

Abstract
Better understanding of electron transfer (ET) taking place at the nano-bio interface can guide design of more effective functional materials used in fuel cells, biosensors, and medical devices. Single-walled carbon nanotube (SWCNT) coupled with biological enzymes serves as a model system for studying the ET mechanism, as demonstrated in the present study. SWCNT enhanced the activity of horseradish peroxidase (HRP) in the solution-based redox reaction by binding to HRP at a site proximate to the enzyme’s activity center and participating in the ET process. ET to and from SWCNT was clearly observable using near-infrared spectroscopy. The capability of SWCNT in receiving electrons and the direct attachment of HRP to the surface of SWCNT strongly affected the enzyme activity due to the direct involvement of SWCNT in ET.
1. Introduction
Biofunctional nanomaterials prepared by coupling redox proteins or enzymes to conducting nanomaterials like carbon nanotubes (CNTs) are of paramount importance for the production of renewable energy and the construction of electrochemical sensors.[1–3] Nanomaterials are employed to increase protein loading and assist with electron transfer (ET) to electrode.[4–10] Efficient ET between the nanomaterials and the attached protein is also imperative to the success of such hybrid materials, and should be evaluated in design of the nano-bio hybrid structure.[11] However, assessment of ET at the nano-bio interface and the control factors are difficult with the nano-bio complex locating on the solid electrode surface, because both attachments of the protein to nanomaterials and the nano-bio complex to electrodes contribute to the observed electrochemical activity.[12] Moreover, biological cells rely on ET for energy production, signal transduction, and cell-cell communication.[13–15] Recent studies have pointed out the possibility of using CNTs and other conducting nanomaterials to generate electrical shortcuts and enhance the performance of cells or tissues.[16–18] Participation of the nanomaterial in ET among biomolecules should be studied for better understanding of the phenomena and for possible manipulation over the biological processes to heal diseases.
Pioneer study of ET at the nano-bio interface reported that binding of the Au nanoparticles to cytochrome c near its activity center could facilitate intermolecular ET between the redox partners, and the Au surface served to increase the local concentration of the redox molecules.[19, 20] The present study revealed that the single-walled CNT (SWCNT) was directly involved in the ET between the enzyme and its substrate besides being a solid support for the enzyme. The peroxidase-SWCNT complex allows the facile monitor of the ET with simple methodologies. It can serve as a model system for study of ET at the nano-bio interface in solution to improve our understanding of the mechanism and the dependence of ET efficiency on nanostructure properties.
Our previous work revealed that both the unmodified and carboxylated SWCNT could enhance the activity of horseradish peroxidase (HRP) in oxidizing the reducing substrates like 2,7-dichlorodihydrofluorescein (H2DCF), a reactive oxygen species (ROS) indicator, and trolox, a water-soluble analog of vitamin E in aqueous solutions.[21] Circular dichroism (CD) spectroscopy showed that HRP was adsorbed by SWCNT. We hypothesized that the adsorption interface would be relevant to the activity enhancement. Hence, in the present study, we employed the crosslinking chemistry coupled with mass spectrometry (MS) to locate peptides near the adsorption sites. Site identification was confirmed by fluorescence spectroscopy, and its relationship to enzyme activity enhancement was studied by visible and near infrared (Vis-NIR) spectroscopy. Finally, impact from pH on enzyme activity enhancement was investigated to further verify our hypothesis.
2. Experimental Section
2.1 Chemicals and Reagents
The carboxylic acid-functionalized SWCNT (SWCNT-COOH, average length of 0.5 – 0.6 μm and diameter of 1.4 ± 0.1 nm) was obtained from Sigma (St. Louis, MO, USA) and used as purchased. HRP, trypsin, bovine serum albumin (BSA), hydrogen peroxide (33%) and Tween 20 were purchased from Sigma. ROS probe 2,7- dichlorodihydrofluorescein diacetate (H2DCF-DA) was obtained from Invitrogen (Eugene, OR, USA). The crosslinker, 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) was from Thermo Fisher Scientific (Rockford, IL, USA).
2.2 Stock Solution Preparation
To ensure good dispersion of SWCNT in the reaction solutions, all SWCNT stocks were prepared in a concentration of 0.5 mg/mL in deionized water with 1-hr ultra-sonication (100 W, 42 kHz) in ice water bath. No aggregation was found in the bottom of the centrifugation tube with a brief centrifugation of the stock solution at 14,000 × g even after several months of storage. HRP was prepared at a concentration of 1.0 mg/mL in deionized water. The trypsin stock solution of 1.0 mg/mL was prepared in 0.01 M hydrogen chloride (HCl). This acid solution was stored at −20 °C and used within two weeks. EDC was dissolved in water just before usage, and diluted to the desired concentrations in 50 mM MOPS buffer.
