

Summary
We have explored a unique combination therapy for metastatic colorectal cancer. This strategy combines a potent and new oncolytic poxvirus expressing a membrane-bound tumor necrosis factor-related apoptosis-inducing ligand (TRAIL or TNFSF10) and oxaliplatin (Ox) chemotherapy. We hypothesized that TRAIL expression would increase the efficacy of the oncolytic poxvirus, and that the therapeutic efficacy would be further enhanced by combination with chemotherapy. The cytotoxicity to cancer cells by Ox, oncolytic VV and trail gene-armed VV alone or in combination was tested in vitro. The trail gene armed oncolytic VV expressed high levels of TRAIL in infected cancer cells and had greater potency as a cytotoxic agent compared to the parent VV. Ox alone exerted concentration-dependent cytotoxicity. In vitro, the combination of the two agents applied at suboptimal concentrations for individual therapy displayed synergy in inducing cancer cells into enhanced levels of apoptosis/necrosis. Western blot analyses confirmed the notion that TRAIL induced cancer cell death mainly through apoptosis, while Ox and vJS6 may induce cell death more through non-apoptotic death pathways. In two aggressive colorectal carcinomatosis models derived from human HCT116 and murine MC38 cells, the combination therapy displayed synergistic or additive antitumor activity and prolonged the survival of the tumor-bearing mice compared to either Ox chemotherapy or vvTRAIL-mediated oncolytic gene therapy alone. This combination strategy may provide a new avenue to treating peritoneal carcinomatosis and other types of metastases of colorectal cancer.
Keywords: virotherapy, vaccinia, gene therapy, chemotherapy, colorectal cancer
Introduction
Colorectal cancer is the third most common type of cancer in Western countries. It is estimated that 148,810 Americans will be diagnosed with colorectal cancer, and 49,960 will die of this disease in 2008.1 About 20% of patients diagnosed with colorectal cancer will have distant metastasis (stage IV) at the time of diagnosis. Following decades during which 5-fluorouracil was the mainstay of chemotherapy for metastatic colorectal cancer, new agents have been introduced into the clinical armamentarium over the past ten years. These include the chemotherapeutic agents oxaliplatin (Ox) and irinotecan, and the biologic agents bevacizumab and cetuximab.2,3 Adding Ox to LV5FU2 regimen significantly improve 5-year disease-free survival and 6-year overall survival in the adjuvant treatment of stage II or III colon cancer.4 Systemic chemotherapy, however, is not particularly effective or has not been completely evaluated for treatment of peritoneal carcinomatosis (PC) in colorectal cancer patients.5 Complete surgical resection of colorectal PC followed by hyperthermic intraperitoneal chemotherapy has resulted in a 2-year survival rate of up to 60%.6-8 Although there has been encouraging progress, there is obviously room for improvement and an impetus for the development of novel targeted biological therapies, using either single agents or agents in combination.
As an experimental regimen to treat cancer, oncolytic viruses have been shown promising in both preclinical models and in clinical trials in cancer patients.9-12 Investigators including us have explored gene therapy, oncolytic virotherapy and chemotherapy, in the form of either mono- or dual therapy for cancer treatment.13-16 We have genetically engineered an oncolytic vaccinia virus (VV) to enhance its tumor-selective replication and retain its efficacious oncolytic potency, as demonstrated previously in murine colorectal cancer models.16-18
We have also sought to improve the efficacy of the oncolytic VV by taking advantage of its powerful promoters to force expression of death ligands or immunostimulatory molecules, thereby eliciting a bystander effect. One death ligand in particular which we have investigated is TRAIL. TRAIL is a transmembrane protein belonging to the tumor necrosis factor (TNF) superfamily, and it has been actively explored for delivery as either a soluble protein or as a gene therapy product for cancer treatment.19,20 TRAIL induces apoptotic or non-apoptotic cell death by cross-linking either of the two functional TRAIL receptors (DR4 or DR5) that each contain a death domain. Activation of these domains leads to formation of the death-inducing signaling complex (DISC). Further activation of signaling molecules downstream results in the activation of a protease cascade, which culminates in cell death. DcR1 and DcR2 serve as decoy receptors, and do not signal DISC formation when ligated by TRAIL.20,21 In mice, there is only one functional receptor.DR522 Using adenoviral vectors for TRAIL expression, we and others have shown that TRAIL displays strong antitumoral activity and can function to kill non-infected cells via bystander effects.23,24 Recent studies have indicated a predominance of TRAIL resistance in primary human tumor cells. The mechanisms of this resistance have been studied.25,26 This resistance can be overcome by sensitizing the cells to TRAIL-induced apoptosis by treatment with biologic agents or chemotherapy.14,15,25,26 Combination strategies are, therefore, often necessary. In developing combination therapies, synergism between agents is sought in order to reduce the dose of each agent, thus minimizing toxicity while maximizing the therapeutic effect of the combination.
Platinum agents are among those chemotherapeutics shown to sensitize cancer cells to TRAIL-induced apoptosis. Ox is a novel diaminocyclohexane platinum derivative with clinical utility in the treatment of patients with colorectal cancer. Ox can block DNA transcription and replication by covalently binding to form interstrand and intrastrand DNA cross-links, ultimately leading to cell death. As a single-agent, Ox is generally well tolerated. Ox combination chemotherapy is the first-line treatment for patients with metastatic colorectal cancer.2,3
In this study, we hypothesized that combination therapy consisting of the oncolytic VV expressing TRAIL and Ox would work synergistically against colorectal cancer models. VV is oncolytic and replicates selectively in cancer cells. As noted above, expression of TRAIL may improve the efficacy of VV by eliciting a bystander effect. Ox may enhance the cytotoxicity further by sensitizing the cancer cells to TRAIL-induced apoptosis.27-31 Our current results from studies with colorectal cancer cells in vitro and with both murine and human colorectal carcinomatosis models in vivo have demonstrated that the combined therapy worked synergistically to inhibit tumor growth and significantly prolonged survival.
Results
Human and murine colorectal cancer cells express functional receptors for TRAIL
Cancer cells can be resistant to TRAIL-mediated killing due to the lack of expression of functional TRAIL receptors. In order to select suitable colorectal cancer models for this study, we first surveyed the expression of TRAIL receptors in human and murine colorectal cancer cell lines (Fig. 1). DLD1 and HCT116, two human colorectal cancer cell lines, express both functional and decoy TRAIL receptors. HCT116 cells express high levels of the functional receptor, DR4, while DLD1 cells express high levels of DR4 and DR5. In terms of decoy receptors, DcR2, but not DcR1, is highly expressed in both human colorectal cancer lines. It has previously been shown that there is only one functional murine TRAIL receptor, DR5.22 Indeed, MC38 murine colon cancer cells express this receptor. The murine decoy receptors DcR1 and DcR2 in MC38 cells were below the level of detection. Treatment with Ox or VV (vJS6) did not alter TRAIL receptor expression in any of these cancer lines (data not shown).
