

Abstract
Expression of BRCA1 is commonly decreased in sporadic breast tumors, and this correlates with poor prognosis of breast cancer patients. Here we show that BRCA1 transcripts are selectively enriched in the Argonaute/miR-182 complex and miR-182 down-regulates BRCA1 expression . Antagonizing miR-182 enhances BRCA1 protein levels and protects them from IR-induced cell death, while overexpressing miR-182 reduces BRCA1 protein, impairs homologous recombination-mediated repair, and render cells hypersensitive to IR. The impaired DNA repair phenotype induced by miR-182 overexpression can be fully rescued by over-expressing miR-182-insensitive BRCA1. Consistent with a BRCA1-deficiency phenotype, miR-182 overexpressing breast tumor cells are hypersensitive to inhibitors of poly (ADP-ribose) polymerase1 (PARP1). Conversely, antagonizing miR-182 enhances BRCA1 levels and induces resistance to PARP1 inhibitor. Finally, a clinical-grade PARP1 inhibitor impacts outgrowth of miR-182 expressing tumors in animal models. Together these results suggest that miR-182-mediated down-regulation of BRCA1 impedes DNA repair, and may impact breast cancer therapy.
INTRODUCTION
Germ-line mutations or deletions in breast cancer susceptibility gene BRCA1 contribute to familial breast tumor formation, but there is limited evidence for direct mutation of the BRCA1 gene in the sporadic form of the disease. However, decreased expression of the BRCA1 gene has been shown to be common (30-65%) in sporadic basal-like breast cancer, and the magnitude of the decrease correlates with disease progression(Mueller and Roskelley, 2003; Thompson et al., 1995; Turner et al., 2004; Turner et al., 2007; Wilson et al., 1999). Because sporadic tumors account for ~90% of the total breast cancer burden, a key question that emerges is how BRCA1 expression is suppressed in these tumors.
DNA methylation, which can be permanent and heritable, is associated with decreased tumor suppressor gene expression in a number of disease contexts (Herman and Baylin, 2000). Although promoter methylation may result in very low levels of BRCA1, aberrant methylation of the BRCA1 promoter is found only in a relatively moderate percentage (10-15%) of sporadic breast tumors (Catteau et al., 1999; Esteller et al., 2000; Matros et al., 2005; Rice et al., 2000) and there is no significant correlation with clinical or pathological parameters of the disease (Matros et al., 2005). Other factors which have been reported to potentially contribute to diminished BRCA1 expression are the transcriptional suppressors ID4 (Turner et al., 2007) and HMGA1(Baldassarre et al., 2003). However it is still unclear how BRCA1 silencing occurs in the majority of sporadic basal-like breast tumors.
From a therapeutic perspective, the expression level of BRCA1 is a major determinant of response to different classes of chemotherapy (Mullan et al., 2006). BRCA1-deficient tumors are hypersensitive to DNA damaging chemotherapeutic agents (such as cisplatin, mitomycin C etc)(Bhattacharyya et al., 2000; Fedier et al., 2003; Moynahan et al., 2001). Based on the principle of ‘synthetic lethality’, BRCA mutation-associated cancers with impaired homologous recombination (HR) mediated repair of DNA double strand break (DSB)s, are being selectively targeted by inhibitors of the DNA repair protein PARP1 (Bryant et al., 2005; Farmer et al., 2005). Conversely, the presence of functional BRCA1 sensitizes tumor cells to antimicrotubule agents (such as vincristine, paclitaxel)(Fedier et al., 2003; Mullan et al., 2001). Therefore, the cellular level of BRCA1 can directly impact malignant transformation and therapeutic response.
Because other DNA repair factors have recently been reported to be regulated by microRNAs,(Crosby et al., 2009; Lal et al., 2009) we hypothesized that specific microRNAs may suppress BRCA1 expression in breast tumors. MicroRNAs (miRNAs) are small (~22 nt) non-coding RNAs that regulate post-transcriptional gene expression by blocking translation of target mRNAs or by accelerating their degradation(Bartel, 2009; Fabian et al., 2010). Although studies addressing their role in cancer pathogenesis are at an early stage, it is apparent that loss- or gain-of-function of specific miRNAs contributes to cellular transformation and tumorigenesis(Chang and Mendell, 2007; Garzon et al., 2009; Ventura and Jacks, 2009). Using the experimental system of in vitro hematopoietic cell differentiation and miRNA expression analysis, we have recently discovered that miRNAs downregulate DSB repair factors and suppress DNA repair in terminally differentiated blood cells (Lal et al., 2009). Our goal was to identify miRNAs targeting BRCA1 and other DSB factors using this same strategy.
RESULTS
Radiation response of microRNA cluster-183 in dividing and post-mitotic cells
In order to identify the differentiation-induced miRNAs that play a role in the DNA damage response, proliferating progenitor K562 cells, and post-mitotic differentiated K562 cells [cells were treated with 12-O-tetradecanoylphorbol-13-acetate (TPA) to produce terminally differentiated megakaryocytes] were exposed to IR and the expression of miRNAs was studied by microarray analysis. We were particularly interested in the IR-response of a subset of miRNAs that have previously been shown to be upregulated in multiple blood lineages(Lal et al., 2009). We hypothesized that in post-mitotic blood cells DNA damage induces apoptosis and miRNAs attenuate the DSB repair machinery promoting cell death. Therefore, the expression of these miRNAs would be indifferent to IR in post-mitotic cells. However, upon irradiation of proliferating progenitor cells, we postulated that these miRNAs are down-modulated to allow increased production of DNA repair proteins and boost the DNA damage response. Following exposure to IR, only 5 miRNAs were down-regulated >2-fold in the proliferating K562 cells but not in the post-mitotic cells (Supplemental Figure 1A). Three of the miRNAs in this set, miR-96, miR-183 and miR-182, are encoded in a ~5-kb gene segment. They are processed from a well conserved polycistronic transcript on human chromosome 7q32.2 (Supplemental Fig. 1B). These miRNAs (termed miRNA cluster-183) have a sensory role (Lewis et al., 2009; Mencia et al., 2009) and are expressed at high levels in mouse retina and sensory hair cells of the ear (Friedman et al., 2009; Jin et al., 2009; Pierce et al., 2008; Xu et al., 2007). miRNA cluster-183 is aberrantly expressed in a variety of tumors (Bandres et al., 2006; Gaur et al., 2007; Hanke et al., 2009; Lowery et al., 2010; Segura et al., 2009; Watkins et al., 2010; Zhang et al., 2006) and is potentially useful for tumor classification (Gaur et al., 2007). We verified the microarray results by qRT-PCR. miR-183, miR-96 and miR-182 were significantly up-regulated during terminal differentiation of HL60 and K562, (Fig. 1A). As expected, there was no significant change in expression of miRNA cluster-183 in differentiated cells exposed to IR (Supplemental Fig. 1C). However there was a sharp decrease in expression of miR-96, miR-183 and miR-182 in undifferentiated HL60 cells (Fig. 1B) within 30 min of IR exposure. Importantly, the expression levels diminished in an IR dosage-dependent manner. The rapid and dramatic change in expression of these miRNAs in response to IR suggests a possible involvement in the DNA damage response.