2.3 Crosslinking of HRP onto SWCNT-COOH
Before crosslinking, 50 μg of HRP and equivalent mass of SWCNT-COOH in deionized water were added into 1 mL of 50 mM MOPS buffer (pH 6.0), and the solution was stirred for three hours. The crosslinker EDC was applied at a molar ratio of 50 to 1 over the enzyme, and the solution was incubated overnight with stirring. The resulting solution was then transferred to a molecular weight cut-off (MWCO)-100 kDa Microcon® centrifugal filter (Millipore, Billerica, MA, USA). The SWCNT-COOH with an average length of 0.5–0.6 μm could not get through the filter pore, so the reaction byproducts and the unconjugated HRP were then washed off with 0.1 % Tween 20 solution and removed by centrifugation at 14,000 × g for 3 times, followed by deionized water for another 3 times. The HRP conjugated with SWCNT-COOH on the filter membrane was redistributed into the phosphate buffer (25 mM, pH 2.0), ready for heme extraction and trypsin digestion.
2.4 Preparation of apo-HRP and Trypsin Digestion
The heme group of the SWCNT-crosslinked or free HRP was removed by 3 rounds of extraction with an equal volume of 2-butanone at 0 °C. The organic phase was removed by SpeedVac and the apo-HRP was re-dispensed into 1 mL of the digestion buffer (NH4HCO3, 50 mM). Appropriate volume of the trypsin stock solution was then added into the digestion buffer such that the molar ratio of trypsin : protein in the sample was 1 : 50. The mixture was incubated overnight. After digestion the peptides and SWCNT-COOH were separated by centrifugation at 14,000 × g with the MWCO-100 kDa Microcon filter. The obtained peptides in the filtrate were purified with ziptip, loaded onto the plate and measured by the Applied Biosystems (ABI) Voyager DE-STR matrix assisted laser desorption ionization (MALDI) - time of flight (TOF) mass spectrometer (Carlsbad, CA, USA). Sequences of peptides 2744.5 and 2545.3 were verified by tandem MS/MS with the ABI Q-STAR XL oMALDI Q-TOF MS/MS instrument and the Mascot search engine provided by Matrix Science Inc. (Boston, MA, USA).
2.5 Control samples for MS
To find out what peptides were crosslinked to the SWCNT, four control samples were prepared along with the SWCNT-crosslinked HRP. The first control was free HRP treated by trypsin digestion and filtration without any SWCNT-COOH. Another control was the HRP passively adsorbed on SWCNT-COOH, which experienced the same steps of surfactant washing, heme removing, and trypsin digestion as the SWCNT-crosslinked HRP. The third control was prepared by mixing 5 μg of the trypsin-digested HRP peptides with 50 μg of SWCNT-COOH, and removing the peptides that bound to the SWCNT by centrifugation at 14,000 × g with the MWCO-100 kDa Microcon filter. The filtrate was analyzed. The last control was HRP itself crosslinked by EDC and digested by trypsin.
2.6 Intrinsic Fluorescence Quenching
Fluorescence measurements were performed in a Spex Fluorolog Tau-3 fluorescence spectrophotometer (HORIBA, CA). For the protein intrinsic fluorescence measurement, SWCNT-COOH were added to the 100 μg/mL HRP solutions at final concentrations of 5, 10, 20, 30 and 40 μg/mL. The mixture was excited at 278 nm, and the emission was measured from 290 to 410 nm. The quenching of apo-HRP fluorescence was also tested with the same procedure. To evaluate any possible interference from SWCNT-COOH, the excitation light intensity were measured after passing through the SWCNT-COOH solution at all tested concentrations. The results were showed in Figure S3 (c), showing very small and inconsistent variation of the light intensity with the presence of SWCNT. Therefore, the interference from SWCNT could be neglected compared to the significant variation of the tryptophan fluorescence.
To test fluorescence quenching of 2,7-dichlorofluorescein (DCF) by SWCNT-COOH, the nanotubes were added to 1 μM DCF dissolved in 25 mM phosphate (pH 7.4). The final concentrations of SWCNT were 2, 5, 10, and 20 μg/mL. The compound was excited at 488 nm, and fluorescent emission was collected from 500 to 580 nm.
2.7 Measurement of H2DCF Oxidation by Fluorescence
Oxidation of H2DCF was measured using the previous procedure with the Victor II microplate reader (Perkin-Elmer, Waltham, MA) equipped with 480 nm filter for excitation and 520 nm filter for emission.[21] The H2DCF stock solution was prepared on a daily basis by de-esterifying 20 nmol H2DCF-DA with 0.25 mL of 0.01 M NaOH. The resulted H2DCF solution was subsequently neutralized by 0.75 mL of 25 mM phosphate buffer (pH 7.4) to 20 μM. Two fold dilution (10 μM) of the stock was used for the measurement in which H2DCF was oxidized to the strongly fluorescent DCF. The consumption of H2DCF could be calculated from the fluorescence intensities measured at various reaction time points based on a calibration curve generated from DCF. Fluorescence quenching caused by SWCNT was adjusted by including SWCNT in the standard DCF solutions when acquiring the calibration curve. The systems we tested included 1.2 μg/mL HRP and 100 μM H2O2 under different pH (4.0 – 12.0) with or without 10 μg/mL SWCNT-COOH; and 1.2 μg/mL HRP and 10 μg/mL BSA mixed in different orders with 10 μg/mL SWCNT-COOH at pH 7.4. HRP activity was monitored by the oxidation of H2DCF within 3 min.
