

Abstract
Background & objectives:
Cardiac malformations in the young constitute a major portion of clinically significant birth defects. Congenital heart disease (CHD) is a common congenital cardiac birth defect, affecting nearly 1 per cent of all live births. Patent ductus arteriosus (PDA) is clinically significant foetal circulation anomaly, second most common form of CHD which constitutes approximately 10 per cent of total CHDs. The study aimed to screen for TFAP2B mutations in CHD patients of Mysore.
Methods:
With informed consent, 100 clinically diagnosed CHD patients and 50 healthy controls in Mysore, south India, were recruited for the analysis of screening of mutations. MassARRAY analysis of 5 prominent mutations of TFAP2B was performed.
Results:
The analysis did not show any of the five mutations of TFAP2B screened by massARRAY in patients and controls, indicating that these mutations were not involved in the manifestation of CHD in the patients at Mysore, south India.
Interpretation & Conclusions:
The findings suggest the lack of involvement of known mutations of TFAP2B with syndromic or nonsyndromic CHDs in Mysore patients.
Keywords: Congenital heart disease, massARRAY – mutations, patent ductus arteriosus, TFAP2B
Congenital heart diseases (CHDs) occurring in 1 per cent of all live births are the most common birth defect and the leading non-infectious cause of mortality in newborns, and account for 25 per cent of all human congenital abnormalities1. Only a minority of CHD is caused by single-gene defects and follows a clear Mendelian inheritance, whereas most malformations are thought to be multigenetic disorders. The available data suggest that foetal heart development is regulated by a group of highly conserved transcription factors, including NKX2.5, TBX5, GATA4, TBX1, CRELD1, ZIC3, TFAP2B, FOG2, and others2.
Patent ductus arteriosus (PDA) is clinically significant foetal circulation anomaly, second most common form of CHD which constitutes approximately 10 per cent of total CHDs. The prevalence of isolated PDA is 1 in 2000 full-term infants3. PDA can be an isolated abnormality or associated with other congenital anomalies or may be part of Char syndrome. It also occurs in most cases randomly with other septal defects. Transcription Factor AP 2 B (TFAP2B) is mapped to chromosome 6p12-p21, a Char syndrome locus4. It is a member of AP-2 family of transcription factors5. Mutations in the gene encoding the TFAP2B, a neural-crest related transcription factor can cause Char syndrome with cardiac, craniofacial, and hand abnormalities. TFAP2 gene family has its role in cardiac morphogenesis, particularly in the outflow tract formation and cardiac septation, by controlling cell proliferation and terminal differentiation5–7. To date, nine mutations associated with CHD, (PDA associated with Char syndrome) have been identified in the TFAP2B (Table I)4,8–10. Studies also suggest that TFAP2B can be a candidate gene for isolated nonsyndromic PDA, an outflow tract defect11. However, there are no studies on such aspects of different forms of CHD and PDA from India. In this study we aimed to identify the known mutations of TFAP2B in the Indian CHD samples in Mysore, south India. We hypothesized that TFAP2B is an essential and conserved gene and the mutations already known would also be present in any of the other cases. Thus, the analysis of mutations of TFAP2B in isolated PDA cases and also in varied forms of CHD was made.
Table I.
Summary of previously reported TFAP2B mutation
Material & Methods
The present investigation was carried out in Mysore, a city in southern India, south-west of Bangalore. The present study was conducted from February 2007 to December 2010 in two major hospitals, K. R. Hospital, and J. S. S. Hospital at Mysore, as and when the cases were registered and were available in the hospitals with ethical consent. These hospitals together receive the majority of children as patients from in and around Mysore due to the nominal fee charged towards the diagnosis and treatment. A detailed family history, for CHD or any congenital malformation, physical examination, chest X-ray and electrocardiogram and Doppler study were considered for recruiting the cases for the study.
A total of 100 clinically diagnosed CHD patients from these hospitals were selected based on the severity of the conditions, viz., patients having a defect of >7 mm were included and also the preterm PDAs and minor defects less than 4 mm were excluded for the present study. The study was approved by the ethical committee of University of Mysore. Informed written consent was obtained from all the patients’ family and controls. Those cases with preterm PDA and minor defects less than 4 mm were excluded from the study as these are not considered as severe forms as these would close with time as the child grows older. The age range was 1 day to 12 yr with 44 females and 66 males. There were Hindus (82%), Muslims (16%) and Christians (2%). The 50 randomly selected controls did not have any clinical history of CHD or other congenital malformations in the family and belonged to the same geographical region. Care was also taken to maintain similarity of ethnic and socio-economic backgrounds of the cases and control groups. The controls were not related to patients in any ways.