Figure 1.
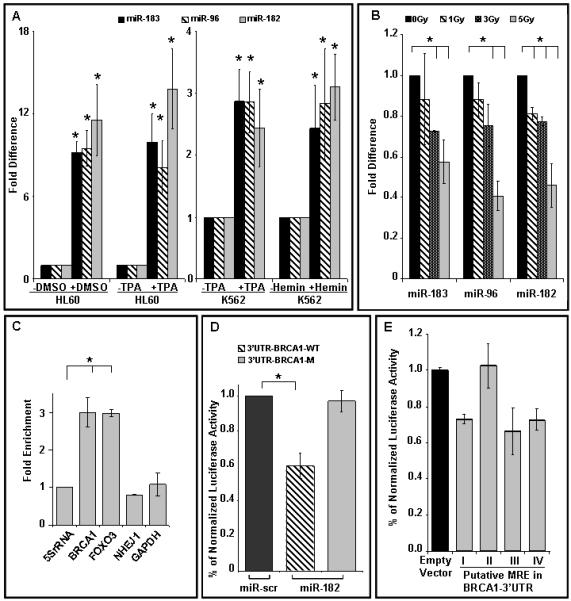
Expression of miRNA cluster-183 is induced during differentiation and suppressed by γ-radiation and BRCA1 is a target of miR-182.
A) Expression of miR-183, miR-96 and miR-182 during differentiation of HL60 cells to neutrophils (DMSO, 8 days, p<0.001) and macrophages (TPA, 3 days, p<0.001); K562 cells to megakaryocytes (TPA, 3 days, p<0.002) and to erythrocytes (Hemin, 4 days, p<0.003).
B) miR-183, miR-96 and miR-182 are rapidly down-regulated with γ-radiation in proliferating cells. HL60 and K562 cells (data not shown) were exposed to indicate doses of γ-radiation, and RNA isolated 30 min after exposure. There was significant reduction in miR-183 (p<0.002), miR-96 (*p<0.003) and miR-182 (*p<0.002) at 3 Gy. In all the above experiments (panel A and B) the expression of miR-183, miR-96, and miR-182 was analyzed with qRT-PCR and normalized to RNU6B, mean ± SD, n=3-6 independent experiments, are shown.
C) Isolation of target transcripts associated with miR-182. Hela cells were co-transfected with expression vectors for HA-tagged AGO1 and miR-182/miR-scr. The immunoprecipitated RNA was analyzed by qRT-PCR using gene-specific primers and normalized to 5S rRNA. FOXO3 and GAPDH served as positive and negative controls, respectively. BRCA1, and not NHEJ1, was significantly (p<0.002) enriched in the pull-down.
D, E) miR-182 targets the 3′UTR of BRCA1 mRNA in a luciferase reporter assay. HeLa cells were co-transfected with BRCA1 3′UTR-luciferase reporter, wildtype (WT, stripped) or mutant (M, grey) (D), or with the four predicted miR-182 MREs in the BRCA1 3′UTR (E) and control mimic (miR-scr, black) or miR-182 mimic (stripped and grey) for 48 h before analysis. Firefly luciferase activity of the reporter was normalized to an internal Renilla luciferase control. Mean ± SD of 3 independent experiments are shown. miR-182 significantly (p<0.001) suppresses lucifererase activity from BRCA1-WT reporter but mutation in the miR-182 recognition sites in BRCA1-M rescues this suppression (p<0.001).
miR-182 targets BRCA1
We had postulated that miRNA cluster-183 was rapidly down-regulated in response to IR in dividing cells to allow increased production of DNA repair factors. In order to identify DNA repair factors targeted by these miRNAs we adopted a computational approach. Several of the available prediction algorithms (such as TargetScan and Pictar) are largely based on evolutionary conservation of target sites across species (Bartel, 2009; Sethupathy et al., 2006). However several critical DSB repair factors (such as MDC1, 53BP1, DNA-PK, BRCA1 and BRCA2) are not found in lower eukaryotes, potentially making these algorithms less effective in identifying DNA repair targets. We used RNA22 which is distinct from other methods in that it obviates the use of a cross-species sequence conservation filter, thus allowing the discovery of miRNA binding sites that may not be present in closely related species (http://cbcsrv.watson.ibm.com/rna22.html) (Miranda et al., 2006). One limitation of this method is that it predicts hundreds or even thousands of potential targets for each miRNA, making it difficult to identify the most important targets. The problem was further compounded by the fact that we had three miRNAs, and there was a cumulative list of targets. We therefore focused on miR-182, which was predicted to target BRCA1.
miR-182 is predicted to target several DSB repair proteins (Supplemental Fig. 1D), which include BRCA1, NHEJ1/XLF etc. However, bioinformatic algorithms have a high margin of error and the majority of predicted genes may not be real targets (Sethupathy et al., 2006). To screen predicted targets we adapted a recently described (Easow et al., 2007; Hendrickson et al., 2008; Karginov et al., 2007) biochemical approach (Fig 1C). miRNAs target their corresponding mRNAs in association with a protein complex that includes the Argonaute proteins, AGO1 and AGO2. This interaction allows for the identification of miRNA–target interactions that occur in vivo. Immunoprecipitation (IP) of a hemagglutinin (HA)-tagged AGO1 can recover miRNA/mRNA complexes. In cells that overexpress a specific miRNA, IP of HA-AGO1 selectively enriches for the overexpressed miRNA and its corresponding target mRNAs. Using this strategy we found that the miR-182/AGO1 complex associates selectively with the BRCA1 transcript (Fig. 1C) at levels comparable with a validated miR-182 target, FOXO3 mRNA (Segura et al., 2009). Levels of BRCA1 mRNA were significantly higher than control transcripts (5S rRNA and GAPDH mRNA) and other predicted targets (NHEJ1 mRNA). The BRCA1 3′UTR is (~1400 nt) long and has four potential miR-182 miRNA recognition elements (MRE) (Supplemental Fig. 1E) and MRE #3 is the most conserved. It is important to note that none of these four sites have perfect complementarity to the ‘seed’ sequence. To verify that BRCA1 is regulated by miR-182, we used the luciferase reporter assay. Luciferase activity was reduced ~2-fold by miR-182 expression in cells transfected with wildtype BRCA1-reporter. Mutation of the predicted MREs completely restored luciferase expression (Fig. 1D). Furthermore we tested the efficacy of each individual MRE and luciferase activity was suppressed to varying degrees by MRE#s 1, 3 and 4 but MRE#2 had no impact on luciferase activity. Together these results suggest that miR-182 targets the BRCA1 transcript directly by interaction with its 3′UTR.