2.8 Vis–NIR spectroscopy of SWCNT-COOH
The absorbance spectrum from 400 to 1,300 nm was obtained by the Cary 500 ultraviolet (UV) Vis IR spectrometer (Varian, Palo Alto, CA). To monitor the electron transfer, the spectrum of 25 μg/mL SWCNT-COOH in the phosphate buffer (25 mM, pH 7.4) was scanned at 1,800 nm/s (0.5 min per scan) upon the addition of HRP and H2O2 to a final concentration of 0.6 μg/mL and 100 μM, respectively, followed by the addition of H2DCF to 20 μM. To avoid any dilution effect, the working solutions of HRP, H2O2 and H2DCF were prepared at 100 μg/mL, 10 mM and 1 mM, so that only very small volumes were added to the measured solutions. In the test of pH effect, the UV-IR spectra of SWCNT-COOH in the buffer solutions from pH 4.0 to 12.0 were also obtained.
2.9 Protein structure and peptide sequences
The protein structure images were prepared using VMD. VMD is developed with NIH support by the Theoretical and Computational Biophysics group at the Beckman Institute, University of Illinois at Urbana-Champaign. The sequence numbers for peptides used here were adopted from PDB, because we used the crystal structure downloaded from PDB to map out the location of these peptides. These numbers were 30 residues different than those reported in UniProtKB/Swiss-Prot, protein ID P00433, Peroxidase C1A. For example, using UniProtKB sequence number, m/z 2545.4 should be T124-R148.
3. Results and Discussion
3.1 Binding site identification by mass spectrometry
Our previous work revealed the adsorption of HRP on SWCNT by CD.[21] To further visualize the close attachment of HRP on SWCNT, AFM was employed in the present study, and the results were displayed in Supplementary Data Figure S1. After incubation with HRP, the surface of SWCNT-COOH became very rough and covered with knots which should be individual HRP molecules. Being adsorbed onto SWCNT-COOH may affect the function of this enzymatic protein, and the effect should be related to where the SWCNT-COOH binds on HRP. To find out the binding site of HRP on SWCNT-COOH, the method of crosslinking chemistry coupled with mass spectrometry was used, which was recently proved to be effective for such purpose in our lab.[22] In the present study, we coupled SWCNT-COOH to HRP using 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC). EDC would link the free amine groups on HRP to the carboxyl group of SWCNT, but it is a zero-length crosslinker so only the ones very close to the SWCNT surface would be affected. Thus, during the subsequent protease digestion, the peptides further away from the SWCNT surface could be released to the supernatant. Those close to the interaction interface would remain attached to the SWCNT surface by the amide bond, and could be recognized as the missing peptides when comparing the digestion patterns of the SWCNT-crosslinked and free HRP obtained with MALDI-TOF-MS. Crosslinking is necessary in this approach, because it can stabilize protein attachment so that the excess protein molecules can be washed off and do not generate high background in supernatant analysis. Furthermore, it can prevent identification of the false positives, i.e. peptides that have high affinity to SWCNT but are not part of the binding site of SWCNT when the protein remains its native folded structure. These peptides could be exposed after protease digestion, adsorbed onto SWCNT surface, and thus not discovered in the supernatant to create confusion.
Figure 1 showed the representative MS results we used to determine the binding site of SWCNT on HRP. After digestion, the free HRP gave out a total of 8 peptide peaks in MALDI-MS (Fig. 1a): m/z 541.4 (amino acid (a. a.) V119-R123), m/z 681.4 (a. a. F179-R183), m/z 743.6 (a. a. I32-R38), m/z 803.5 (a. a. G76-R82), m/z 959.6 (a. a. D20-R27), m/z 1475.9 (a. a. S160-K274), m/z 2545.4 (a. a. T94-R118), and m/z 2744.6 (a. a. D125-K149). The SWCNT-crosslinked HRP was prepared by adding EDC to the mixture of HRP and SWCNT-COOH which had been incubated for 3 hours in MOPS buffer (50 mM, pH 6.0). The unbound HRP was washed off the tubes with 0.1% Tween 20 before the bound protein was digested by trypsin. Two peptides, m/z 2545.4 and m/z 2744.6, were consistently missing from this sample (Fig. 1b). We chose MOPS as the incubation buffer because the structure and activity of HRP are stable at this pH in room temperature,[23] and it is the optimal reaction pH for EDC. Confirmed by MALDI-TOF-MS-MS (Supplementary Data Figure S2), the sequence of m/z 2545.4 was (R93)TVSCADLLTIAA QQSVTLAGGPSWR, with a lysine K84 residue close by; and that of m/z 2744.6 was (R124)DSLQAFLDLAN ANLPAPFFTLPQLK(14 9). These two peptides could have located close to the SWCNT surface during HRP adsorption, and both the K84 and K149 residues were coupled to the –COOH groups of SWCNT by EDC and no longer cleavable by trypsin. Although between K84 and T94 there was a digestion site of R93, SWCNT-COOH may have generated big steric hindrance to prevent the approaching of the protease and inhibit the cleavage at R93. On the contrary, without crosslinking, the adsorbed HRP was completely washed off the SWCNT-COOH by the Tween 20 solution, and no peptide was obtained after digestion (Fig. 1c). To exclude the possibility that the loss of peptides was mainly due to their own high affinity to SWCNT-COOH, we digested HRP and mixed the resulted peptides with the SWCNT-COOH. After removing the tubes from the solution, m/z 2545.4 and m/z 2744.6 were still found in the supernatant instead of being completely taken away by the SWCNT-COOH via adsorption (Supplementary Data Figure S3a). Trypsin digestion of HRP treated with EDC when no SWCNT-COOH was in the sample resulted in a completely different MS pattern (Fig. S3b). No peptide belonging to HRP could be identified from this MS result due to the extensive intramolecular crosslinking. The protein self-crosslinking did not occur to such a large extend in the sample containing SWCNT-COOH because the tubes provided sufficient -COOH groups for EDC to prevent it from activating those on the protein.