Peripheral venous blood samples (2 ml) were collected in EDTA tubes and stored in -80°C until use. Genomic DNA was extracted from peripheral leukocytes by using Wizard genomic DNA purification kit (Promega, USA) and was checked for purity. MassARRAY analysis was carried out by using Sequenom-iPLEXR Gold SNP genotyping platform with Spectro CHIPR and MALDI-TOF Mass spectrometer, USA. The technique, massARRAY is one of the most promising mutation analysis tools to analyze mutations in disease-causing genes involving the MALDI TOF. The introduction of matrix-assisted laser desorption/ionization (MALDI) time-of-fight (TOF) mass spectrometry (MS) has resulted in the direct and rapid identification of intact biomolecules12–14. The primers/probes employed in the mass spectrometry analysis are given in Table II.
Table II.
Sequences and molecular weights of unextended (UEP) and extended (EP) TFAP2B primer

Mutations were genotyped using the Sequenom® MassARRAY technology (Sequenom®, San Diego, CA, USA) with the help of Vimta Laboratories, Hyderabad15. The iPLEX™ assay was followed according to manufacturers instructions using 5 ng of genomic DNA. In brief, Massarray analysis was done as follows: The fragments of 300-400 nucleotides including the mutant site of the samples and the control DNA were amplified with the primers that were designed by MassARRAY® Designer software, USA. The amplified products were used as templates for the extension of an oligonucleotide probe over a mutation site and terminated using dideoxynucleotides, to produce different size products for each allele of a mutation. The extended products were analyzed by SEQUENOM MALDI-TOF mass spectrometry, and the time-of-flight is proportional to mass, permitting precise determination of the size of products generated, which was converted into genotype information.
Results & Discussion
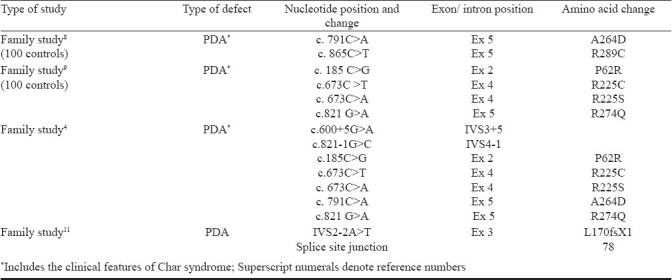
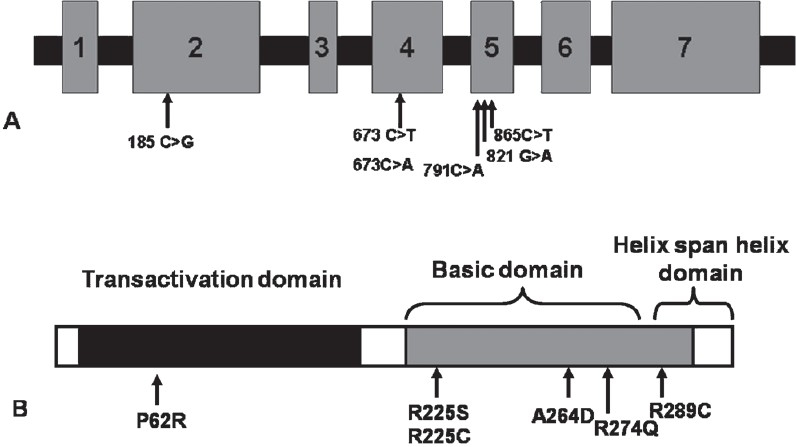
TFAP2B is a 28.8 kb gene consisting of seven exons, having a transcript size of 5770 bps and coding for 460 amino acids (Fig.). In the present investigation, five mutations of TFAP2B c.185C>G, c.673C>T, c.673 C>A, c.791C>A, c.821 G>A, c.865C>T were screened Table IIIin 100 CHD cases and 50 controls, which included 18 PDA cases, 56 ventricular septal defect (VSD), 21 atrial septal defect (ASD), 3 atrioventricular septal defect (AVSD), 10 ASD with VSD, 4 tetralogy of fallot (TOF), and 7 complex CHDs Table IV. Four of the eighteen patients with PDA showed the presence of facial dysmorphisms Table V. To date, there are nine mutations identified in TFAP2B associated with PDA, and only six of which are associated with Char syndrome-PDA Table I. The location of these mutations is presented in the Fig.. The analysis did not show any of the mutations of TFAP2B screened by massARRAY (Table III) indicating that these mutations were not present in CHD patients investigated.
Fig. (A).
Gene structure of TFAP2Bindicating the mutations involved in CHD. Numbers 1-7 show seven exons. (B): Transcription factor indicating the domains of TFAP2Band the change in amino acid involved in CHD. [Source: Ref.4].
Table III.