miR-182 mediated suppression of BRCA1 impedes DNA repair
Next, we looked at BRCA1 expression levels after ectopic overexpression of miR-182 and observed a significant decrease in BRCA1 protein (Fig. 2A). Importantly, overexpression of miR-183 and miR-96, which are co-expressed with miR-182, did not affect BRCA1 protein levels. The physiological relevance of the BRCA1/miR-182 interaction was further established by evaluating the endogenous expression pattern of miR-182 and BRCA1. miR-182 is overexpressed in the course of hematopoietic differentiation. If miR-182 is regulating BRCA1 levels, then the prediction would be that BRCA1 levels diminish in parallel with increasing expression of miR-182. We differentiated HL60 cells to granulocytes using DMSO and monitored the expression levels of miR-182 and BRCA1 over 8 days. Consistent with our prediction, we observed a striking inverse correlation of BRCA1 protein levels, with miR-182 expression (Fig. 2B). Together these results suggest that BRCA1 is a physiologically relevant target of miR-182.
Figure 2.
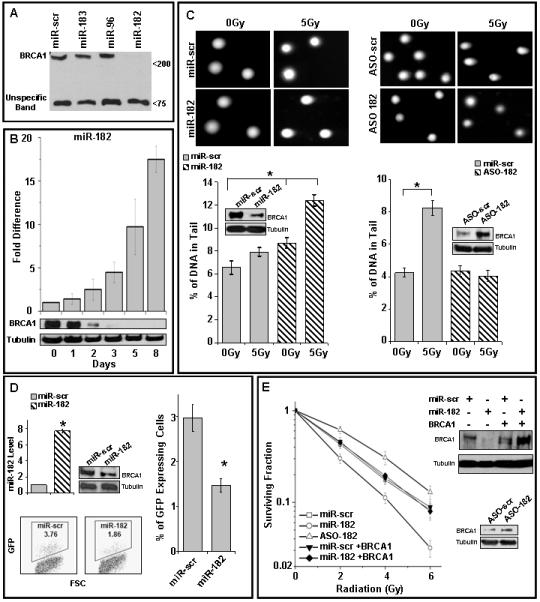
Reduction of BRCA1 protein levels by miR-182 impacts DNA repair.
A) miR-182 suppresses BRCA1 expression. HL60 cells transiently transfected with expression vectors for miR-183, miR-96, miR-182 and control (miR-scr), were harvested after 3 days and cell lysates analyzed by immunoblot after normalization for total protein. The indicated non-specific band served as a visual representation for loading control
B) Kinetics of miR-182 and BRCA1 protein expression in DMSO-treated HL60 cells. miR-182 expression and BRCA1 protein during DMSO-induced differentiation of HL60 cells. miR-182 was quantified by qRT-PCR normalized to RNU6B. Tubulin served as a loading control for the immunoblot.
C) Altering miR-182 levels impacts the amount of unrepaired DSB by comet assay. Proliferating HL60 cells (left panel) were transfected with control or miR-182 mimic and differentiated HL60 cells (right panel) were transfected with control or miR-182 antagomir (ASO). Transfected cells were irradiated, allowed to repair for 18 h and analyzed by single-cell gel electrophoresis (comet assay). BRCA1 protein is compared to tubulin levels in the immunoblots. Representative images are shown in the upper panel and the mean ± SD for each condition below. Residual DNA damage after irradiation is significantly altered in miR-182 mimic (p<0.001) or miR-182 ASO (p<0.001) transfected cells.
D) Overexpression of miR-182 impedes homologous recombination-mediated repair of DSBs. U2OS cells carrying the recombination substrate (DR-GFP) were stably transfected with expression vectors for miR-182 or control. I-SceI expression plasmid was transiently transfected and the GFP positive cells analyzed 48 h later by flow cytometry (FACS). miR-182 expression and representative FACS profiles are shown. HR repair was significantly (p<0.002) impaired (lower panel). Mean ± SD of 3 independent experiments is shown.
E) BRCA1 mediates the DNA damage sensitivity induced by miR-182. Cells were mock transfected; transfected with miR-182 antagomir (ASO); transfected with miR-182 mimic or BRCA1 cDNA lacking the 3′UTR or both. Cell viability was assayed after indicated doses of γ-radiation by clonogenic cell survival assay. Curves were generated from 3 independent experiments. miR-182 mimic significantly (p<0.003) enhanced sensitivity to IR, whereas miR-182 ASO reduced (p<0.001) IR-sensitivity. Representative immunoblots for D and E are shown.
BRCA1 is an integral component of the cellular DNA damage response (Boulton, 2006; Huen et al., 2010). To determine whether miR-182-mediated BRCA1 down-regulation affects DNA repair, we measured the persistence of DSBs after IR, as an indicator of unrepaired damaged DNA, by single-cell gel electrophoresis (neutral comet assay) (Fig. 2C). HL60 cells with ectopic overexpression of miR-182 had lower levels of BRCA1 protein and significantly higher residual DNA damage relative to control cells (Fig. 2C, left panel). Conversely, HL60 cells differentiated with DMSO, and transfected with miR-182 antisense oligonucleotides (termed antagomirs, ASO) had significantly lower amounts of DNA breaks and higher levels of BRCA1 protein (Fig. 2C, right panel).
miR-182 expression impacts HR-mediated repair
DSBs are one of the most deleterious types of damage caused by radiation (Jackson and Bartek, 2009). Two major pathways, HR and non-homologous end joining (NHEJ) have evolved to deal with DSBs and the critical components of these pathways are conserved from yeast to mammals (Shrivastav et al., 2008). BRCA1 is involved in the HR-mediated DSB repair pathway (Moynahan et al., 1999) and recent reports suggest that it may impact the pathway choice (Bouwman et al., 2010; Bunting et al., 2010; Cao et al., 2009). To test whether expression of miR-182 impacts HR repair, we assayed for HR-mediated repair of an I-SceI-induced DSB, in U2OS cells, using a recombination substrate DR-GFP (Nakanishi et al., 2005). Consistent with the role of BRCA1 in HR (Moynahan et al., 1999; San Filippo et al., 2008), cells overexpressing miR-182 had significantly reduced HR-efficiency (Fig. 2D). The cumulative impact of BRCA1-deficiency on cellular DNA damage response is increased sensitivity to IR and other DNA damaging agents (Moynahan et al., 2001; Scully et al., 1999). Manipulating miR-182 levels (via mimics or antagomirs) in HeLa cells impacts sensitivity to IR (Fig. 2E). Importantly, the effect of miR-182 on IR sensitivity was fully rescued by over-expressing miR-182-insensitive BRCA1. Together these results suggest that miR-182 mediated downregulation of BRCA1 impedes the DNA damage response.