Figure 1.

MS spectra for the analysis of (a) free HRP digest; (b) the supernatant after the SWCNTs-COOH adsorbed and crosslinked to HRP, washed with 0.1% Tween 20, subject to trypsin digestion, and finally removed from the mixture; (c) the supernatant after the SWCNTs-COOH went through the same treatment as (b) except for crosslinking.
3.2 Binding site verification
Even though we obtained reproducible MS results for HRP digestion before and after it being crosslinked to the SWCNT-COOH, one must be very careful in using ion intensities to compare peptide abundances. The ion intensities are strongly affected by sample preparation, ion suppression, deposition on the MALDI plate, etc. Therefore, we searched for other evidences to verify the close location of these peptides towards the SWCNT surface during HRP adsorption. The pI of m/z 2545.4 is 5.5 and that of m/z 2744.6 is 4.2, computed by the tool available on ExPASy, the SIB (Swiss Institute of Bioinformatics) Bioinformatics Resource Portal. They were partially charged positively at pH 6.0, rendering weak electrostatic interaction with the negatively charged SWCNT-COOH. They also contain more than 50% non-polar amino acids, indicating that strong hydrophobic interaction between HRP and the hydrophobic carbon surface of SWCNT-COOH is possible. The consecutive phenylalanine residues on peptide D125-K149 (m/z 2744), F142 and F143, could enhance the binding through π-π stacking; and the Thr and Ser residues on peptide T94-R118 could form H-bonds with the carboxyl groups, both drawing the SWCNT-COOH closer to these peptides. In fact, peptide D125-K149 contains residues (N135, L138, A140, P141, F142, F143, T144 and Q147) that have been predicted by computational modeling to locate within 5 Å distance from SWCNT-COOH.[24]
Moreover, peptide T94-R118 (m/z 2545) has the only tryptophan residue, W117, of the entire HRP molecule.[25] This residue can be utilized to verify the close position of peptide T94-R118 to the SWCNT-COOH surface, because the proximate location to SWCNT may allow SWCNT to quench the intrinsic fluorescence of tryptophan through energy transfer. We thus examined the fluorescence spectra of HRP and apo-HRP with SWCNT-COOH at different concentrations (Figure 2). When the sample was excited at 278 nm, low fluorescence was observed in the native HRP with the maximum emission close to 350 nm (Fig. 2a). Apo-HRP has the heme group removed and eliminates the intermolecular energy transfer between W117 and the heme which are 18 Å away from each other. Therefore, the fluorescent intensity of the apo-HRP increased by 7 times compared to that of the native HRP (Fig. 2b). Nevertheless, the fluorescence intensity of both the native HRP and the apo-HRP gradually decreased with increase concentration of SWCNT-COOH. The SWCNT-COOH itself had almost no interference on the excitation light (Supplementary Data Figure S4). The fluorescence quenching effect can be studied using the Stern-Volmer equation:
| (1) |
in which [Q0] represents the concentration of the quencher, If is the intensity of fluorescence with the quencher, and If0 is the intensity without the quencher, kq is the quencher rate co-efficient, and τ0 is the fluorescence lifetime of molecule without a quencher present. For biopolymer like HRP, τ0 is 10−8 s. Using this value, we calculated If0/If τ0 of both the native HRP and the apo-HRP and plotted them verse concentration of SWCNT-COOH, the quencher (Fig. 2c). The curves displayed nice linear relationship (R2 > 0.95) between If0/If τ0 and SWCNT concentration with kq values for HRP and apo-HRP of 5.51 × 109 and 7.15 × 109, respectively. This result well confirmed the fluorescence of HRP was quenched by SWCNT-COOH because of the close location of the only tryptophan residue W117 to the SWCNT surface.