List of mutations of TFAP2B analyzed by massARRAY in the present study
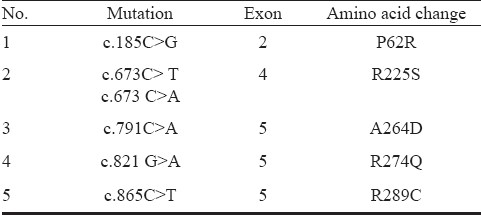
Table IV.
Mutation screening by massARRAY analysis of TFAP2B of 100 congenital heart disease patients and 50 controls in Mysore, south India
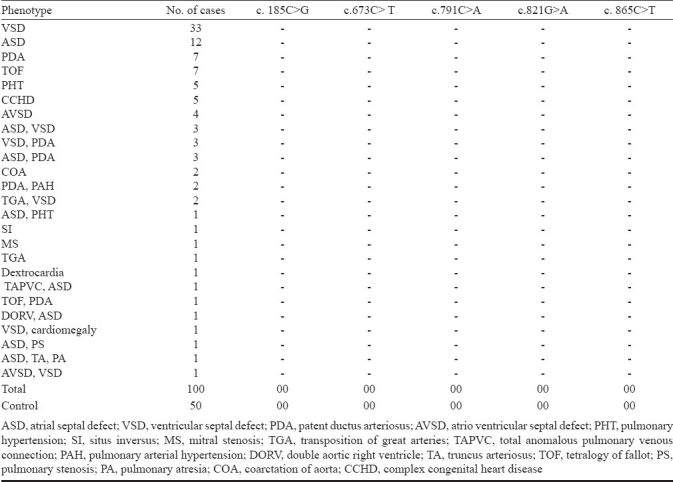
Table V.
List of clinical features of the 18 PDA cases involved in the present investigation

CHD usually refers to abnormalities in the heart's structure or function that arise before birth2. Clinically significant anomalies range from persistence of foetal circulation like PDA to complex defects such as transposition of the great vessels, single ventricle anomalies; hypoplastic left heart syndrome, and complex variants of heterotaxy16,17. PDA occurs in autosomal dominant and recessive disorders that are nonsyndromic and constitutes 10 per cent of all CHDs3. PDA can occur as an isolated abnormality or in association with syndrome like Char syndrome, which is characterized by the triad of typical facial features, PDA, and aplasia or hypoplasia of the middle phalanges of the fifth fingers. Typical facial features are flat midface, flat nasal bridge and broad flat nasal tip, wideset eyes, downslanting palpebral fissures, mild ptosis, short philtrum resulting in a triangular mouth, and thickened (patulous) everted lips. Studies suggest that mutations of TFAP2B are associated with CHAR syndrome related PDA4–6. A recent study has identified mutations of TFAP2B in nonsyndromic PDA cases having none of the features of Char syndrome11.
Since TFAP2 gene family has an important role in cardiac morphogenesis, mainly outflow tract formation and cardiac septation5–7, thus we carried out this analysis to determine the frequency of TFAP2B mutations and to define the subset of CHDs that is associated with mutations in TFAP2B, if any, other than PDA cases. PDA is known occur randomly with septal defects, therefore, we hypothesized that TFAP2B mutations could also be involved in septal and other forms of CHDs. TFAP2B, a neural-crest related transcription factor, has three main functional domains, the N-terminal portion of the protein contains a transactivation domain with a conserved phosphotyrosine motif, the C terminus contains a highly conserved basic domain, which is necessary for DNA binding, and the helix-span-helix domain is involved in DNA binding and dimerization. The TFAP2 transcription factors comprise a family of proteins that are closely related to sequence-specific DNA-binding molecules. Eight TFAP2B mutations have been reported in six unrelated individuals and families with Char syndrome Table I4,8,9. Five of the mutations affect the basic domain. The sixth missense mutation, p.Pro62Arg, alters the PY motif in the transactivation domain. All of these missense changes affect highly conserved residues. Among the five basic domain mutations, four affect arginine residues. Another splice site mutation has been identified in nonsyndromic isolated case of PDA, indicating that the mutations of TFAP2B are involved in those patients who do not exhibit any features related to Char syndrome11.
In the present investigation, five of the six missense mutations were analysed by massARRAY. The analysis did not identify any of the five mutations among the 18 PDA cases and also in the varied CHD cases analyzed for mutations in TFAP2B. Thus, the mutations screened were not involved in the manifestation of PDA in all the isolated and also syndromic related PDA cases analyzed in the present study.
This study has certain limitations. Only prominent mutations were screened and not all the mutations reported. Since this is a targeted mutation based study, the unknown mutations in all the exons were not studied. The sample size was small due to the selection criteria and lack of consent from the patients. It is also possible that Indian populations may harbour mutations distinct from other populations previously reported.
Thus, identification and analysis of specific nuclear families with Char syndrome or having more than two children affected with PDA in a family and further a large scale screening of PDA cases would help substantiate the role of TFAP2B mutations in the manifestation of the disease.