miR-182 levels correlate with BRCA1 expression in breast tumor lines
BRCA1 expression affects both breast tumor development and therapy (Mullan et al., 2006; Narod and Foulkes, 2004; Palacios et al., 2008). Based on molecular profiling of tumors, breast cancer types have been divided into those with high expression of the ER gene (luminal) and those that do not express ER (basal) (Perou et al., 2000). ER-negative status is an intrinsic feature of BRCA1-related breast cancer (Atchley et al., 2008; Foulkes et al., 2004). We investigated BRCA1 protein and miR-182 expression in a panel of breast cancer lines derived from ER positive and negative sporadic tumors (Fig. 3A). Consistent with clinical data, the basal-like ER-negative cell lines 21NT, BT549, HS578T and HCC38 had relatively low levels of BRCA1 protein (Neve et al., 2006). Interestingly, in five of the six ER negative cell lines there was inverse correlation of BRCA1 protein and miR-182 expression (Fig. 3B). To test whether the cell cycle profile contributes to this correlation we compared the cell cycle profile of HS578T (no correlation of BRCA1 protein and miR-182) with three other ER-negative cell lines (Fig. 3C). Interestingly, HS578T has a very distinct profile with a mixed diploid and polyploid cell population (Zajac et al., 2008). Finally, in synchronized 21NT cells we examined the relative expression of miR-182 and BRCA1 in the course of the cell cycle (Fig. 3D). As described previously (Gudas et al., 1996; Shrivastav et al., 2008) BRCA1 expression increases with entry into-S-phase. Interestingly, miR-182 is co-expressed with BRCA1 during the cell cycle. This is consistent with a report by Bartel and colleagues (Shkumatava et al., 2009) where they show that miRNAs and their biologically important target transcripts are co-expressed. Together these results suggest that miR-182 may regulate BRCA1 expression during the cell cycle and their correlation may depend on cellular DNA content.
Figure 3.
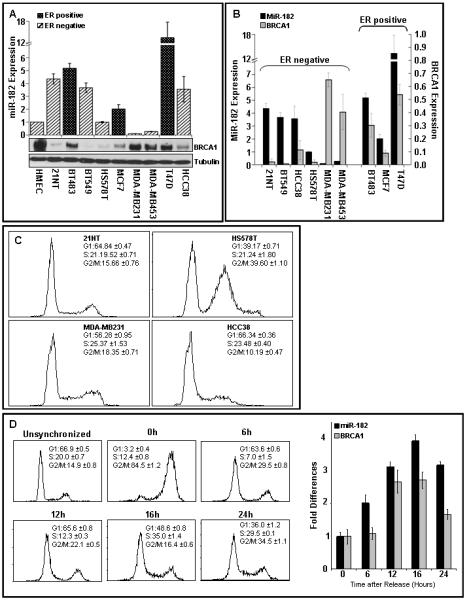
Cell cycle dependent correlation of miR-182 and BRCA1 in breast tumor lines.
A, B) miR-182 and BRCA1 protein in breast tumor cell lines. miR-182 quantified by qRT-PCR (normalized to RNU6) and relative to non-tumorigenic breast epithelial cell, HMEC, is shown on the upper panel. Mean ± SD, n=3 independent experiments are shown. A representative immunoblot for BRCA1 (tubulin as control) from the indicated cells are shown in the lower panel. Relative BRCA1 expression was quantified by densitometry using tubulin as control and normalized to expression in HMEC. The estrogen receptor (ER) expression status of the different tumor lines has been indicated.
C) Cell cycle profile of the indicated ER-negative cell lines. Asynchronously growing cells were fixed, stained with PI and analyzed by flow cytometry. The cell cycle distribution was assessed using FloJo. Representative images from 3 independent experiments are shown.
D) Expression of miR-182 and BRCA1 in synchronized cells. 21NT cells were synchronized using a combination of double thymidine block and nocadozole, and the relative amount of miR-182 and BRCA1 mRNA determined by qRT-PCR (normalized to 5S rRNA). Representative images of the cell cycle profile at indicated times after release are shown on the left panel. Mean ± SD, n=3 independent experiments are shown on the right panel.
BRCA1 is targeted by miR-182 in breast tumor lines
The physiological relevance of the BRCA1/miR-182 interaction was further established by evaluating the endogenous expression pattern of miR-182 and BRCA1 in post-mitotic breast epithelial cells. It has been reported that TPA-treatment of breast cancer lines leads to a post-mitotic state (Cunliffe et al., 2003), we confirmed this result (Supplemental Fig. 2). We treated MCF7 cells with TPA and monitored the expression levels of miR-182 and BRCA1 over 3 days. Consistent with our expectation, we observed a striking inverse correlation of BRCA1 protein levels, with miR-182 expression (Fig. 4A). Like blood cells, upon IR treatment there was rapid and dosage-dependent decrease of miR-182 expression in proliferating MCF7 cells, but not post-mitotic MCF7 cells (Fig. 4B). Relative decrease in miR-182 levels with IR exposure in MCF7 cells is comparable to levels in primary human mammary epithelial cells (HMECs, data not shown). Interestingly the decrease in miR-182 is specific to IR and exposure of MCF7 cells to other DNA damaging agents, UV (single strand breaks/ crosslinks) and camptothecin (replication stress) does not impact miR-182 levels (Fig. 4C).
Figure 4.
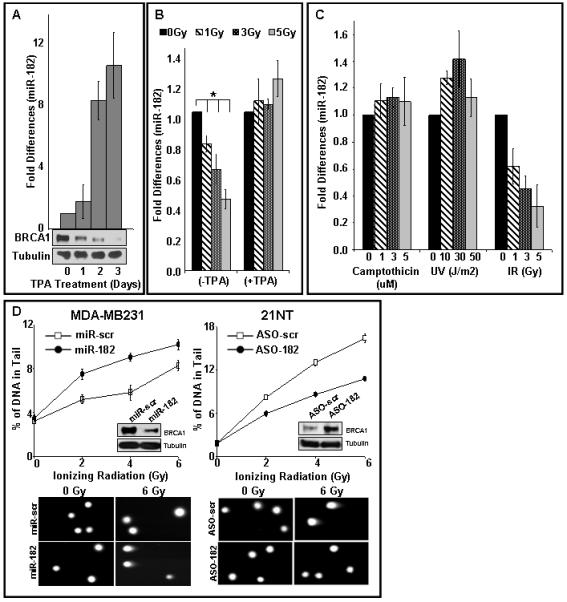
miR-182 targets BRCA1 in breast cancer cell lines and impacts function.