Figure 2.

Intrinsic fluorescence spectra of 100 μg/mL (a) HRP or (b) apo-HRP at the present of 0, 5, 10, 20, 30, 40 μg/mL SWCNT-COOH in 25 mM phosphate buffer (pH 7.4). (c) Stern-Volmer plots for dependence of fluorescence quenching and SWCNT-COOH concentration.
We mapped these two peptides on the crystal structure of HRP (PDB ID: 1H5H, Figure 3). Interestingly, residues of P139, A140, and P141 on peptide D125-K149 were adjacent to the heme group. Especially, the distance between the carbonyl oxygen of P139 and the Fe atom on the heme was measured to be 7.44 Å by VMD. This carbonyl oxygen is important to the catalytic activity of HRP, because it stabilizes substrate binding during catalytic reactions.[26] N135-A140, P141, F142 and F143 are part of the substrate binding pocket as well as the access channel towards the heme center for substrates.[26–29] From the above discussion and Fig. 3, we also know that peptide T94-R118 is also quite close to the heme group because its W117 residue is only 18 Å from the heme group, a distance short enough for intermolecular energy transfer and significant quench of the fluorescence from tryptophan (Fig. 3). Locating near to both the heme and the substrate binding site, SWCNT-COOH could possibly participate in the ET between HRP and the substrate.
Figure 3.

Crystal structure of HRP (PDB: 1H5H) with the heme center shown in cyan and the two peptides close to the SWCNTs-COOH surface during adsorption displayed in yellow and purple.
3.3 Electron transfer between SWCNT and HRP
Within the catalytic cycle of HRP, hydrogen peroxide first oxidizes the ferric peroxidase to the enzyme intermediate compound I, i.e. an oxoferryl porphyrin π cation radical, which could then accept two consecutive electrons from the reducing substrate and return to the ferric state. With one binding site close to the heme group, the electron from the reducing agent could be accepted by SWCNT-COOH and then transferred to the oxoferryl porphyrin. Owing to the excellent conductivity of SWCNT, electron transfer could be accelerated, resulting in enhanced enzyme activity. Even though the intramolecular electron transfer process is typically completed within a few femtoseconds at the single molecule level and hard to capture by the analytical techniques available to us, the overall redox status of the SWCNT-COOH and the reducing agent could be observed using optical spectroscopy. It has been reported by several research groups that the Vis-NIR absorption spectroscopy is a reliable tool for monitoring electron transfer between redox reagents and SWCNT.[30–34] The SWCNT-COOH sample we used has absorption features at three regions: S11 (1500–1800 nm), S22 (900–1100 nm) and M11 (600–700 nm), that correspond to the energy transfer between electronic states of the semiconducting and metallic tubes.[21] Our previous work indeed proved that the absorption intensity of these regions would increase if the SWCNT were reduced, with the S22 region centered at 1050 nm displaying the most obvious change.[21] Therefore, we sequentially added the HRP-H2O2 mixture and the reducing substrate (H2DCF) to the solution of SWCNT-COOH, and assessed the redox state of the tubes using the absorbance at 1,050 nm. The Vis-NIR spectrum spanned from 400 nm to 1200 nm, and thus generation of the oxidation product of H2DCF, 2,7-dichlorofluorescein (DCF), could also be monitored in the same spectrum at its distinct absorption wavelength of 500 nm (Supplementary Data Figure S5).
The change in the absorbance (Abs) at 1,050 nm over the entire reaction course was summarized in Figure 4. We could see that the Abs at 1,050 nm decreased from 0.42 to 0.36 upon being mixed with HRP and H2O2 (step 1), but increased instantly back to 0.43 when H2DCF was added (step 2). The drop of Abs at 1,050 in step 1 indicates that SWCNT-COOH was first oxidized by HRP, which should be activated by H2O2 and in the form of compound I. The increase of Abs. 1050 upon addition of H2DCF in step 2 could be attributed to the tubes accepting electrons from H2DCF, which also coincided with the continuous Abs increase at 500 nm, i.e. generation of the oxidation product, DCF (Fig. S5). The direct ET between H2DCF and SWCNT-COOH, could be made possible by close attachment of H2DCF on the tube surface. H2DCF has the aromatic ring structure and could interact strongly with SWCNT-COOH through π-π stacking. The adsorption could induce energy transfer between the molecule and SWCNT-COOH, and indeed, the fluorescence of DCF, the oxidation product of H2DCF, was quenched significantly by SWCNT-COOH (Supplementary Data Figure S6). Such quench is difficult to be observed in H2DCF because it barely fluoresces. However, its structure is close enough to DCF, except that DCF is more rigid after the removal of 2 hydrogen atoms, and the quenching result is strong evidence that H2DCF and its oxidation product are adsorbed on the surface of SWCNT-COOH and electrons can be transferred from H2DCF to SWCNT-COOH.
Figure 4.

