

Abstract
New drugs with enhanced electron donor properties that target the ryanodine receptor from skeletal muscle sarcoplasmic reticulum (RyR1) are shown to be potent inhibitors of single-channel activity. In this article, we synthesize derivatives of the channel activator 4-chloro-3-methyl phenol (4-CmC) and the 1,4-benzothiazepine channel inhibitor 4-[-3{1-(4-benzyl) piperidinyl}propionyl]-7-methoxy-2,3,4,5-tetrahydro-1,4-benzothiazepine (K201, JTV519) with enhanced electron donor properties. Instead of activating channel activity (∼100 μM), the 4-methoxy analog of 4-CmC [4-methoxy-3-methyl phenol (4-MmC)] inhibits channel activity at submicromolar concentrations (IC50 = 0.34 ± 0.08 μM). Increasing the electron donor characteristics of K201 by synthesizing its dioxole congener results in an approximately 16 times more potent RyR1 inhibitor (IC50 = 0.24 ± 0.05 μM) compared with K201 (IC50 = 3.98 ± 0.79 μM). Inhibition is not caused by an increased closed time of the channel but seems to be caused by an open state block of RyR1. These alterations to chemical structure do not influence the ability of these drugs to affect Ca2+-dependent ATPase activity of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase type 1. Moreover, the FKBP12 protein, which stabilizes RyR1 in a closed configuration, is shown to be a strong electron donor. It seems as if FKBP12, K201, its dioxole derivative, and 4-MmC inhibit RyR1 channel activity by virtue of their electron donor characteristics. These results embody strong evidence that designing new drugs to target RyR1 with enhanced electron donor characteristics results in more potent channel inhibitors. This is a novel approach to the design of new, more potent drugs with the aim of functionally modifying RyR1 single-channel activity.
Introduction
The sarcoplasmic reticulum (SR) is an internal membrane system that controls the myoplasmic Ca2+ concentration and hence controls the contractile state of the muscle cell. A large number of chemically diverse compounds have been shown to either activate or inhibit the SR Ca2+ release channel. The common characteristic of most channel activators is their ability to act as electron acceptors, and common to the channel inhibitors are their electron donor characteristics. Moreover, there is a strong correlation between the strength of the electron donor/acceptor and its potency as a channel inhibitor/activator (Marinov et al., 2007). It occurred to us that this could serve as a basis and direction for development of new drugs targeting the RyR.
4-Chloro-3-methyl phenol (4-CmC) is a disinfectant and preservative that activates ryanodine binding and single-channel activity in skeletal and cardiac muscle SR at concentrations ranging from 50 to 400 μM (Herrmann-Frank et al., 1996). It also inhibits the Ca2+ pump protein from SR at low millimolar concentrations (Al-Mousa and Michelangeli, 2009). A large number of derivatives of 4-CmC, most of which are commercially available, have been shown to activate the RyR1 at various concentrations (Jacobson et al., 2006).
4-[-3{1-(4-Benzyl) piperidinyl}propionyl]-7-methoxy-2,3,4,5-tetrahydro-1,4-benzothiazepine) (K201, JTV519) is a benzothiazepine derivative that shows both antiarrhythmic and cardioprotective properties. These beneficial effects to the heart seem to be caused by its ability to decrease the Ca2+ leak mediated by the cardiac ryanodine receptor (RyR2). However, it is not specific in targeting the SR. K201 alters the gating of the dihydropyridine receptor (Kohno et al., 2003), inhibits annexin V-dependent Ca2+ fluxes (Kaneko et al., 1997), and has a natriuretic effect on the glomerular filtration rate (Lisy and Burnett, 2006). K201 also blocks the delayed rectifying K+ channel, which results in prolongation of the cardiac action potential (Kiriyama et al., 2000).