A) Kinetics of miR-182 and BRCA1 protein expression in TPA-treated MCF7 cells. miR-182 expression and BRCA1 protein levels during TPA-induced differentiation of MCF7 cells. miR-182 was quantified by qRT-PCR normalized to RNU6B. Mean ±SD, n=3 independent experiments, p<0.0091 are shown. Tubulin served as a loading control for the immunoblot.
B, C) miR-182 is rapidly down-regulated with IR, and not other DNA damaging agents, in proliferating breast epithelial cells. Proliferating MCF7 cells (B, left panel and C, right panel) and TPA-treated post-mitotic MCF7 cells (B, right panel) were exposed to indicate doses of IR, UV (C, middle panel) and camptothecin (C, left panel) for 1 h. RNA isolated 30 min after exposure. There was significant reduction of miR-182 in MCF7 cells (*p<0.007). The expression of miR-182 was analyzed with qRT-PCR and normalized to RNU6B and in all panels, mean ± SD, n=3-6 independent experiment, are shown.
D) Altering miR-182 levels impacts the amount of unrepaired DSB by comet assay in ER negative tumor cells. MDA-MB231 cells (left panel) were transfected with either control mimic (miR-scr) or 182 mimic (miR-182). Conversely, 21NT cells (right panel) were transfected with antagomirs (ASO), either control (AS0-scr) or ASO-182. Transfected cells were irradiated at indicated doses, allowed to repair for 18 h and analyzed by single-cell gel electrophoresis (comet assay). BRCA1 protein is compared to tubulin levels in the immunoblots. Representative images are shown in the upper panel and the mean ±SD, n=3 independent experiments are shown below. Residual DNA damage after irradiation is significantly altered in 182 mimic (miR-182, p<0.001) or ASO-182 (p<0.001) transfected cells.
miR-182 influences the DNA damage response in breast tumor lines
To determine whether miR-182-mediated BRCA1 down-regulation in breast cancer lines affects DNA repair, we overexpressed miR-182 in MDA-MB231 cells (low endogenous miR-182, high BRCA1 protein) and reduced miR-182 expression in 21NT cells (high endogenous miR-182, low BRCA1 protein). MDA-MB231 cells with ectopic overexpression of miR-182 had lower levels of BRCA1 protein and significantly higher residual DNA damage relative to control cells (Fig. 4D, left panel). Conversely, 21NT cells transfected with miR-182-(ASO) had higher levels of BRCA1 protein and significantly lower amounts of DNA breaks (Fig. 4D, right panel). Together these results suggest that BRCA1 is a physiologically relevant target of miR-182 in breast cancer cells.
To determine the therapeutic impact of miR-182 mediated regulation of BRCA1 in breast cancer, we overexpressed miR-182 in MDA-MB231 cells and reduced miR-182 expression in 21NT cells and assessed their sensitivity to IR. Consistent with the DNA repair assays, MDA-MB231 cells overexpressing miR-182 were significantly more sensitive to different doses of IR (Fig. 5A, left panel). Importantly, the effect of miR-182 on IR sensitivity was fully rescued by over-expressing miR-182-insensitive BRCA1, further confirming that miR-182 impacts the radio-sensitivity of breast tumor cells via BRCA1. Furthermore, antagonizing miR-182 expression in 21NT by miR-182-(ASO) induced significant radio-resistance in these cells (Fig. 5A, right panel). Together these results strongly suggest that miR-182 mediated down-regulation of BRCA1 significantly impacts the radiation response of breast cancer cells.
Figure 5.
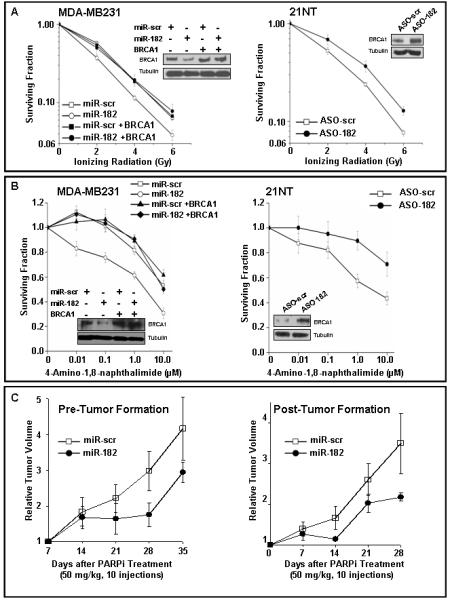
miR-182 mediated regulation of BRCA1 impacts sensitivity to radiation and PARP1 inhibitor in breast cancer cells.
A, B) BRCA1 mediates the radiosensitivity, and sensitivity to PARP1 inhibitor, induced by miR-182 in breast cancer cell lines. MDA-MB231 cells (left panels) were transfected with either control mimic or 182 mimic or BRCA1 cDNA lacking the 3′UTR or both. Conversely, 21NT cells (right panels) were transfected with either control ASO or 182 ASO. Cell viability was assayed by clonogenic cell survival assay after indicated doses of γ-radiation (A) or in the presence of PARP1 inhibitor (ANI) at indicated concentrations (B). Curves were generated from 3 independent experiments. miR-182 mimic significantly enhanced sensitivity to IR (p<0.004) and to ANI (p<0.001), whereas miR-182 ASO reduced sensitivity to IR (p<0.001) and ANI (p<0.002). Representative immunoblots for A and B are shown.
C) miR-182 impacts PARP inhibitor sensitivity in xenograft mouse models. MDA-MB231 cells stably expressing miR-scr or miR-182 were implanted in each thigh of nude mice (n=5) and olaparib treatment was initiated either 2 days post-implantation (left panel) or tumors outgrown (4-7 weeks) were treated (right panel). Tumor volume was determined in 7 days intervals, and the median fold differences after olaparib treatment represented graphically.