Absorbance change of 25 μg/mL SWCNT-COOH at 1,050 nm with the addition of 0.6 μg/mL HRP followed by 20 μM H2DCF.
Oxidation of H2DCF by SWCNT-COOH completed in less than 1.5 minutes with Abs at 500 nm barely changing afterwards; and Abs at 1,050 nm decreased to and remained stable at 0.38 when the SWCNT-COOH passed most of the electrons to HRP (Step 3 in Fig. 4). The HRP molecules that were reduced in this step could be the excess HRP compound I not being reduced in step 1; or those returned to the ferric state by SWCNT-COOH in step 1 being oxidized again by the 100 μM H2O2 in solution. More study on how the amounts and molar ratio of HRP and H2O2 affect the three reaction steps shown in Fig. 4 can be carried out in the future to shed more light on this process.
Nevertheless, our results strongly supports that SWCNT-COOH mediated the electron transfer between HRP and the reducing agent. Since such an effect was enabled by the close location of the SWCNT-COOH to the enzyme activity center, it was dependent on the direct attachment between HRP and the SWCNT-COOH. If the SWCNT-COOH was coated by BSA before mixed with HRP, activity enhancement of the SWCNT-HRP system was inhibited, probably because BSA blocked their interaction and effectively prevented the electron transfer (Supplementary Data Figure S7).
3.4 Impact from solution pH
SWCNT-COOH participates in the ET between HRP and the reducing substrate, and its excellent electron conductivity could accelerate the transportation of electrons, which leads to faster catalytic reaction. If this was the case, the enzyme activity enhancement should be closely related to the capability of SWCNT in accepting and conducting electrons. Indeed, the Abs. at 1,050 nm increased with pH, while the degree of the SWCNT-induced HRP activity enhancement decreased with pH (Figure 5 and Supplementary Data Figure S8). At the more basic pH, SWCNT was in a more reduced state with poorer capability to accept electron, as indicated by the higher Abs at 1,050 nm. It has also been reported by Lee and Cui that the conductance of SWCNT-COOH decreases exponentially with the increase of pH due to the hole doping/un-doping effects.[35] Both the more reduced state and the decrease in conductance of SWCNT-COOH could hold up the ET mediated by SWCNT-COOH and thus result in smaller enzyme activity enhancement at basic pH. Under extreme basic condition, SWCNT-COOH even acted as an inhibitor probably by obstructing the substrate approaching the heme group.
Figure 5.

Percent change of 1.2 μg/mL HRP activity by 10 μg/mL SWCNTs-COOH and absorbance of 25 μg/mL SWCNTs-COOH at 1,050 nm under different pHs. Ten μM H2DCF was used as substrate for HRP.
4. Conclusions
Our study reveals that SWCNT-COOH could mediate ET from the reducing substrate to HRP, because it located close to the heme center when HRP was attached (Figure 6). Therefore, by docking the conductive nanomaterial near the enzyme activity center and by choosing appropriate reaction conditions to ensure nanomaterials have good access to and large capacity for electrons, it is possible to maximize the performance of the functional hybrid materials as sensors or in fuel cells. The docking site could be quickly explored by the approach of crosslinking chemistry coupled with MS. With the availability of diverse crosslinking agents, this method could be applied to study the binding interface between diverse nanomaterials and proteins.
Figure 6.
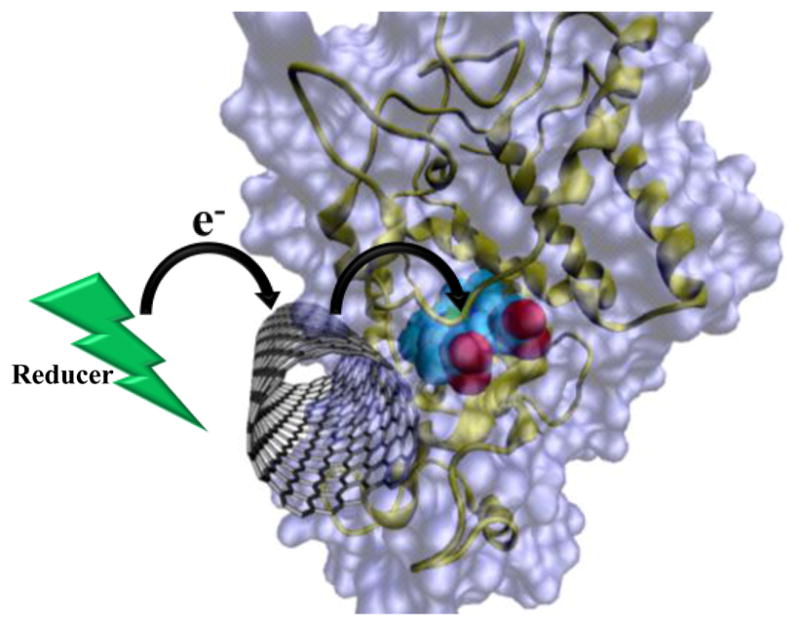
Docking of SWCNT close to the enzyme activity center enables the participation of SWCNT in ET between the reducing substrate and the enzyme, which results in the enhanced enzyme activity.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences Grant No. 1R21ES017870-01A1 to W. Zhong. L. Ren thanks the sponsorship from the Environmental Toxicology Program of UCR.
