI] [PubMed] [Google Scholar]
- 58.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 59.Murray EJ, Leaman DP, Pawa N, Perkins H, Pickford C, Perros M, Zwick MB, Butler SL. A low-molecular-weight entry inhibitor of both CCR5- and CXCR4-tropic strains of human immunodeficiency virus type 1 targets a novel site on gp41. J. Virol. 2010;84:7288–99. doi: 10.1128/JVI.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lacy DB, Mourez M, Fouassier A, Collier RJ. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem. 2002;277:3006–10. doi: 10.1074/jbc.M109997200. [DOI] [PubMed] [Google Scholar]
- 61.Jancarik J, Kim SH. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Cryst. 1991;24:409–411. [Google Scholar]
- 62.McPherson A., Jr. The growth and preliminary investigation of protein and nucleic acid crystals for X-ray diffraction analysis. Methods Biochem. Anal. 1976;23:249–345. doi: 10.1002/9780470110430.ch4. [DOI] [PubMed] [Google Scholar]
- 63.MacDowell AA, Celestre RS, Howells M, McKinney W, Krupnick J, Cambie D, Domning EE, Duarte RM, Kelez N, Plate DW, Cork CW, Earnest TN, Dickert J, Meigs G, Ralston C, Holton JM, Alber T, Berger JM, Agard DA, Padmore HA. Suite of three protein crystallography beamlines with single superconducting bend magnet as the source. J. Synchrotron Rad. 2004;11:447–55. doi: 10.1107/S0909049504024835. [DOI] [PubMed] [Google Scholar]
- 64.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology. Academic Press, Inc.; New York: 1997. pp. 307–326. Vol. 276: Macromolecular Crystallography, part A. [DOI] [PubMed] [Google Scholar]
- 65.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 66.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 67.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lang PT, Ng HL, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci. 2010;19:1420–31. doi: 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 71.Collaborative Computational. Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 72.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 73.Christensen KA, Krantz BA, Collier RJ. Assembly and disassembly kinetics of anthrax toxin complexes. Biochemistry. 2006;45:2380–6. doi: 10.1021/bi051830y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 75.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–9. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 76.Stark H, Mueller F, Orlova EV, Schatz M, Dube P, Erdemir T, Zemlin F, Brimacombe R, van Heel M. The 70S Escherichia coli ribosome at 23 A resolution: fitting the ribosomal RNA. Structure. 1995;3:815–21. doi: 10.1016/s0969-2126(01)00216-7. [DOI] [PubMed] [Google Scholar]
- 77.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 78.White HE, Saibil HR, Ignatiou A, Orlova EV. Recognition and separation of single particles with size variation by statistical analysis of their images. J. Mol. Biol. 2004;336:453–60. doi: 10.1016/j.jmb.2003.12.015. [DOI] [PubMed] [Google Scholar]