miR-182 impacts PARP inhibitor sensitivity
HR-deficiency of BRCA-mutation associated breast tumors selectively sensitizes them to PARP1 inhibitors(Bryant et al., 2005; Farmer et al., 2005) which is a working therapeutic strategy to eliminate these tumors (Fong et al., 2009; Tutt et al., 2010). Since miR-182 influences HR-mediated repair (Fig. 2D) we speculated that miR-182 expression will impact cellular sensitivity to PARP1 inhibitors. Overexpressing miR-182, diminished BRCA1 levels and sensitized MDA-MB231 cells to PARP1 inhibition (Fig. 5B, left panel) using the PARP1 inhibitors, 4-Amino-1, 8-naphthalimide (ANI) and ABT-888 (data not shown). This effect is reverted by expressing miR-182 insensitive BRCA1 transcripts. Conversely, reducing miR-182 enhanced BRCA1 expression and induced resistance to ANI (Fig. 5B, right panel) and ABT-888 (data not shown) in 21NT cells.Next we tested if the clinical PARP inhibitor olaparib can retard outgrowth of miR-182 expressing tumors. We treated mice 2 days after injection or those bearing xenografts, derived from MDA-MB-231 cells either stably expressing miR-182 or scramble control (Supplemental Fig. 3), with olaparib or vehicle control for 10 days. As anticipated, a 10 day treatment with olaparib retarded tumor outgrowth in animals treated both pre- or post tumor formation (Fig 5C). In untreated animals the tumors grew at a comparable rate in the presence or absence of miR-182 (Supplemental Fig. 4). We harvested the tumors and confirmed the expression of miR-182 (data not shown). Altogether, these results convincingly demonstrate that miR-182 is a mediator of the cellular response to PARP inhibitors. Importantly, the observation that the DNA repair deficient phenotype induced by miR-182 was largely rescued by miR-182 resistant BRCA1 transcripts, suggests that the key target of miR-182 in DNA repair is BRCA1.
DISCUSSION
We have previously used the experimental system of in vitro hematopoietic cell differentiation to find miRNAs involved in DSB repair (Lal et al., 2009), and the same strategy was applied to identify miR-182. A rapid, IR dosage-dependent decrease in miR-182 expression further implicated a role in the DNA damage response. This IR-induced change in miR-182 is independent of p53, as it occurs in both p53-proficient (MCF7, HMEC) as well as p53-deficient (K562, HL60) cells. Using a combination of computational, and biochemical, methods we found that BRCA1 was targeted by miR-182. It is feasible that other factors in the DNA damage response are affected by miR-182 expression. However, the observation that the DNA repair deficient phenotype induced by miR-182 was largely rescued by miR-182 resistant BRCA1 transcripts, suggests that the key target of miR-182 in DNA repair is BRCA1.
Decreased expression of the BRCA1 has been shown to be common in sporadic basal-like breast cancer (Mueller and Roskelley, 2003; Turner et al., 2004). Although the magnitude of the decrease correlates with disease progression, the molecular mechanism of BRCA1 suppression in sporadic tumors is unclear. We anticipate that overexpression of miRNAs such as miR-182, may play a role in BRCA1 downregulation in sporadic breast tumors. Manipulation of miR-182 expression in multiple breast tumor lines impacts BRCA1 levels and sensitivity to PARP1 inhibition, both in cultured cells or in xenograft models. This observation has potential clinical relevance. Although familial breast cancer patients with BRCA-mutations are currently being treated with PARP inhibitors, majority of patients with breast cancer (~90%) have the sporadic form of the disease and do not have BRCA mutations. It is currently unclear if patients with sporadic breast cancer benefit from the therapeutic approach with PARP inhibitors. Our results suggest that sporadic tumors overexpressing miRNAs which target BRCA proteins (such as miR-182) may also be susceptible to PARP inhibition or other strategies based on synthetic lethality with BRCA. Future studies will determine whether expression levels of miR-182 and possibly other miRNAs that regulate BRCA1 can serve as determinants of therapeutic strategy, and of clinical outcome for patients with breast cancer.
EXPERIMENTAL PROCEDURES
Cell culture and differentiation
HL60 cells and K562 cells were grown and differentiated into different lineages as previously described(Lal et al., 2009). MCF-7 cells (106 cells/6 cm dish) were differentiated with 100 nM TPA for 3 days in DMEM with 10% (v/v) FBS (Cunliffe et al., 2003). The breast cancer lines and human mammary epithelial cells used in Fig. 3, were cultured in media according to protocols from ATCC (http://www.atcc.org/).
miRNA microarray, RNA Isolation and Quantitative PCR
Undifferentiated and differentiated K562 cells were untreated or exposed to 2 Gy of γ-radiation. Total RNA was extracted after 2 h with Trizol (Invitrogen) according to manufacturer’s manual and treated with 10 U DNase I for 30 min at 37°C in 50 μl. RNAse-free water) was added and DNase I removed with phenol-choloroform (Ambion). The TaqMan Human MicroRNA Array v 1.0 (Early Access) platform was used for quantitative miRNA analysis and was carried out in Dana-Farber Cancer Institute core facility. In subsequent experiments TaqMan MicroRNA Assay from Applied Biosystems was used as per the manufacturer’s instructions. mRNA expression was anlayzed by qPCR using SYBR Green master mix (Applied Biosystems) according to the manufacture’s manual and the BioRad iCycler. Gene specific primers:
BRCA1 F: CAACATGCCCACAGATCAAC
R: ATGGAAGCCATTGTCCTCTG
NHEJ1 F: AGTGCCAAGTGAGGGAGCTA
R: CCACTTGGACCTCTTGTGT
FOXO3 F: GATAAGGGCGACAGCAACAG
R: CCAGTTCCCTCATTCTGGAC
5S rRNA F: GCC CGA TCT CGT CTG ATC T
R: AGC CTA CAG CAC CCG GTATT
GAPDH F: TGCACCACCAACTGCTTAGC
R: GGCATGGACTGTGGTCATGAG.
FNDC3A F: CTTGGAGCTGGTCCTTTCAG
R: CCTTCCCCAGCTTCATTACAAlgorithm tools to predict targets of miR-183 cluster
We applied RNA22 algorithm (http://cbcsrv.watson.ibm.com/rna22_targets.html) to find targets of miR-182. 3′UTR of BRCA1 was further analyzed by RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) and PITA algorithm http://genie.weizmann.ac.il/pubs/mir07 /mir07_prediction.html).
Immunoprecipitation of miR-182 targets
miR-182 was cloned from HL60 cDNA in pcDNA3.1-Puro expression vector (Invitrogen) using the following primers,
miR-182:
F: CGGCGGCCGCGATATGAGGGGAAGGGAGGA
R: CGGCGGCCGCGAGAAGGTTCACCACCCAGA
Cells were co-tranfected with HA-AGO1 (Addgene) and miR-182 or miR-scr (expression vectors). After 2 days cells were harvested in 400 μl lyses buffer (100 mM KCl, 5 mM MgCl2, 10 mM Hepes, pH 7.0, 0.5% Nonidet P-40) containing freshly added RNaseOUT (Invitrogen) and Protease Inhibitor Cocktail (Roche). After centrifugation, a 50 μl aliquot was taken as input for subsequent RNA extraction. The remaining supernatant was gently shaken with HA-beads (HA-probe Santa Cruz sc-7392) for 4 h at 4° C in spin columns (Pierce Spin Columns-Screw Cap). The columns were drained, washed and the retained beads were treated with 5 U DNaseI in NT2 buffer (50 mM Tris, pH7.4, 150 mM NaCl, 1 mM MgCl2, 0.05 % Nonidet P-40) for 10 min at 37°C, washed, treated with Proteinase K. Finally the beads were re-suspended and RNA extracted using acid phenol-choloroform (Ambion). The analysis was done as follows:
Scrambled Control: Pull-Down / Input (say =A)
miR-182: Pull-Down / Input (say =B) . Fold Enrichment= B/A.