Figure 1. TRAIL receptor expression in human and murine colorectal cancer cells.
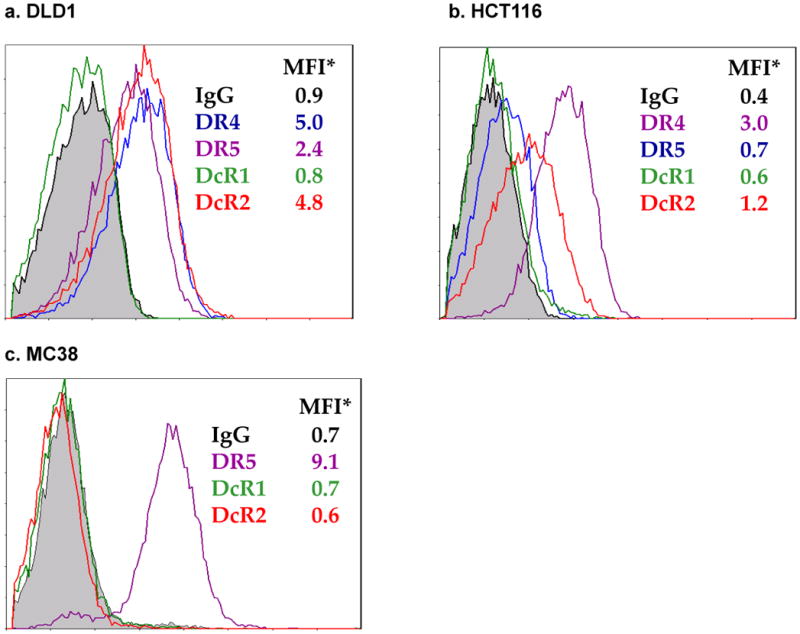
Expression of TRAIL receptors was analyzed by antibody labeling followed by flow cytometry. Representatives of 3 independent experiments with similar results are shown. Treatment with Ox or VJS6 did not alter receptor expression (data not shown). MFI: mean fluorescence intensity.
Construction of vvTRAIL and TRAIL expression in the virus-infected cancer cells
We have constructed a new recombinant VV expressing the membrane-associated TRAIL molecule. Using flow cytometry, we confirmed viral expression of the membrane-associated TRAIL molecule in infected DLD1 cells (Fig. 2). As shown, the DLD1 colorectal cancer cells did not express TRAIL. Infection of the cells with vvTRAIL at MOI of 0.25 led to significant levels of TRAIL on the cell surface, with peak at 18 hpi. Cotreatment with Ox (at 1.0 μg/ml) did not affect the expression of TRAIL during the duration of the experiment (24 h). Viral infection along with an apoptosis inhibitor (Z-VAD) did not lead to significant increase of TRAIL. Neither treatment of cells with the control virus vJS6 nor with Ox led to overexpression of TRAIL. A similar pattern of TRAIL expression was seen whether viewed as mean fluorescence intensity (Fig. 2A) or percentage of TRAIL-positive cells (Fig. 2B). These results clearly indicate that the virus expressed the cell surface-associated TRAIL protein in infected cancer cells.
Figure 2. Expression of TRAIL in vvTRAIL-infected cancer cells.

TRAIL expression in DLD1 cancer cells was evaluated by antibody labeling followed by flow cytometry. Designated cells were treated with vvTRAIL or vJS6 at an MOI of 0.25. Ox concentration was 1.0 μg/ml. The concentration of the pan-caspase inhibitor ZVAD was 20 μM. Cells were harvested for flow cytometry at the indicated time points and labeled with a phycoerythrin-conjugated antibody specific for TRAIL. Results are expressed in terms of MFI (A) or % TRAIL-positive cells (B). *Cells treated with vvTRAIL + Z-VAD, vJS6 or Ox alone were analyzed only at the 24 h time point.
Combination of vvTRAIL and Ox enhanced the cytotoxicity to cancer cell in vitro
We assessed the in vitro cytotoxicities of both single agent and combination therapies in two human and one murine colorectal cancer lines (Fig. 3). The effect on viral cytotoxicity to cancer cells by TRAIL expression was examined by comparing the killing effect of vvTRAIL to that of vJS6, a control VV. The cancer cell lines varied in their susceptibility to VV-induced cytotoxicity, evidenced by the IC50 values for vJS6. DLD1 and HCT116 are relatively resistant to vJS6, whereas MC38 cells are quite sensitive (Fig. 3A). Thus lower ranges of viral doses were used in MC38 cells.
Figure 3. Reduction of cell viability mediated by single-agent or combination treatments.




(A). Dose-dependent enhancement of viral cytotoxicity by Ox in the three colorectal cancer cell lines. The cell viability at 96 h after treatment was determined by MTS assay. Data are expressed as the mean ± SEM of at least 3 independent experiments. (B, C) .The cell viability (B) and the associated combination index (CT) (C) after cancer cells treated with specific combination of drugs. For DLD1 and HCT116 human colorectal cancer cells, Ox [0.25 μg/ml] and VV [0.5 MOI] at indicated doses were used. For MC38 colon cells, Ox [0.25 μg/ml] and VV [0.1 MOI] were used. CI values are expressed as mean ± SD from at least 2 experiments for which the CI was calculable. (D). The half maximal inhibitory concentration (IC50) values of vvTRAIL for the 3 colorectal cancer cell lines, estimated from the dose-response curves normalized for the cytotoxic effect of Ox. Data are expressed as the mean ± SEM of at least 3 independent experiments.
Choosing the suboptimal doses for singular agents Ox, vJS6 or vvTRAIL, we compared the inhibitory effects of cell viability by mono and dual treatments (Figure 3B). A few interesting observations were made. DLD1 cells were sensitive to cytotoxicity induced by vvTRAIL, but not to either Ox alone or oncolytic lysis by vJS6 itself. HCT11 cancer cells were quite sensitive to Ox alone, but not sensitive to VJS6 or vvTRAIL. In contrast, MC38 cancer cells were sensitive to vJS6 alone, but not to Ox. Based on the formulation by Chou and Talalay,32 we have calculated the CI values of the combination effects by Ox and VV (Figure 3C). In DLD1 cancer cells, CIs from both combinations (Ox + vJS6, and Ox + vvTRAIL) were less than 1.0, indicating a synergism between the two agents. In HCT116 cancer cells, Ox and vvTRAIL produced a synergistic effect, while combining Ox and vJS5 might generate the antagonistic effect (CI > 1.0). Interestingly, in MC38 cells, the two combinations, most likely generate additive effects (CI values close to 1.0 with large standard deviation values).