Acknowledgments
The authors thank patients and patients’ family, the clinicians and the PGs in Department of Pediatrics, Cheluvamba Hospital, Mysore Medical College, and JSS Hospital, Mysore. Thanks are also extended to CSIR, New Delhi and UGC-RFSMS for providing the financial assistance and also Vimta Laboratries, Hyderabad for providing massARRAY facility. Authors also thank Prof. H.A. Ranganath for his encouragement.
References
- 1.Nemer M. Genetic insights into normal and abnormal heart development. Cardiovasc Pathol. 2008;17:48–54. doi: 10.1016/j.carpath.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–8. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 3.Mani A, Meraji SM, Houshyar R, Radhakrishnan J, Mani A, Ahangar M, et al. Finding genetic contributions to sporadic disease: a recessive locus at 12q24 commonly contributes to patent ductus arteriosus. Proc Natl Acad Sci USA. 2002;99:15054–9. doi: 10.1073/pnas.192582999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mani A, Radhakrishnan J, Farhi A, Carew KS, Warnes CA, Nelson-Williams C, et al. Syndromic patent ductus arteriosus: evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder. Proc Natl Acad Sci USA. 2005;102:2975–9. doi: 10.1073/pnas.0409852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckert D, Buhl S, Weber S, Jager R, Schorle H. The AP-2 family of transcription factors 2005; 6 : 246. Genome Biol. 2005;6:246.1–246.8. doi: 10.1186/gb-2005-6-13-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18:101–10. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammer S, Toenjes M, Lange M, Fischer JJ, Dunkel I, Mebus S, et al. Characterization of TBX20 in human hearts and its regulation by TFAP2. J Cell Biochem. 2008;104:1022–33. doi: 10.1002/jcb.21686. [DOI] [PubMed] [Google Scholar]
- 8.Satoda M, Zhao F, Diaz GA, Burn J, Goodship J, Davidson HR, et al. Mutations in TFAP2B cause char syndrome a familial form of patent ductus arteriosus. Nat Genet. 2000;25:42–6. doi: 10.1038/75578. [DOI] [PubMed] [Google Scholar]
- 9.Zhao F, Weismann CG, Satoda M, Pierpont ME, Sweeney E, Thompson EM, et al. Novel TFAP2B mutations that cause Char syndrome provide a genotype- phenotype correlation. Am J Hum Genet. 2001;69:695–703. doi: 10.1086/323410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark KL, Yutzey KE, Benson DW. Transcription factors and congenital heart defects. Annu Rev Physiol. 2006;68:97–121. doi: 10.1146/annurev.physiol.68.040104.113828. [DOI] [PubMed] [Google Scholar]
- 11.Khetyar M, Syrris P, Tinworth L, Abushaban L, Carter N. Novel TFAP2B mutation in nonsyndromic patent ductus arteriosus. Genet Test. 2008;12:457–60. doi: 10.1089/gte.2008.0015. [DOI] [PubMed] [Google Scholar]
- 12.Wilke RA, Simpson RU, Mukesh BN, Bhupati SV, Dart RA, Ghebranious NR, et al. Genetic variation in CYP27B1 is associated with congestive heart failure in patient with hypertension. Pharmacology. 2009;10:1789–97. doi: 10.2217/pgs.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soderlund-Strand A, Dillner J, Carlson J. High-throughput genotyping of oncogenic human papilloma viruses with MALDI-TOF mass spectrometry. Clin Chem. 2008;54:86–92. doi: 10.1373/clinchem.2007.092627. [DOI] [PubMed] [Google Scholar]
- 14.Zou CC, Liang L, Wang CL, Fu JF, Zhao ZY. Polymorphisms of the Ghrelin/Obestatin gene and ghrelin levels in Chinese children with short stature. Clin Endocrinol (Oxf) 2008;69:99–104. doi: 10.1111/j.1365-2265.2007.03171.x. [DOI] [PubMed] [Google Scholar]
- 15.2010. [accessed on August 30, 2010]. Available from: www.sequenom.com .
- 16.Gelb BD, Zhang J, Sommer RJ, Wasserman JM, Reitman MJ, Willner JP. Familial patent ductus arteriosus and bicuspid aortic valve with hand anomalies: a novel heart-hand syndrome. Am J Med Genet. 1999;87:175–9. doi: 10.1002/(sici)1096-8628(19991119)87:2<175::aid-ajmg9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell ME, Sander TL, Klinkner DB, Tomita-Mitchell A. The molecular basis of congenital heart disease. Semin Thorac Cardiovasc Surg. 2007;7:231–7. doi: 10.1053/j.semtcvs.2007.07.013. [DOI] [PubMed] [Google Scholar]