A substructure of K201, 7-methoxy-4-methyl-2,3,4,5-tetrahydro-1,4-benzothiazepine (S107) has been shown to enhance binding of FKBP12.6 to a R2474S mutant form of RyR2, inhibit the Ca2+ leak from RyR2 channels, and prevent cardiac arrhythmias. It was also shown that this drug fails to interact with other cardiac ion channels at concentrations up to 10 μM (Lehnart et al., 2008). Moreover, S107 prevents dissociation of the FKBP12-RyR1 complex and prevents a decline in exercise performance in skeletal muscle (Bellinger et al., 2008).
Exercise intolerance and skeletal muscle weakness are major limiting factors in humans with chronic heart failure. Protein kinase A hyperphosphorylation of RyR1, and the dissociation of the FKBP12-RyR1 complex have been implicated in defects in skeletal muscle intracellular Ca2+ handling and early fatigue in heart failure muscle (Wehrens et al., 2005). K201 has been shown to inhibit the reconstituted solubilized RyR1 with an IC50 of ∼25 μM and to induce subconductance states at positive holding potentials but not at negative potentials. In permeabilized skeletal muscle fibers, K201 also decreased spark frequency but increased the frequency of embers (Almassy et al., 2008).
In this study, we design two new derivatives of 4-CmC and K201 with enhanced electron donor properties and demonstrate that both new drugs act as potent inhibitors of RyR1, independent of the absence or presence of FKBP12. Moreover, these new drugs have no significant effect on channel closed time (τc). They primarily inhibit channel activity by decreasing the open time (τo) of the channel.
Materials and Methods
Sarcoplasmic Reticulum Preparation.
SR vesicles were isolated from rabbit fast-twitch skeletal muscle according to the method of MacLennan (1970) with small modifications; 50 μM dithiothreitol and 0.2 μg/ml leupeptin were added to all buffers except the final resuspension buffer. SR vesicles were then further fractionated on a discontinuous sucrose gradient (Salama and Abramson, 1984). The following sucrose solutions (percentage by weight) plus 10 mM HEPES, pH 7.0, were layered sequentially in an SW28 centrifuge tube (Beckman Coulter, Fullerton, CA): 4 ml of 45%, 7 ml of 40%, 12 ml of 35%, 7 ml of 30%, and 4 ml of 27%. Thirty milligrams of unfractionated SR were layered on top of the gradient and then spun at 22,000 rpm overnight. The heavy fraction was used in all single-channel experiments. All SR was stored in liquid N2 immediately after its preparation. The Portland State University institutional animal care and use committee approved all animal care and use protocols.
ATPase Activity.
Ca2+-dependent ATPase activity of SR vesicles in the presence of the Ca2+ ionophore A23187 was determined spectrophotometrically in the presence of various concentrations of drugs. The standard assay medium contained 100 mM KCl, 20 mM HEPES, 1 mM MgCl2, 1 mM EGTA, 500 μM NADH, 1 mM phosphoenolpyruvate, 5 units of lactate dehydrogenase, and 0.5 mM Mg-ATP at pH 7.0. The rate of NADH oxidation (equivalent to the rate of ATP hydrolysis) was monitored in a stepwise fashion by recording the decrease in absorbance at 340 nm as a function of time as described previously (Favero et al., 1995). The reaction was initiated by the addition of 0.1 mg/ml SR to the standard assay medium containing 1.0 mM EGTA (Ca2+-independent ATPase activity). ATPase activity was then recorded after the subsequent addition of CaCl2 to yield a free Ca2+ concentration of 8 μM and the Ca2+ ionophore A23187 (1 μg/ml). Ca2+ independent ATPase activity was subtracted from total activity (typically 20% of total activity) to yield Ca2+ dependent ATPase activity.
FKBP12 Protein.
The FKBP12 protein was overexpressed in Escherichia coli BL21 (DE3) cells and purified by Ni2+-agarose affinity chromatography using standard protocols (Van Lanen et al., 2005).
Planar Lipid Membranes.