Finally, we also estimated the half maximal inhibitory concentration (IC50) values of vvTRAIL in combination with increasing doses of Ox (Figure 3D). We observed a gradual reduction of the viral dose of vvTRAIl with increasing doses of Ox in both DLD1 and HCT116 cancer cells, but not in MC38 cancer cells. These results are consistent with those for cell viability and CI values (Figure 3B and C).
In summary, Ox sensitized DLD1 and HCT116 cells to the cytotoxic effects of vvTRAIL, as evidenced by a reduction in the IC50's of vvTRAIL for these cell lines. The combination of the two agents generated synergistic effects in these two cancer cell lines. In contrast, such a synergistic effect was not clearly seen for the MC38 cell line. It is most like an additive effect in MC38 cancer cells.
Combination treatment induced enhanced cell death in cancer cells
To study cell death in the single agent or combination-treated cells, we first used annexin V-PE and 7AAD staining (Fig. 4). Positive staining with annexin V denotes the early stages of apoptosis, while staining with 7AAD indicates late stage of apoptosis or necrosis. As we see for DLD1 cells, a baseline total of up to 11.0% cells were in apoptosis/necrosis in a healthy population of mock-treated cells. This percentage was only 7.3% in Ox-treated cells, but increased to 21.7% in vJS6-infected cells, and 52.5% in vvTRAIL-treated cells. While combination treatment of Ox plus vJS6 increased this percentage to 29.7%, the other combination of Ox and vvTRAIL increased this number more dramatically, to 71.5%. We have also conducted similar studies with HCT116 and MC38 cancer cells, and the same patterns of apoptosis/necrosis were observed (Supplemental Fig. 1).
Figure 4. Apoptosis/necrosis in the colorectal cancer cells treated with the single agents or combinations.
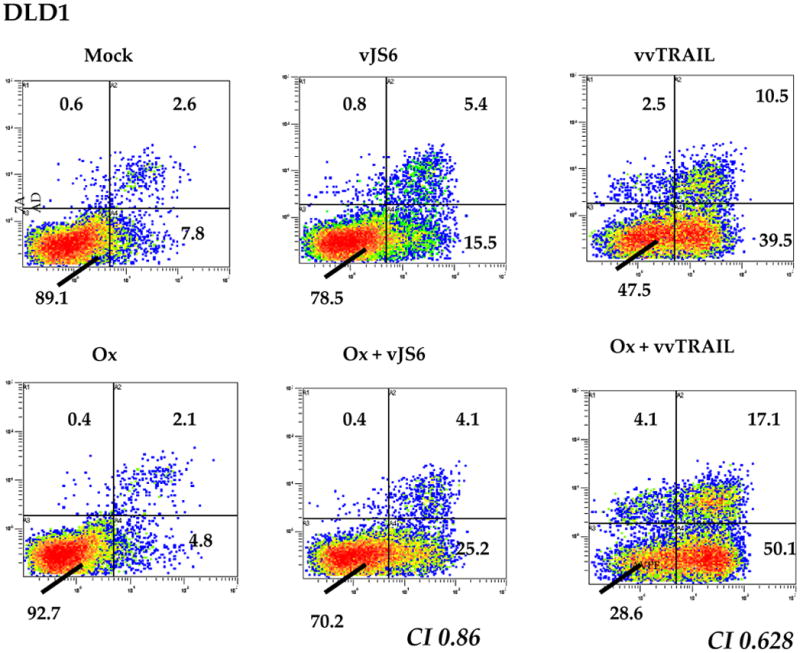
Ox [0.5 μg/ml] and virus [MOI = 0.25] were used for DLD1 cells. Ox [0.5 μg/ml] and virus (MOI = 0.1) were used for HCT116 cells. For MC38 cells, Ox [1.0 μg/ml] and virus (MOI = 0.1) were used. Shown are data from DLD1 cancer cells. Data for HCT116 and MC38 cells are presented as supplemental Figure 1. Apoptosis/necrosis was evaluated using Annexin V-PE and 7AAD. X-axis: Annexin V-staining; Y-axis: 7AAD staining. Representative data of 3 independent experiments are shown.
Since it has previously been shown that the mitochondria-dependent (type II) signaling pathway plays a major role in TRAIL or cisplatin-TRAIL combination-induced apoptosis of cancer cells,25,33,34 we decided to explore the potential regulation of a panel of anti-apoptotic and pro-apoptotic proteins involved in the signaling pathway in our mono- and combination therapies. We selected four representative molecules in the type II signaling pathway: Bcl-xL for anti-apoptotic proteins, Bak for pro-apoptotic proteins, and cleavage of caspase-8 and PARP for the execution of the apoptosis process. The cleavage of both caspase-8 and PARP are quite early in apoptotic signaling and thus involved in both type I (mitochondria-independent) and type II (mitochondria-dependent) apoptotic signaling pathways.
DLD1 cancer cells were mock treated or treated with Ox, vJS6, vvTRAIL, or Ox in combination with either vJS6 or vvTRAIL, for 12, 18, 24 and 48 h. Cancer cells treated with etoposide were used as a positive control for apoptosis. The express levels or activation were examined by Western blots (Figure 5A). At 24 h after treatment, bcl-xL was reduced in cancer cells treated with vvTRAIL, Ox + vvTRAIL or the control drug etoposide (lanes 4, 6 and 7, respectively). In contrast, the pro-apoptotic protein Bak was induced in these three treatments. At this point, neither Bcl-xL nor Bak was up or down-regulated in cells treated with Ox, vJS6 or the combination. The transient burst and then proteolytic cleavage of PARP by caspases is a hallmark of apoptosis and it prevents induction of necrosis during apoptosis and ensures appropriate execution of caspase-mediated programmed cell death. A significant increase of cleaved PARP (89 kDa) was observed in cells treated with vvTRAIL or Ox + vvTRAIL (lanes 4 and 6).
Figure 5. Western blot analyses of cell death-related proteins.