Luciferase assay
The wild type (WT) and mutated (M) miR-182 recognition elements of 3′UTR-BRCA1 were annealed (sequence see below) and cloned in pMIR-REPORT (Ambion) downstream to Firefly Luciferase. The luciferase assays were done in HeLa cells as described previously (Lal et al., 2009).
WT F:
CTAGTAGAAGAGATTTCTAAAAGTCTGAGATATATTTGCTAGATTTCTAAAGAATGTGTTCTAAAACAGCAGAAGATTTTCAAGAACCGGTTTCCAAAGACAG
WT R:
AGCTCTGTCTTTGGAAACCGGTTCTTGAAAATCTTCTGCTGTTTTAGAACACATTCTTTAGAAATCTAGCAAATATATCTCAGACTTTTAGAAATCTCTT
M F:
CTAGAAGAGACGATACCCGTCTGAGATATATTTGCTAGGCGATACCCGGGTGTGTTCTAAAACAGCAGAAGCCGATACCCGGCCGGCGATACCCGACAG
M R:
AGCTCTGTCGGGTATCGCCGGCCGGGTATCGCTTCTGCTGTTTTAGAACACACCCGGGTATCGCCTAGCAAATATATCTCAGACGGGTATCGCTCTCTTCTAG
The mutant residues have been indicated.
Primers for the individual sites:
Site I
F: CTAGGGAAAATGAAACTAGAAGAGATTTCTAAAAGTCT
R: AGCTAGACTTTTAGAAATCTCTTCTAGTTTCATTTTCC
Site II
F: CTAGAAAGTCTGAGATATATTTGCTAGATTTCTAAAGAATG
R: AGCTCATTCTTTAGAAATCTAGCAAATATATCTCAGACTTT
Site III
F: CTAGAGAATGTGTTCTAAAACAGCAGAAGATTTTCAAGAACCG
R: AGCTCGGTTCTTGAAAATCTTCTGCTGTTTTAGAACACATTCT
Site IV
F: CTAGAAAACAGCAGAAGATTTTCAAGAACCGGTTTCCAAAGACAG
R: AGCTCTGTCTTTGGAAACCGGTTCTTGAAAATCTTCTGCTGTTTT
Immunoblots
The immunoblots were done as described previously (Lal et al., 2009; Lee et al.) with BRCA1 antibodies 1:500 dilution (CALBIOCHEM, MS110) and Tubulin antibodies 1:10000 dilution (Sigma, Clone B-5-1-2).
Single-cell gel electrophoresis (comet) assay
21NT cells were transfected with 100 nM, control or miR-182 antagomir (Ambion) whereas MDA-MB231 cells were transfected with 10 nM, control or miR-182 mimic (Ambion). After 3 days, cells were irradiated and allowed to recover for 18 h prior to analysis. The single cell comet assays (Trevigen) was carried out as previously described (Lal et al., 2009; Lee et al.).
Homologous Recombination Reporter Assay
HR assay was done as described (Chowdhury et al., 2008; Lee et al., 2010). Briefly, 2×105 cells plated overnight in 24-well plates were transfected with 0.8 μg of I-SceI expression plasmid (pCBA Sce) using Lipofectamine 2000. 2 days later, GFP positive cells were assayed by FACScan.
Clonogenic assay
Hela cells (0.35 × 106 cells/well), MDA-MB231 cells ( 0.4 × 106 cells/well), 21NT cells ( 0.5 ×106) were seeded overnight and transfected with 10 nM of miRNA mimics or antagomirs (Ambion). In rescue experiments miR-182 or control mimics were co-transfected with 1 μg/ml BRCA1 (pcDNA3.1 vector). After 2 days, 1000 cells in 4 ml DMEM media (10% FBS v/v) were seeded on 6 cm dishes in four replicates and incubated overnight before treatment. PARP inhibitors (4-amino-1,8-naphthalimide (Sigma) or ABT-888 or olaparib [(ChemieTek) in DMSO] were added to the growth media at indicated concentrations . Irradiated cells as well as cells in the presence of PARP inhibitor were allowed to form colonies for 14 days. For evaluation, formed colonies were stained with crystal violet and surviving colonies containing >50 cells were counted. Plating efficiency was 30–50%.
Cell Cycle Synchronization
21NT cells were seeded on 6 well plate (2.5 ×105/well) in DMEM 10% FBS media and treated twice with 2 mM thymidine for 16 h with a 9 h interval between treatments. Subsequently the cell were released into media containing 100 ng/ml Nocadozole and incubated for 12 h. The cells were washed and released into DMEM 20% FBS media and were collected for FACS analysis and RNA extraction after indicated time intervals. For FACS the cell were fixed in 70% ethanol, washed twice with PBS buffer and analyzed in PI/RNase staining buffer (BD Pharmingen).
Xenograft Experiments
MDA-MB231 cells (2.5 X 106/injection) stably expressing miR-scr or miR-182 from pcDNA3.1(+)Neo were injected subcutaneously into CD1 nude mice. Olaparib (50 mg/kg, obtained in collaboration with AstraZeneca) or vehicle (PBS+10% DMSO+ 10% HPCB) treatment was initiated either at 2 days post injection or at detectable tumor volume (~70 mm3) between 4-7 weeks later. Each animal obtained 10 intra-peritoneal injections on 10 consecutive days. Measurement of tumor volume by calipers was commenced 7 days post the subcutaneous injection and continued in 7 days intervals.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by R01CA142698 (NCI), JCRT and a Barr Award (DC), the Medical Research Council (TH) and by the NIHR Biomedical Research Centre, Oxford (ALH, RAS). MG and KA were supported by the NIA-IRP, NIH, and RAS is supported by the Higher Education Funding Council for England. We thank members of the Harris, Helleday and Chowdhury laboratories for useful discussions.