Bilayers were made from a mixture of 5:3:2 phosphatidylethanolamine-phosphatidylserine-phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) painted across a 150-μm whole separating two compartments. The cis chamber to which 5 to 10 μg/ml SR was added contained 400 mM Cs+CH3O3S− and 20 mM HEPES, pH 7.4, whereas the trans side of the membrane contained 40 mM Cs+CH3O3S− and 20 mM HEPES, pH 7.4. After fusion of an SR vesicle to the bilayer, 4 M Cs+CH3O3S− and 20 mM HEPES, pH 7.4, were added to the trans side of the membrane to equalize the salt concentration on the two sides of the membrane to 400 mM. Channel output was filtered at 800 to 1000 Hz, and traces of not less than 3 min were recorded after addition of various concentrations of channel modulators. Under these filtering conditions, channel open and closed times can be resolved down to ∼0.5 ms. The average open time of our control single-channel recordings was 0.6 ms. Given these experimental conditions, we were not able to directly measure decreases in τo. However,
where Po is the open probability, To is the total open time, and Tc is the total closed time. Moreover, To = Nτo, and Tc = Nτc, where N is the number of channel openings and the number of channel closings, τo is the average open time, and τc is the average closed time. Therefore,
By measuring Po and τc, one can calculate the corresponding value of τo. If a channel inhibitor has no significant effect on τc, as Po decreases, τo must decrease. All single-channel analysis was performed using the ClampFit program from the pClamp software suite (Molecular Devices, Sunnyvale, CA). Changes in τc were directly measured, and the corresponding changes in τo were calculated under conditions in which Po had not decreased to <50% of control. Changes in τc and Po were considered significant using a Student's t test if p < 0.05.
Electron Donor Measurements.
Electron donor measurements were performed as described by Marinov et al. (2007). All measurements were performed under continuous illumination by visible light in a buffer containing 1 mM Tris, 10 μM methylene blue, and 100 μM XTT, pH 7.4, at room temperature. Visible light excited the dye (methylene blue), which led to the sequential formation of a singlet and then a triplet state. Two triplets combined to form a dye anion and dye cation radical pair. Electron donors donated electrons to the dye cation radical and decreased their concentration. This increased the lifetime of the dye anion radical. In the presence of O2, the dye anion radical reduced O2, which yielded superoxide. XTT is a probe that readily reacts with superoxide (Sutherland and Learmonth, 1997). Its reduction by superoxide resulted in a time-dependent increase in absorbance at 470 nm (Olojo et al., 2005). More potent electron donors increased the rate and amount of reduction of XTT.
The electron acceptor properties of a compound were assayed in the presence of a sacrificial electron donor (i.e., 100 μM EGTA). Continuous illumination of methylene blue in the presence of EGTA resulted in the time-dependent production of superoxide and a large time-dependent reduction of XTT. In the presence of an increasing concentration of an electron acceptor, the rate of reduction of O2 to form superoxide decreased, and the XTT signal (Abs470 nm) decreased. The initial rate of reduction of XTT (in the presence of 100 μM EGTA) was plotted versus the concentration of the electron acceptor, and the data were fit to a four-parameter logistic curve (SigmaPlot version 10). The IC50 derived corresponds to the concentration of the electron acceptor at which half-maximal donor activity (referenced to EGTA) was observed.
Assay for Dissociation of the FKBP12-RyR1 Complex.