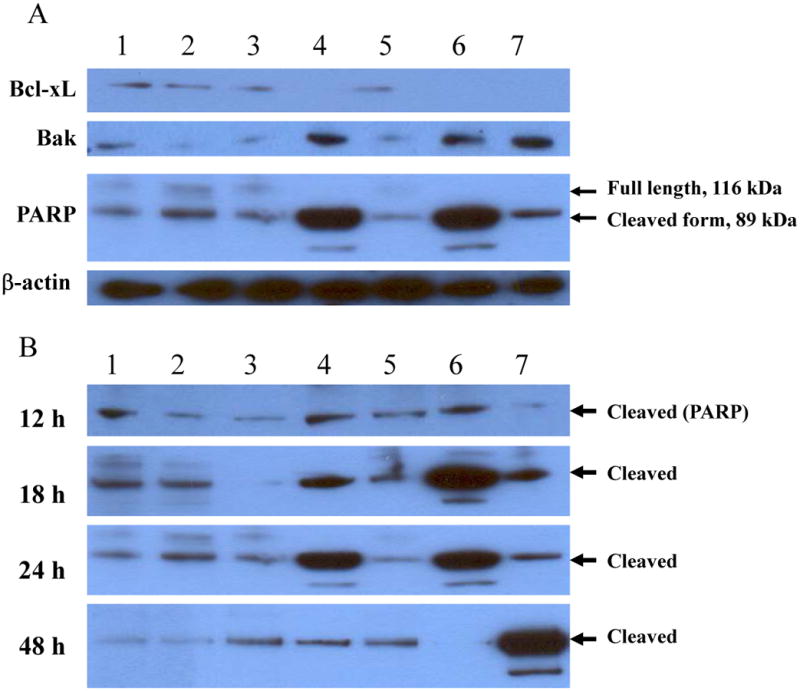
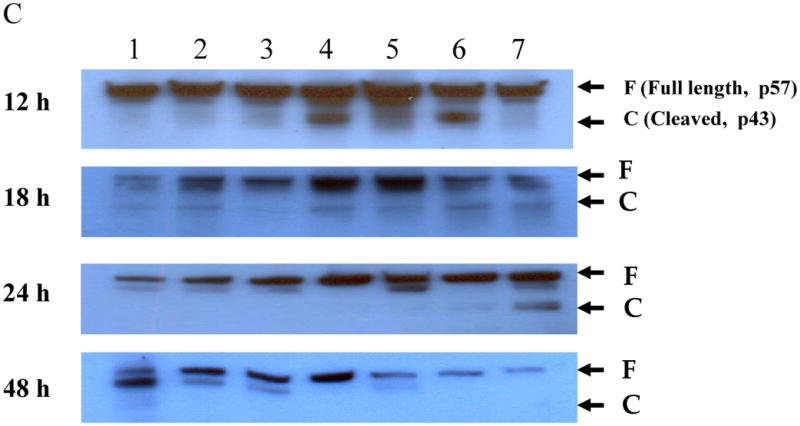
DLD1 cells were grown in 6-well plates overnight and then treated with the agents as indicated for 12, 18, 24 and 48 h. Cells were lyzed and subject to Western blot analysis as described in Materials and Methods. The cells were treated with, lane 1: mock-treated; lane 2: Ox; lane 3: vJS6; lane 4: vvTRAIL; lane 5: Ox + vJS6; lane 6: Ox + vvTRAIL; and lane 7: etoposide (100 μM) (positive control). (A). The regulation of representative anti-apoptotic and pro-apoptotic proteins in cancer cells under various treatments. The anti-PARP antibody was against the cleaved form of PARP. (B). Kinetics of cleaved PARP. The antibody mainly recognizes the cleaved form of PARP. The time points were 12, 18 24 and 48 h. (C). Kinetics of cleaved caspase-8.
We further examined the kinetics of the PARP cleavage in the cancer cells at multiple time points (Figure 5B). At 12 h, there were low levels of cleaved PARP in all samples. At 18 h, there was an increased cleave in vvTRAIL-treated cells. However, Ox + vvTRAIL-treated cells showed a dramatic enhanced cleave (lane 6). This process was at plateau by 24 h. At 48 h, it reduced to basal level in vvTRAIL-treated cells, and it was undetectable in Ox + vvTRAIL-treated cells because essentially all cells were dead. The control drug etoposide-treated cancer cells showed a peak cleaved PARP at 48 h, suggesting the slower kinetics of apoptosis-induction by this agent. We have then repeated the same experiments with for cleavage of caspase-8 (Fig. 5C). The exact same pattern of cleavage was observed as in PARP in variously treated cells. We have seen very little, if any, cleaved caspase-8 in cells treated with Ox, vJS6, or the combination.
It is interesting to observe that cancer cells treated with Ox, vJS6 or the combination did not show much change with Bcl-xL and Bak, and not much cleavage of either caspase-8 or PARP. We were initially surprised at these results. However, upon close examination, we found that two reasons for explanation. First, the single agent (Ox or vJS6) at the suboptimal doses did not cause a lot of cell death in DLD1 cells (Fig. 3B). Secondly, it has been shown that Ox triggers more necrosis than apoptosis in cancer cells including colorectal cancer cells.35,36 As for vJS6, our previous study has shown that oncolytic VV can induce both necrosis and apoptosis.18. Thus, it is not surprising that no much change in those apoptotic proteins in cancer cells treated with either Ox or vJS6. In summary, Single agent of Ox or vJS6 caused small quantities of cell death at suboptimal doses, while the combination of these agents may enhance cell death, but they may induce cell death via non-apoptotic as well as apoptotic signaling pathways.
Regional treatment with vvTRAIL and Ox increased survival in in vivo carcinomatosis models
Colorectal peritoneal carcinomatosis (PC) is a frequent and very lethal event.6-8 In order to assess the efficacy of treatment modalities in a setting close to human patients, we established two tumor models which mimic the aggressiveness and the late stage of cancer often found in colorectal cancer patients. Thus, we have chosen two aggressive colorectal PC models (MC38 and HCT116) and treated them at a stage when tumors were well established. Both the HCT116 and MC38 carcinomatosis models are very aggressive and lethal, resulting in 100% mortality of the untreated mice within 30 days. In both models, we observed peritoneal carcinomatosis within one week of intraperitoneal tumor cell inoculation, and we began treatment of MC38 tumor at day 7, and HCT116 tumor on day 12 (Fig. 6).
Figure 6. The therapeutic effects of the mono-, dual and triple combination therapy on two models of colorectal carcinomatosis.