Appendix A: Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:xx-xxxx:J.carbon.2011.xx.xxx.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang J. Carbon-nanotube based electrochemical biosensors: a review. Electroanal. 2005;17(1):7–14. [Google Scholar]
- 2.Willner B, Katz E, Willner I. Electrical contacting of redox proteins by nanotechnological means. Curr Opin Biotechnol. 2006;17(6):589–96. doi: 10.1016/j.copbio.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Willner I, Baron R, Willner B. Integrated nanoparticle-biomolecule systems for biosensing and bioelectronics. Biosens & Bioelectron. 2007;22(9–10):1841–52. doi: 10.1016/j.bios.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Tsai T, Heckert G, Neves LF, Tan Y, Kao D, Harrison RG, et al. Adsorption of glucose oxidase onto single-walled carbon nanotubes and its application in layer-by-layer biosensors. Anal Chem. 2009;81(19):7917–25. doi: 10.1021/ac900650r. [DOI] [PubMed] [Google Scholar]
- 5.Zhang B, Xing Y, Li Z, Zhou H, Mu Q, Yan B. Functionalized Carbon Nanotubes Specifically Bind to alpha -Chymotrypsin’s Catalytic Site and Regulate Its Enzymatic Function. Nano Lett. 2009;9(6):2280–4. doi: 10.1021/nl900437n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, Jia H, Kim J, Wang P. Kinetic limitations of a bioelectrochemical electrode using carbon nanotube-attached glucose oxidase for biofuel cells. Biotechnol & Bioengineer. 2009;104(6):1068–74. doi: 10.1002/bit.22496. [DOI] [PubMed] [Google Scholar]
- 7.Zhang P, Henthorn DB. Comparison of different functionalization routes for the fabrication of enzyme-single wall carbon nanotube conjugates. J Nanosci & Nanotechnol. 2009;9(8):4747–52. doi: 10.1166/jnn.2009.1274. [DOI] [PubMed] [Google Scholar]
- 8.Baravik I, Tel-Vered R, Ovits O, Willner I. Electrical Contacting of Redox Enzymes by Means of Oligoaniline-Cross-Linked Enzyme/Carbon Nanotube Composites. Langmuir. 2009;25(24):13978–83. doi: 10.1021/la902074w. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Joshi PP, Hobbs KL, Johnson MB, Schmidtke DW. Nanostructured Biosensors Built by Layer-by-Layer Electrostatic Assembly of Enzyme-Coated Single-Walled Carbon Nanotubes and Redox Polymers. Langmuir. 2006;22(23):9776–83. doi: 10.1021/la060857v. [DOI] [PubMed] [Google Scholar]
- 10.Withey GD, Lazareck AD, Tzolov MB, Yin A, Aich P, Yeh JI, et al. Ultra-high redox enzyme signal transduction using highly ordered carbon nanotube array electrodes. Biosens & Bioelectron. 2006;21(8):1560–5. doi: 10.1016/j.bios.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Dumitrescu I, Unwin PR, MacPherson JV. Electrochemistry at carbon nanotubes: perspective and issues. Chem Commun. 2009;(45):6886–901. doi: 10.1039/b909734a. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Jia H, Wang P. Challenges in biocatalysis for enzyme-based biofuel cells. Biotechnol Adv. 2006;24(3):296–308. doi: 10.1016/j.biotechadv.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Kovacic P, Hall ME. Bioelectrochemistry, reactive oxygen species, receptors, and cell signaling: how interrelated? J Receptors & Signal Transduction. 2010;30(1):1–9. doi: 10.3109/10799890903517939. [DOI] [PubMed] [Google Scholar]
- 14.Rich PR, Marechal A. The mitochondrial respiratory chain. Essays in Biochem. 2010;47:1–23. doi: 10.1042/bse0470001. [DOI] [PubMed] [Google Scholar]
- 15.Klingenberg M. Wanderings in bioenergetics and biomembranes. Biochim Biophys Acta, Bioenergetics. 2010;1797(6–7):579–94. doi: 10.1016/j.bbabio.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Lovat V, Pantarotto D, Lagostena L, Cacciari B, Grandolfo M, Righi M, et al. Carbon nanotube substrates boost neuronal electrical signaling. Nano Lett. 2005;5(6):1107–10. doi: 10.1021/nl050637m. [DOI] [PubMed] [Google Scholar]
- 17.Yeh S-R, Chen Y-C, Su H-C, Yew T-R, Kao H-H, Lee Y-T, et al. Interfacing Neurons both Extracellularly and Intracellularly Using Carbon-Nanotube Probes with Long-Term Endurance. Langmuir. 2009;25(13):7718–24. doi: 10.1021/la900264x. [DOI] [PubMed] [Google Scholar]