SR vesicles (1.0 mg/ml) in buffer containing 250 mM KCl, 15 mM NaCl, 20 mM Pipes, pH 7.1, and 50 μM CaCl2 were incubated with 30 μM hexadecahydro-5,19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone (FK-506), 30 μM FK-506 + 20 μM K201, or an equivalent amount of dimethyl sulfoxide as a control for 30 min at 37°C. Samples were then pelleted at 167,000g for 20 min in a Airfuge (Beckman Coulter). The supernatant and pellets were brought up to equivalent volumes. Samples were then solubilized in nonreducing sample buffer containing 62.5 mM Tris, pH 6.8, 10% sucrose, 4% SDS, and 0.02% bromphenol blue and run onto a 4 to 12% SDS-polyacrylamide gel electrophoresis gradient gel. Proteins were then electrophoretically transferred onto polyvinylidene difluoride membranes at a constant 15 V for 60 min using a Trans-Blot Semi-Dry Transfer Cell Apparatus (Bio-Rad Laboratories, Hercules, CA). Membranes were incubated for 1 h in blocking buffer (5% nonfat dry milk in TBST) at 23°C with agitation. They were then incubated overnight with a monoclonal antibody to FKBP12 (MAB3777; R&D Systems, Minneapolis, MN) at 1 μg/ml in TBST. Membranes were then washed three times for 10 min in TBST and incubated for 2 h with an anti-rat IgG horseradish peroxidase-linked secondary antibody at a 1:10,000 dilution (HAF005; R&D Systems). Secondary antibody was detected using a SuperSignal West Pico Luminescent Kit (Thermo Fisher Scientific, Waltham, MA). Western Blot imaging and integration were performed using an Alpha Innotech Fluorchem SP system (Quansys Biosciences, Logan, UT).
Preparation of K201, K201 Dioxole Derivative, and 4-Methoxy-3-Methyl Phenol.
K201 was prepared according to the procedure reported by Wehrens et al. (2004). The dioxole derivative of K201 (3) was synthesized as shown in Scheme 1, following the same procedure as that for K201, but modifying the last step in which now the brominated thiazepine (1) reacts with commercially available 4-benzo[1,3]dioxol-5-ylmethyl-piperidine (2). For 3-(4-(benzo[d][1,3]dioxol-5-ylmethyl)piperidin-1-yl)-1-(7-methoxy-2,3-dihydrobenzo[f][1,4]thiazepin-4(5H)-yl)propan-1-one (3), the reaction mixture of 1 (0.100 g), 4-benzo[1,3]dioxol-5-ylmethyl-piperidine (2, 0.086 g), and Na2CO3 (0.086 g) in dimethylformamide (5 ml) was stirred at 60°C for 12 h. Dimethylformamide was evaporated under vacuum, and the product was extracted with ethyl acetate, washed with H2O, and dried over anhydrous Na2SO4. After filtration and removal of solvents, the crude product was purified by column chromatography. 1H NMR (400 mHz, CDCl3) δ (ppm): 1.11 to 2.95 (17H, m), 3.78 (3H, s), 3.92 to 4.15 (2H, m), 4.70 (2H, s), 5.92 (2H, s), 6.57 to 7.50 (6H, m). 1H NMR spectra were acquired on an ARX-400 Advance spectrometer (Bruker, Newark, DE).
Scheme 1.

Synthesis of the dioxole derivative of K201. Inset shows the structure of 4-MmC.
4-MmC was prepared following the procedure reported by Higgins et al. (2001), starting from 3-methyl-p-anisaldehyde. Purification of the product was achieved by flash chromatography and confirmed by 1H NMR. The structure of 4-MmC is shown in the inset of Scheme 1.
Results
We have previously shown that activators of the RyR from sarcoplasmic reticulum are electron acceptors, whereas RyR inhibitors are electron donors. Moreover, more potent channel activators are stronger electron acceptors and more potent channel inhibitors are stronger electron donors (Marinov et al., 2007). In Fig. 1, we demonstrate that 4-CmC is an electron acceptor. In Fig. 1A, 100 μM EGTA is used as a sacrificial electron donor. As described under Materials and Methods, illumination of methylene blue with visible light results in the production of anion/cation radical pairs. EGTA readily donates electrons to cation radicals, which increases the lifetime of the dye anion radical. The dye anion radical passes electrons to O2, which results in the formation of superoxide (O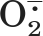 ). When XTT is reduced by superoxide, the absorbance at 470 nm increases (Sutherland and Learmonth, 1997). In the presence of an electron acceptor, such as 4-CmC, the dye anion radical passes electrons to the electron acceptor and the amount of superoxide generated decreases. The time-dependent reduction of XTT was fit to an exponential rise to a maximum value (y = A1 · (1 − e−k1t) + A2 · (1 − e−k2t) (Fig. 1A), and the initial rate of reduction of XTT was determined by the product of k1 · A1 + k2 · A2. In Fig. 1B, the decrease in the initial rate of reduction of XTT is plotted versus [4-CmC] and fit to a four-parameter logistic curve. The IC50 derived, which corresponds to the concentration of 4-CmC, at which half-maximal electron acceptor activity is observed is 21.2 ± 2.0 μM. This value is similar to the concentration of 4-CmC at which the RyR1 is half-maximally activated (Herrmann-Frank et al., 1996).