(A). Treatment of human colorectal cancer HCT116 carcinomatosis in nude mice. Athymic nude mice (n=10 mice/group) were injected intraperitoneally (i.p.) with 1 × 107 HCT116 cells. Twelve (12) days after tumor cell injection, the mice were treated with i.p. administration of Ox [2.5 mg/Kg], vJS6 [1.0 × 106 pfu], vvTRAIL [1.0 × 106 pfu], or combinations of Ox with either of the viruses. Viruses were injected on day 12 and Ox was administered on days 13, 15, 17, and 19. Ox in combination with either vJS6 (p < 0.045) or vvTRAIL (p < 0.0025) resulted in significantly longer survival compared to controls. Combination treatment with Ox and vvTRAIL achieved significantly longer survival than treatment with vvTRAIL alone (p < 0.0075) or treatment with the combination of vJS6 and Ox (p < 0.0001). (B). Treatment of MC38 syngeneic murine colorectal carcinomatosis in C57BL/6 mice. C57BL/6 mice were injected intraperitoneally (i.p.) with 2 × 105 MC38 cells, followed on day 7 by i.p. administration of Ox, vJS6 [3.0 × 106 pfu], vvTRAIL [3.0 × 106 pfu] or combinations. Viruses were injected on day 7 and Ox was administered on days 9, 11, 13, and 15. Combination treatment with vvTRAIL and Ox (p < 0.0075 vs. control) resulted in survival advantages significantly better than with Ox (p < 0.019), vJS6 (p < 0.031), or vvTRAIL (p < 0.01) alone. Comparison of vvTRAIL and Ox treatment to the combination of Ox and vJS6 did not reveal a significant survival advantage (p < 0.093).
Even with such aggressive, well established tumors, we observed a survival advantage both in some single agent-treated groups and in the combination treated groups. In the HCT116 model in the athymic nude mice (Figure 6A), treatment with single agents Ox (p < 0.837), vJS6 (p < 0.401), or vvTRAIL (p < 0.636) did not result in any survival advantages compared to control (untreated) mice. However, Ox in combination with either vJS6 (p < 0.045) or vvTRAIL p < 0.0025) resulted in significantly longer survival compared to no treatment. Furthermore, the greatest benefit in terms of survival was seen with combination treatment with Ox and vvTRAIL, which achieved significantly longer survival than treatment with vvTRAIL alone (p < 0.0075) or treatment with the combination of Ox and vJS6 (p < 0.0001). It is worth noting that the in vivo results agreed with what we observed in vitro (Fig. 3C).
In the MC38 tumor model in syngeneic C57BL/6 mice (Figure 6B), the greatest survival advantage was seen with combination treatment with Ox and vvTRAIL (p < 0.0075 vs. control). This difference in survival was significantly better than with Ox (p < 0.019), vJS6 (p < 0.031), or vvTRAIL (p < 0.01) alone. However, more complex results have been obtained in this tumor model. It seems that there may be some degree of antagonism between chemotherapy and oncolytic virotherapy (Ox and vJS6), since mice treated by vJS6 + Ox exhibited decreased survival compared to those treated by vJS6 alone. These data are also supported by the in vitro results (Fig. 3C). In MC38 cancer cells in vitro, there was a small antagonism between vJS6 and Ox (CI = 1.04), while a small synergism between vvTRAIL and Ox (CI = 0.90). It is likely that these effects are playing a role in the observed lack of statistically significant difference in mice treated with “vvTRAIL + Ox” versus those treated with “vJS6 + Ox” (p < 0.093). Nevertheless, the combination of vvTRAIL + Ox was still far better than any monotherapy in this tumor model.
Discussion
Genetically engineered tumor-selective oncolytic VV has been shown to be effective in both tumor models and in a phase I clinical trial.9,10,12,17,18,37-40 To further enhance its oncolytic potency, the oncolytic VV can be armed with genes such as a death ligand to produce a bystander effect. TRAIL is a death ligand which has been extensively studied during the last decade. Many studies have explored a strategy of direct induction of apoptosis of cancer cells by targeting death receptors using a recombinant soluble TRAIL protein or agonistic anti-DR4 or anti-DR5 monoclonal antibodies. This approach has been quite appealing, and a few clinical trials for the treatment of cancer using such strategies are underway.19-21 An alternative therapeutic strategy is to deliver the TRAIL gene to cancer cells using tumor-selective viral vectors.13,24
We and others have previously tested some forms of dual therapy on various tumor models. For example, the combination of TRAIL and chemotherapeutic agents has been examined on cancer cells and in tumor models.14,15,29 We and others have examined the effect of an oncolytic adenovirus expressing TRAIL in various tumor models.13,24,34 In this context, it is important to point out that adenoviruses (Ad) expressing TRAIL have been used at an MOI of 10 to 100 (pfu/cell) in order to achieve significant infection of human cancer cells.13 However, this relative high viral dosage might not be clinically practicable or achievable. In contrast, oncolytic VV, with its great capacity to carry genes and its powerful promoters to drive gene expression, is an excellent vector to achieve forced expression of membrane-bound TRAIL on target cancer cells. One great advantage of using a replicating VV is its rapid replication and spread among cells in tumor tissue.
We have attempted to take advantage of these observations by combining chemotherapy with an oncolytic VV armed with TRAIL gene. We have demonstrated that vvTRAIL plus Ox mediated profound cytotoxicity in cultured colorectal cancer cells of both human and murine origins. The combination of Ox and vvTRAIL displayed significantly enhanced apoptosis/necrosis in all three colorectal cancer cell lines tested. Ox sensitized the cancer cells to vvTRAIL-mediated cytotoxicity. Further analysis of key proteins in the apoptosis pathways in cells treated with various agents lead to somewhat surprising results. The expression of TRAIL on the cell surface of cancer cells lead to down-regulation of anti-apoptotic protein Bcl-xL, up-regulation of pro-apoptotic protein Bak, and increase of cleaved caspase 8 and PARP, hallmarks of apoptosis. However, we did not observe little, if any, these changes in cancer cells treated with Ox or vJS6, the parental oncolytc poxvirus. There are two reasons for that. First, the dose for Ox was puporsely suboptimal and it did not cause a lot of cell death. Secondly, Ox induced cell death more frequently through necrosis than apoptosis. (Ref) The oncolytic poxvirus encodes a number of anti-apototic proteins and may induce cancer cell into necrosis too. (Guo 2005). Despite different mechanisms of cell death, this combination therapy of Ox and vvTRAIL has achieved significant therapeutic effects in two colorectal carcinomatosis models in mice.
The carcinomatosis models used in this study are essentially confined to peritoneal cavity, and we have treated the disease with regional delivery of vvTRAIL and/or Ox. This regional delivery may be much more effective than intravenous administration for treating peritoneal carcinomatosis.41 It is important to point out that there are frequently disseminated metastases in multiple organs at late stage of the disease in cancer patients, and one great advantage of oncolytic viruses such as VV, is that they could be delivered systemically to target metastases in multiple organs.9,11,12,42
Combination of oncolytic virotherapy, chemotherapy, and/or immunotherapy has been investigated in recent pre-clinical and clinical studies in order to maximize the efficacy.43-48 Indeed some promising results have been obtained in animal models and in clinical studies with human patients.43-48 We believe that further optimizations of each component of the therapeutic modality would maximize the overall therapeutic effects. Towards this goal, investigators have been working to improve oncolytic virotherapy not only by genetic engineering of the oncolytic viruses themselves, but also by modulating the host tumor microenvironment for optimal oncolytic effects from these viruses.43,49-51 Finally, a small-molecule drug-regulatable expression of cancer cell death-inducing molecules such as TRAIL in conjunction with a replicating oncolytic poxvirus would exert a better synergistic effect in such a combination therapy.52
Materials and Methods
Cell culture
The colorectal cancer cell lines of both human (HCT116 and DLD1) and murine (MC38) origins have been used in our laboratories previously.17,18,27 HeLa and CV-1 cells have been used extensively in our laboratories for viral plaque assays and the production of viral stocks.