REFERENCES
- Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN, Arun BK. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol. 2008;26:4282–4288. doi: 10.1200/JCO.2008.16.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, Fedele M, Pierantoni G, Croce CM, Fusco A. Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol. 2003;23:2225–2238. doi: 10.1128/MCB.23.7.2225-2238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzo M, Garcia-Foncillas J. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer. 2006;5:29. doi: 10.1186/1476-4598-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- Boulton SJ. Cellular functions of the BRCA tumour-suppressor proteins. Biochem Soc Trans. 2006;34:633–645. doi: 10.1042/BST0340633. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–1965. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- Chang TC, Mendell JT. microRNAs in vertebrate physiology and human disease. Annu Rev Genomics Hum Genet. 2007;8:215–239. doi: 10.1146/annurev.genom.8.080706.092351. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA Regulation of DNA Repair Gene Expression in Hypoxic Stress. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunliffe HE, Ringner M, Bilke S, Walker RL, Cheung JM, Chen Y, Meltzer PS. The gene expression response of breast cancer to growth regulators: patterns and correlation with tumor expression profiles. Cancer Res. 2003;63:7158–7166. [PubMed] [Google Scholar]
- Easow G, Teleman AA, Cohen SM. Isolation of microRNA targets by miRNP immunopurification. Rna. 2007;13:1198–1204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fedier A, Steiner RA, Schwarz VA, Lenherr L, Haller U, Fink D. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53-deficient cells. Int J Oncol. 2003;22:1169–1173. [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT, Ghadirian P, Tung N, Olopade OI, Weber BL, McLennan J, et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 2004;10:2029–2034. doi: 10.1158/1078-0432.ccr-03-1061. [DOI] [PubMed] [Google Scholar]
- Friedman LM, Dror AA, Mor E, Tenne T, Toren G, Satoh T, Biesemeier DJ, Shomron N, Fekete DM, Hornstein E, Avraham KB. MicroRNAs are essential for development and function of inner ear hair cells in vertebrates. Proc Natl Acad Sci U S A. 2009;106:7915–7920. doi: 10.1073/pnas.0812446106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- Gudas JM, Li T, Nguyen H, Jensen D, Rauscher FJ, 3rd, Cowan KH. Cell cycle regulation of BRCA1 messenger RNA in human breast epithelial cells. Cell Growth Differ. 1996;7:717–723. [PubMed] [Google Scholar]
- Hanke M, Hoefig K, Merz H, Feller AC, Kausch I, Jocham D, Warnecke JM, Sczakiel G. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol. 2009 doi: 10.1016/j.urolonc.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, Herschlag D, Ferrell JE, Brown PO. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS ONE. 2008;3:e2126. doi: 10.1371/journal.pone.0002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Baylin SB. Promoter-region hypermethylation and gene silencing in human cancer. Curr Top Microbiol Immunol. 2000;249:35–54. doi: 10.1007/978-3-642-59696-4_3. [DOI] [PubMed] [Google Scholar]
- Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZB, Hirokawa G, Gui L, Takahashi R, Osakada F, Hiura Y, Takahashi M, Yasuhara O, Iwai N. Targeted deletion of miR-182, an abundant retinal microRNA. Mol Vis. 2009;15:523–533. [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Conaco C, Xuan Z, Schmidt BH, Parker JS, Mandel G, Hannon GJ. A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci U S A. 2007;104:19291–19296. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Pan Y, Navarro F, Dykxhoorn DM, Moreau L, Meire E, Bentwich Z, Lieberman J, Chowdhury D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol. 2009;16:492–498. doi: 10.1038/nsmb.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Pan Y, Kanner S, Sung P, Borowiec JA, Chowdhury D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat Struct Mol Biol. 2010;17:365–372. doi: 10.1038/nsmb.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MA, Quint E, Glazier AM, Fuchs H, De Angelis MH, Langford C, van Dongen S, Abreu-Goodger C, Piipari M, Redshaw N, et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet. 2009;41:614–618. doi: 10.1038/ng.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery AJ, Miller N, Dwyer RM, Kerin MJ. Dysregulated miR-183 inhibits migration in breast cancer cells. BMC Cancer. 10:502. doi: 10.1186/1471-2407-10-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matros E, Wang ZC, Lodeiro G, Miron A, Iglehart JD, Richardson AL. BRCA1 promoter methylation in sporadic breast tumors: relationship to gene expression profiles. Breast Cancer Res Treat. 2005;91:179–186. doi: 10.1007/s10549-004-7603-8. [DOI] [PubMed] [Google Scholar]
- Mencia A, Modamio-Hoybjor S, Redshaw N, Morin M, Mayo-Merino F, Olavarrieta L, Aguirre LA, del Castillo I, Steel KP, Dalmay T, et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41:609–613. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- Mueller CR, Roskelley CD. Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. 2003;5:45–52. doi: 10.1186/bcr557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullan PB, Gorski JJ, Harkin DP. BRCA1--a good predictive marker of drug sensitivity in breast cancer treatment? Biochim Biophys Acta. 2006;1766:205–216. doi: 10.1016/j.bbcan.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Mullan PB, Quinn JE, Gilmore PM, McWilliams S, Andrews H, Gervin C, McCabe N, McKenna S, White P, Song YH, et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene. 2001;20:6123–6131. doi: 10.1038/sj.onc.1204712. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios J, Robles-Frias MJ, Castilla MA, Lopez-Garcia MA, Benitez J. The molecular pathology of hereditary breast cancer. Pathobiology. 2008;75:85–94. doi: 10.1159/000123846. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Pierce ML, Weston MD, Fritzsch B, Gabel HW, Ruvkun G, Soukup GA. MicroRNA-183 family conservation and ciliated neurosensory organ expression. Evol Dev. 2008;10:106–113. doi: 10.1111/j.1525-142X.2007.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis. 2000;21:1761–1765. doi: 10.1093/carcin/21.9.1761. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, Zakrzewski J, Blochin E, Rose A, Bogunovic D, et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009;106:1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3:881–886. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- Shkumatava A, Stark A, Sive H, Bartel DP. Coherent but overlapping expression of microRNAs and their targets during vertebrate development. Genes Dev. 2009;23:466–481. doi: 10.1101/gad.1745709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, Savage K, Gillett CE, Schmitt FC, Ashworth A, Tutt AN. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–2132. doi: 10.1038/sj.onc.1210014. [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins AJ, Hamoudi R, Liu H, Zhang J, de Leval L, Isaacson P, Wotherspoon A, Du MQ. Characterisation of 7q32 deletion of splenic marginal zone lymphoma demonstrates frequent miR-182 somatic mutations in a wide range of lymphoma subtypes. Haematologica. [DOI] [PMC free article] [PubMed] [Retracted]
- Wilson CA, Ramos L, Villasenor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet. 1999;21:236–240. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]
- Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007;282:25053–25066. doi: 10.1074/jbc.M700501200. [DOI] [PubMed] [Google Scholar]
- Zajac M, Moneo MV, Carnero A, Benitez J, Martinez-Delgado B. Mitotic catastrophe cell death induced by heat shock protein 90 inhibitor in BRCA1-deficient breast cancer cell lines. Mol Cancer Ther. 2008;7:2358–2366. doi: 10.1158/1535-7163.MCT-08-0327. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.