). When XTT is reduced by superoxide, the absorbance at 470 nm increases (Sutherland and Learmonth, 1997). In the presence of an electron acceptor, such as 4-CmC, the dye anion radical passes electrons to the electron acceptor and the amount of superoxide generated decreases. The time-dependent reduction of XTT was fit to an exponential rise to a maximum value (y = A1 · (1 − e−k1t) + A2 · (1 − e−k2t) (Fig. 1A), and the initial rate of reduction of XTT was determined by the product of k1 · A1 + k2 · A2. In Fig. 1B, the decrease in the initial rate of reduction of XTT is plotted versus [4-CmC] and fit to a four-parameter logistic curve. The IC50 derived, which corresponds to the concentration of 4-CmC, at which half-maximal electron acceptor activity is observed is 21.2 ± 2.0 μM. This value is similar to the concentration of 4-CmC at which the RyR1 is half-maximally activated (Herrmann-Frank et al., 1996).
Fig. 1.

4-Chloro-3-methyl phenol is an electron acceptor. A, time-dependent reduction of XTT in the presence of 100 μM EGTA (●) plus the following concentrations of 4-CmC: 5 μM (○), 10 μM (▾), 20 μM (▵), 50 μM (■), 100 μM (□), or 200 μM (♦). B, initial rate of XTT reduction versus [4-CmC]: data derived from A. Data shown are means ± S.E. (n = 3).
In Fig. 2 it is shown that replacement of the chloro in position 4 with a methoxy group converts 4-CmC from an electron acceptor (Fig. 1) into 4-methoxy-3-methyl phenol (4-MmC), which acts as a potent electron donor. A comparison of the electron donor properties of several relevant compounds at 50 μM is shown in Fig. 2. The same order of electron donor effectiveness (FKBP12 > 4-MmC > dioxole derivative of K201 > K201) was observed at lower concentrations tested (10 μM). Moreover, as shown in Fig. 3, 4-MmC is a potent inhibitor of single-channel activity of RyR1. Figure 3A shows representative single-channel traces as a function of 4-MmC concentration, whereas Fig. 3B shows the normalized Po as a function of concentration added to the cis chamber of the bilayer. The calculated IC50 for inhibition of single-channel activity by 4-MmC is 0.34 ± 0.08 μM. Conversion of an arene such as 4-CmC from an electron acceptor into an electron donor results in the conversion of a channel activator into a potent channel inhibitor.
Fig. 2.

Comparison of electron donor properties of control (no addition; ●), 50 μM K201 (○), 50 μM dioxole derivative of K201 (▾), 50 μM 4-MmC (▵), and 50 μM FKBP12 (■). All time-dependent scans were repeated at least three times under continuous illumination. Values shown are means ± S.E (n = 3). Curves shown are best fits to an exponential rise to maximum.
Fig. 3.

RyR1 is inhibited by 4-MmC. A, representative single-channel traces as a function of [4-MmC]: 400 mM CsCH3O3S, 20 mM HEPES, pH 7.4, on both sides of the bilayer, 50 μM Ca2+ cis at a holding potential of −36 mV. Channel openings are shown as downward deflections (C, closed state; O, open state). B, open probability normalized to 1.0 versus [4-MmC] (●). C, closed time histogram normalized to 1 versus [4-MmC]. Data shown are means ± S.E. (n = 11). Data are expressed relative to 0 μM 4-MmC with Po = 0.27 ± 0.06 and τc = 1.22 ± 0.44 ms.