Construction of vvTRAIL
In order to generate cDNA encoding murine TRAIL (approved gene symbol tnfsf10), total RNA was isolated from splenocytes isolated from a C57BL/6 mouse using TRIzol reagent according to manufacturer's instruction (Invitrogen). Primers for PCR were generated from the published murine tnfsf10 mRNA sequence (GenBank accession no: NM_009425) with flanking SalI and EcoRI sites. RT-PCR using these primers and total RNA from mouse splenocytes was performed to obtain the TRAIL cDNA. The cDNA was then isolated from an agarose gel and digested with SalI and EcoRI and inserted into pCB023-II, a vaccinia shuttle plasmid with flanking regions covering thymidine kinase (TK) homologous regions. The pCB023-II-TRAIL plasmid was amplified and purified from E. coli DH5α cells. The inserted DNA was sequenced to confirm its identity.
vvTRAIL was produced in CV-1 cells by a procedure of homologous recombination of transfected plasmid DNA pCB023-II-TRAIL and infection with the parental TK-deleted virus vJS6.16,17 Homologous recombination resulted in the insertion of the TRAIL gene into the tk locus of the virus, removing the β-gal gene in the parental vJS6. The caspase inhibitor z-VAD-fmk (20 μM) was included in the media of the transfection mixture and subsequent clone isolation. PCR was then used to confirm the correct viral construct in selected clones. The vvTRAIL virus was expanded in HeLa cells without the presence of z-VAD-fmk in the medium. All viral titers were determined by plaque assays on CV1 cells. All virus samples were sonicated for 2 min prior to being used in any experiment.
Flow cytometry
For flow cytometric analysis of TRAIL receptors, cancer cells were mock-treated or treated for 24 h as specified with Ox (Sigma-Aldrich, St. Louis, MO), vJS6, or the combination of Ox and vJS6. Cells were then harvested by trypsinization and washed with 1× PBS containing 0.5% BSA. The human colon adenocarcinoma cells were then incubated for 30 min on ice with phycoerythrin-conjugated mouse anti-human antibodies against DR4, DR5, DcR1, and DcR2 (R & D Systems, Minneapolis, MN). Excess unbound antibody was removed by one wash with PBS with 0.5% BSA. Cells were then submitted for flow cytometry. MC38 cells were similarly evaluated for expression using antibodies against the murine functional TRAIL receptor DR5 (R & D Systems, Minneapolis, MN) and the murine decoy receptors DcR1 and DcR2 (Abcam Cambridge, MA).
For flow cytometric analysis of murine TRAIL expression, DLD1 cells were infected with vvTRAIL at an MOI of 0.25, and samples were harvested at 6 h intervals to evaluate the expression of murine TRAIL in these cells. Comparative expression of murine TRAIL in cells treated with vvTRAIL in combination with Ox at 1.0 μg/ml was also evaluated. Cells were seeded in 10-cm plates at predetermined densities. After an overnight incubation, the cells were incubated with the virus for 2 hours in medium supplemented with 2% FBS. The medium was then changed to medium supplemented with 10% FBS, with or without Ox. Cells were harvested by trypsinization and washed with PBS with 0.5% BSA. The cells were then incubated for 30 min on ice with phycoerythrin-conjugated mouse antibodies against mouse TRAIL or the appropriate isotype control (Biolegend, San Diego, CA). Excess unbound antibody was removed by one wash with PBS containing 0.5% BSA. Cells were then submitted for flow cytometry. Untreated cells, Ox-treated cells, and cells treated with the parental virus (vJS6) served as controls. In anticipation that there would be apoptotic cell death with the expression of murine TRAIL and that such cell death would limit evaluation of TRAIL expression, some samples were treated with ZVAD 20 μM (R & D Systems, Minneapolis, MN) beginning 2 h prior to viral infection.
Cell proliferation assay
Cells were seeded in 96-well plates at predetermined densities appropriate for each cell line (from 5.0 × 103 to 1.0 × 104 cells per well). After an overnight incubation, the cells in designated wells were infected with various MOI's of vJS6 or vvTRAIL. The cells were incubated with the virus for 2 h in medium supplemented with 2% FBS. At the end of this incubation, medium containing 10% FBS and various concentrations of Ox was applied to the designated wells. After 24 h, the medium in all wells was replaced with maintenance medium without Ox. Cell viability was assessed 96 h after viral infection using an MTS assay kit (Promega, Madison, WI).
Apoptosis assay
Cells were seeded in 10-cm plates at predetermined densities. After an overnight incubation, cells were incubated for 2 h with virus (vJS6 or vvTRAIL) in DMEM containing 2% FBS. The medium was then replaced with maintenance medium with or without Ox. The medium in all plates was replaced with maintenance medium 24 h after the addition of Ox. Forty-eight (48) hours after viral infection, cells were stained with annexin V-phycoerythrin (PE) and 7AAD using an Annexin V-PE Apoptosis Detection kit (BD Pharmingen, San Diego, CA) according to the manufacturer's instructions.
Western blots
Cancer cells were grown in 6-well plates and mock-treated or treated with one of the following agents or combinations: Ox, vJS6, vvTRAIL, or Ox in combination with either vJS6 or vvTRAIL. Cancer cells in another well were treated with 100 μM of etoposide (Sigma-Aldrich Corp., St Loius, MO) was used as a positive control for apoptosis. Cells were harvested at various time points and whole cell extracts were prepared in cell lysis buffer supplemented with protease inhibitors (Roche Applied Science, Mannheim, Germany). Protein concentration was determined by Bradford method using a kit (Pierce Biotechnology, Rockford, IL). Ten micrograms of proteins from the extracts were separated using SDS-PAGE, and Western blots were performed as described previously.18 The antibodies against human Bak, Bax, Bcl-xL, cleaved PARP and caspase-8 were obtained from Cell Signaling Technology (Beverly, MA).