- 18.Kotov NA, Winter JO, Clements IP, Jan E, Timko BP, Campidelli S, et al. Nanomaterials for Neural Interfaces. Adv Mat. 2009;21(40):3970–4004. [Google Scholar]
- 19.Bayraktar H, You C-C, Rotello VM, Knapp MJ. Facial Control of Nanoparticle Binding to Cytochrome c. J Am Chem Soc. 2007;129(10):2732–3. doi: 10.1021/ja067497i. [DOI] [PubMed] [Google Scholar]
- 20.Carver AM, De M, Bayraktar H, Rana S, Rotello VM, Knapp MJ. Intermolecular Electron-Transfer Catalyzed on Nanoparticle Surfaces. J Am Chem Soc. 2009;131(11):3798–9. doi: 10.1021/ja806064t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren L, Zhong W. Oxidation Reactions Mediated by Single-Walled Carbon Nanotubes in Aqueous Solution. Environ Sci Technol. 2010:377–9. doi: 10.1021/es101821m. [DOI] [PubMed] [Google Scholar]
- 22.Li N, Zeng S, He L, Zhong W. Exploration of Possible Binding Site of Nanoparticles on Protein by Cross-linking Chemistry Coupled with Mass Spectrometry. Anal Chem. 2011;83:6929–34. doi: 10.1021/ac201889j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chattopadhyay K, Mazumdar S. Structural and conformational stability of horseradish peroxidase: effect of temperature and pH. Biochem. 2000;39(1):263–70. doi: 10.1021/bi990729o. [DOI] [PubMed] [Google Scholar]
- 24.Allen BL, Kotchey GP, Chen Y, Yanamala NV, Klein-Seetharaman J, Kagan VE, et al. Mechanistic investigations of horseradish peroxidase-catalyzed degradation of single-walled carbon nanotubes. J Am Chem Soc. 2009;131(47):17194–205. doi: 10.1021/ja9083623. [DOI] [PubMed] [Google Scholar]
- 25.Brunet JE, Gonzalez GA, Sotomayor CP. Intramolecular tryptophan heme energy transfer in horseradish peroxidase. Photochem Photobiol. 1983;38(2):253–4. doi: 10.1111/j.1751-1097.1983.tb03871.x. [DOI] [PubMed] [Google Scholar]
- 26.Henriksen A, Schuller DJ, Meno K, Welinder KG, Smith AT, Gajhede M. Structural interactions between horseradish peroxidase C and the substrate benzhydroxamic acid determined by X-ray crystallography. Biochem. 1998;37(22):8054–60. doi: 10.1021/bi980234j. [DOI] [PubMed] [Google Scholar]
- 27.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat Struct Mol Biol. 1997;4(12):1032–8. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 28.Veitch NC. Horseradish peroxidase: a modern view of a classic enzyme. Phytochem. 2004;65(3):249–59. doi: 10.1016/j.phytochem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 29.Veitch NC, Williams RJ. Two-dimensional 1H-NMR studies of horseradish peroxidase C and its interaction with indole-3-propionic acid. Eur J Biochem. 1990;189(2):351–62. doi: 10.1111/j.1432-1033.1990.tb15496.x. [DOI] [PubMed] [Google Scholar]
- 30.Itkis ME, Perea DE, Niyogi S, Rickard SM, Hamon MA, Hu H, et al. Purity evaluation of as-prepared single-walled carbon nanotube soot by use of solution-phase near-IR spectroscopy. Nano Lett. 2003;3(3):309–14. [Google Scholar]
- 31.Strano MS, Huffman CB, Moore VC, OConnell MJ, Haroz EH, Hubbard J, et al. Reversible, band-gap-selective protonation of single-walled carbon nanotubes in solution. J Phys Chem B. 2003;107(29):6979–85. [Google Scholar]
- 32.Xu Y, Pehrsson PE, Chen L, Zhao W. Controllable redox reaction of chemically purified DNA- single walled carbon nanotube hybrids with hydrogen peroxide. J Am Chem Soc. 2008;130(31):10054–5. doi: 10.1021/ja802743h. [DOI] [PubMed] [Google Scholar]
- 33.Zheng M, Diner BA. Solution redox chemistry of carbon nanotubes. J Am Chem Soc. 2004;126(47):15490. doi: 10.1021/ja0457967. [DOI] [PubMed] [Google Scholar]
- 34.Zheng M, Rostovtsev VV. Photoinduced charge transfer mediated by DNA-wrapped carbon nanotubes. J Am Chem Soc. 2006;128(24):7702–3. doi: 10.1021/ja061464l. [DOI] [PubMed] [Google Scholar]
- 35.Lee D, Cui T. pH-dependent conductance behaviors of layer-by-layer self-assembled carboxylated carbon nanotube multilayer thin-film sensors. J Vac Sci Technol, B: Microelectron Nanometer Struct--Process, Meas, Phenom. 2009;27:842. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.