Analysis of the effects of combined drug and biological treatment
The Chou and Talalay equation was used for the analysis. When the two agents were administered in combination, the dose of the combination required to produce fractional survival (f) could be separated into the components (D)1 and (D)2 of drugs 1 and 2, respectively. For each level of cytotoxicity (f=0.95, 0.90,…0.05), a parameter called the combination index (CI) was calculated using the CalcuSyn software (Biosoft, Cambradge, MA) according to the Chou and Talalay equation:32
A CI < 1 indicates synergism, while a CI = 1 indicates an additive effect, and CI > 1 indicates antagonism.
Animal studies
The animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Female athymic nude mice and C57BL/6 mice of 5-6 weeks old, were obtained from Taconic Corporation (Germantown, NY). They were housed in standard conditions and given food and water ad libitum.
For the human HCT116 colorectal carcinomatosis model, athymic nude mice (n =10 mice/group) were injected intraperitoneally (i.p.) with 1 × 107 HCT116 cells. Twelve (12) days after tumor cell injection, the mice were treated with i.p. administration of Ox [2.5 mg/Kg], vJS6 [1.0 × 106 pfu], vvTRAIL [1.0 × 106 pfu], or combinations of Ox with either of the viruses. Virus (either vJS6 or vvTRAIL) were administered on day 12 with non virally treated mice receiving equivalent injections of vehicle. Ox [2.5mg/Kg] was given starting the next day (day 13) with repeat treatments every other day for a total of four injections (days 13, 15, 17, and 19). Animals receiving no chemotherapy were given equivalent injections of vehicle. For the murine model, C57BL/6 mice were injected intraperitoneally (i.p.) with 2 × 105 MC38 cells, followed in 7 days by i.p. administration of Ox, vJS6 [3.0 × 106 pfu], vvTRAIL [3.0 × 106 pfu] or combinations as described above. All animals were observed for survival, and survival data were plotted on Kaplan-Meier curves. Significance of survival differences between groups was tested using the log-rank test.
Statistical analysis
The differences among the treatment groups were assessed by using an analysis of variance (ANOVA). A p-value < 0.05 was considered significant. The analysis of the combined effects of multiple agents was performed with CalcuSyn software 2.0 (Biosoft, Cambridge, UK). For in vivo studies log rank tests were used to determine survival differences between treatment groups.
Supplementary Material
Acknowledgments
We thank Ms. Hui Xu for technical assistance, and Ms. Eve Blasko for excellent administrative assistance.
This work was supported in part by the NIH R01 grant CA100415, the Intramural Research Program of the NIH, and by David C. Koch Regional Therapy Cancer Center.
Footnotes
Conflicts of Interest: DLB is a consultant of Jennerex Biotherapeutics, a company developing oncolytic viruses.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Kelly C, Cassidy J. Chemotherapy in metastatic colorectal cancer. Surg Oncol. 2007;16:65–70. doi: 10.1016/j.suronc.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Lee JJ, Chu E. An update on treatment advances for the first-line therapy of metastatic colorectal cancer. Cancer J. 2007;13:276–281. doi: 10.1097/PPO.0b013e3181570062. [DOI] [PubMed] [Google Scholar]
- 4.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27:3109–3116. doi: 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 5.Royal RE, Pingpank JF., Jr Diagnosis and management of peritoneal carcinomatosis arising from adenocarcinoma of the colon and rectum. Semin Oncol. 2008;35:183–191. doi: 10.1053/j.seminoncol.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Elias D, Delperro JR, Sideris L, Benhamou E, Pocard M, Baton O, et al. Treatment of peritoneal carcinomatosis from colorectal cancer: impact of complete cytoreductive surgery and difficulties in conducting randomized trials. Ann Surg Oncol. 2004;11:518–521. doi: 10.1245/ASO.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP. Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Ann Surg. 2006;243:212–222. doi: 10.1097/01.sla.0000197702.46394.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franko J, Gusani NJ, Holtzman MP, Ahrendt SA, Jones HL, Zeh HJ, 3rd, et al. Multivisceral resection does not affect morbidity and survival after cytoreductive surgery and chemoperfusion for carcinomatosis from colorectal cancer. Ann Surg Oncol. 2008;15:3065–3072. doi: 10.1245/s10434-008-0105-x. [DOI] [PubMed] [Google Scholar]
- 9.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 11.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 12.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo W, Zhu H, Zhang L, Davis J, Teraishi F, Roth JA, et al. Combination effect of oncolytic adenovirotherapy and TRAIL gene therapy in syngeneic murine breast cancer models. Cancer Gene Ther. 2006;13:82–90. doi: 10.1038/sj.cgt.7700863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziauddin MF, Yeow WS, Maxhimer JB, Baras A, Chua A, Reddy RM, et al. Valproic acid, an antiepileptic drug with histone deacetylase inhibitory activity, potentiates the cytotoxic effect of Apo2L/TRAIL on cultured thoracic cancer cells through mitochondria-dependent caspase activation. Neoplasia. 2006;8:446–457. doi: 10.1593/neo.05823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YH, Lee YJ. Time sequence of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and cisplatin treatment is responsible for a complex pattern of synergistic cytotoxicity. J Cell Biochem. 2006;98:1284–1295. doi: 10.1002/jcb.20844. [DOI] [PubMed] [Google Scholar]
- 16.Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000;7:66–73. doi: 10.1038/sj.cgt.7700075. [DOI] [PubMed] [Google Scholar]
- 17.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 18.Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 19.Griffith TS, Stokes B, Kucaba TA, Earel JK, Jr, VanOosten RL, Brincks EL, et al. TRAIL gene therapy: from preclinical development to clinical application. Curr Gene Ther. 2009;9:9–19. doi: 10.2174/156652309787354612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 21.Duiker EW, Mom CH, de Jong S, Willemse PH, Gietema JA, van der Zee AG, et al. The clinical trail of TRAIL. Eur J Cancer. 2006;42:2233–2240. doi: 10.1016/j.ejca.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 22.Grosse-Wilde A, Kemp CJ. Metastasis suppressor function of tumor necrosis factor-related apoptosis-inducing ligand-R in mice: implications for TRAIL-based therapy in humans? Cancer Res. 2008;68:6035–6037. doi: 10.1158/0008-5472.CAN-08-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagawa S, He C, Gu J, Koch P, Rha SJ, Roth JA, et al. Antitumor activity and bystander effects of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene. Cancer Res. 2001;61:3330–3338. [PubMed] [Google Scholar]
- 24.Sova P, Ren XW, Ni S, Bernt KM, Mi J, Kiviat N, et al. A tumor-targeted and conditionally replicating oncolytic adenovirus vector expressing TRAIL for treatment of liver metastases. Mol Ther. 2004;9:496–509. doi: 10.1016/j.ymthe.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 26.Song JJ, An JY, Kwon YT, Lee YJ. Evidence for two modes of development of acquired tumor necrosis factor-related apoptosis-inducing ligand resistance. Involvement of Bcl-xL. J Biol Chem. 2007;282:319–328. doi: 10.1074/jbc.M608065200. [DOI] [PubMed] [Google Scholar]
- 27.Zhu H, Zhang L, Huang X, Davis JJ, Jacob DA, Teraishi F, et al. Overcoming acquired resistance to TRAIL by chemotherapeutic agents and calpain inhibitor I through distinct mechanisms. Mol Ther. 2004;9:666–673. doi: 10.1016/j.ymthe.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Gourdier I, Crabbe L, Andreau K, Pau B, Kroemer G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene. 2004;23:7449–7457. doi: 10.1038/sj.onc.1208047. [DOI] [PubMed] [Google Scholar]
- 29.Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005;4:2026–2036. doi: 10.1158/1535-7163.MCT-05-0262. [DOI] [PubMed] [Google Scholar]
- 30.Jalving M, de Jong S, Koornstra JJ, Boersma-van Ek W, Zwart N, Wesseling J, et al. TRAIL induces apoptosis in human colorectal adenoma cell lines and human colorectal adenomas. Clin Cancer Res. 2006;12:4350–4356. doi: 10.1158/1078-0432.CCR-05-2487. [DOI] [PubMed] [Google Scholar]
- 31.Toscano F, Fajoui ZE, Gay F, Lalaoui N, Parmentier B, Chayvialle JA, et al. P53-mediated upregulation of DcR1 impairs oxaliplatin/TRAIL-induced synergistic anti-tumour potential in colon cancer cells. Oncogene. 2008;27:4161–4171. doi: 10.1038/onc.2008.52. [DOI] [PubMed] [Google Scholar]
- 32.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 33.Ndozangue-Touriguine O, Sebbagh M, Merino D, Micheau O, Bertoglio J, Breard J. A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene. 2008;27:6012–6022. doi: 10.1038/onc.2008.197. [DOI] [PubMed] [Google Scholar]
- 34.Shamimi-Noori S, Yeow WS, Ziauddin MF, Xin H, Tran TL, Xie J, et al. Cisplatin enhances the antitumor effect of tumor necrosis factor-related apoptosis-inducing ligand gene therapy via recruitment of the mitochondria-dependent death signaling pathway. Cancer Gene Ther. 2008;15:356–370. doi: 10.1038/sj.cgt.7701120. [DOI] [PubMed] [Google Scholar]
- 35.Arnould S, Guichard S, Hennebelle I, Cassar G, Bugat R, Canal P. Contribution of apoptosis in the cytotoxicity of the oxaliplatin-irinotecan combination in the HT29 human colon adenocarcinoma cell line. Biochem Pharmacol. 2002;64:1215–1226. doi: 10.1016/s0006-2952(02)01291-1. [DOI] [PubMed] [Google Scholar]
- 36.Lim SC, Choi JE, Kang HS, Iy Han S. Ursodeoxycholic acid switches oxaliplatin-induced necrosis to apoptosis by inhibiting reactive oxygen species production and activating p53-caspase 8 pathway in HepG2 hepatocellular carcinoma. Int J Cancer. 2009 doi: 10.1002/ijc.24853. [DOI] [PubMed] [Google Scholar]
- 37.Yang S, Guo ZS, O'Malley ME, Yin X, Zeh HJ, Bartlett DL. A new recombinant vaccinia with targeted deletion of three viral genes: its safety and efficacy as an oncolytic virus. Gene Ther. 2007;14:638–647. doi: 10.1038/sj.gt.3302914. [DOI] [PubMed] [Google Scholar]
- 38.Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67:10038–10046. doi: 10.1158/0008-5472.CAN-07-0146. [DOI] [PubMed] [Google Scholar]
- 40.Foloppe J, Kintz J, Futin N, Findeli A, Cordier P, Schlesinger Y, et al. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008;15:1361–1371. doi: 10.1038/gt.2008.82. [DOI] [PubMed] [Google Scholar]
- 41.Kulu Y, Dorfman JD, Kuruppu D, Fuchs BC, Goodwin JM, Fujii T, et al. Comparison of intravenous versus intraperitoneal administration of oncolytic herpes simplex virus 1 for peritoneal carcinomatosis in mice. Cancer Gene Ther. 2009;16:291–297. doi: 10.1038/cgt.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8:301–313. [PubMed] [Google Scholar]
- 43.Kaufman HL, Lenz HJ, Marshall J, Singh D, Garett C, Cripps C, et al. Combination chemotherapy and ALVAC-CEA/B7.1 vaccine in patients with metastatic colorectal cancer. Clin Cancer Res. 2008;14:4843–4849. doi: 10.1158/1078-0432.CCR-08-0276. [DOI] [PubMed] [Google Scholar]
- 44.Song CK, Han HD, Noh KH, Kang TH, Park YS, Kim JH, et al. Chemotherapy enhances CD8(+) T cell-mediated antitumor immunity induced by vaccination with vaccinia virus. Mol Ther. 2007;15:1558–1563. doi: 10.1038/sj.mt.6300221. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann D, Bangen JM, Bayer W, Wildner O. Synergy between expression of fusogenic membrane proteins, chemotherapy and facultative virotherapy in colorectal cancer. Gene Ther. 2006;13:1534–1544. doi: 10.1038/sj.gt.3302806. [DOI] [PubMed] [Google Scholar]
- 46.Harrop R, Drury N, Shingler W, Chikoti P, Redchenko I, Carroll MW, et al. Vaccination of colorectal cancer patients with TroVax given alongside chemotherapy (5-fluorouracil, leukovorin and irinotecan) is safe and induces potent immune responses. Cancer Immunol Immunother. 2008;57:977–986. doi: 10.1007/s00262-007-0428-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiocca EA, Smith KM, McKinney B, Palmer CA, Rosenfeld S, Lillehei K, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16:618–626. doi: 10.1038/sj.mt.6300396. [DOI] [PubMed] [Google Scholar]
- 48.Kumar S, Gao L, Yeagy B, Reid T. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther. 2008;10:371–379. [PubMed] [Google Scholar]
- 49.Hu W, Hofstetter W, Guo W, Li H, Pataer A, Peng HH, et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008;15:616–624. doi: 10.1038/cgt.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99:1768–1781. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 51.Fong Y, Kim T, Bhargava A, Schwartz L, Brown K, Brody L, et al. A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectal cancer. Mol Ther. 2009;17:389–394. doi: 10.1038/mt.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo ZS, Li Q, Bartlett DL, Yang JY, Fang B. Gene transfer: the challenge of regulated gene expression. Trends Mol Med. 2008;14:410–418. doi: 10.1016/j.molmed.2008.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.