It has recently been shown that the two RyR2 inhibitors, tetracaine and flecainide, differ in their mode of action. Whereas tetracaine inhibits channel activity by increasing channel closed time, flecainide decreases channel open time but hase little effect on the average closed time of RyR2 (Watanabe et al., 2009; Hilliard et al., 2010). In Fig. 3C, we show that 4-MmC, in a manner similar to that of flecainide, shows no significant effect on τc of RyR1 at concentrations as high as 1.0 μM (where Po has decreased to ∼30% of control). Under these conditions, using eq. 3, τo decreases to 16% of the control value before addition of 4-MmC.
A similar approach to developing new drugs with enhanced electron donor properties targeting RyR1 focused on the drug K201. K201 and its dioxole derivative were synthesized as described under Materials and Methods. As expected, the dioxole derivative of K201 is a more potent electron donor than is K201 (Fig. 2). Moreover, as shown in Fig. 4, representative traces at the single-channel level (Fig. 4, A and B) and a plot of the normalized Po versus concentration (Fig. 4C) demonstrates that the dioxole derivative of K201 (IC50 = 0.24 ± 0.05 μM) is approximately 16 times more potent in closing down RyR1 than is K201 (IC50 = 3.98 ± 0.79 μM). In a manner similar to that of 4-MmC, both K201 (Fig. 4D) and its dioxole derivative (Fig. 4E) decrease RyR1 open probability without significantly increasing the closed time of the channel.
Fig. 4.

Dioxole derivative of K201 is a more potent inhibitor of the RyR1 than is K201. A, representative single-channel traces as a function of [K201]. B, Single-channel traces as a function of [dioxole derivative of K201]. C, open probability normalized to 1.0 for control is plotted versus [K201] (●) and versus the [dioxole derivative of K201] (○). D, closed time histogram normalized to 1 versus [K201]. E, versus [dioxole derivative of K201]. Data shown are means ± S.E. (n = 8). D, data are expressed relative to 0 μM K201 with Po = 0.11 ± 0.06 and τc = 4.50 ± 1.37 ms. E, data are expressed relative to 0 μM dioxole derivative of K201 with Po = 0.05 ± 0.02 and τc = 1.31 ± 0.33 ms. Experimental conditions are identical to those described in the legend to Fig. 3.
The proposal that K201 stabilizes the RyR by causing the reassociation of the FKBP12-RyR1 complex in skeletal muscle and the FKBP12.6-RyR2 complex in cardiac muscle (Marx et al., 2000) has been challenged by a number of laboratories (Yano et al., 2003; Hunt et al., 2007; Blayney et al., 2010). In Figs. 5 and 6, it is shown that the addition of 30 μM FK-506 results in complete dissociation of the FKBP12-RyR1 complex (Fig. 5). The FKBP12 protein moves from the pellet fraction in the control (lane 2) to the supernatant fraction (lane 3) after treatment with 30 μM FK-506. Addition of 30 μM FK-506 + 20 μM K201 does not result in reassociation of the RyR1/FKBP12 complex. The FKBP12 remains in the supernatant fraction (lane 5). At the single-channel level, the addition of 30 μM FK-506 causes a 4- to 8-fold stimulation of the channel Po (Fig. 6), yet the Ki for inhibition of the normalized single-channel activity in the absence (Ki = 3.98 ± 0.79 μM) or presence of FK-506 (4.6 ± 0.43 μM) is unchanged. Inhibition of channel activity by K201 is not affected by the presence or absence of FKBP12.
Fig. 5.

FK-506 dissociates the FKBP12-RyR1 complex independent of K201. SR vesicles were treated with 30 μM FK-506 ± 20 μM K201. Pellets and supernatants were run on a 4 to 12% polyacrylamide gel, transferred to polyvinylidene difluoride and treated with a monoclonal antibody to FKBP12 and a horseradish peroxidase secondary antibody as described under Materials and Methods. Lanes 1 and 2, control; lanes 3 and 4, treated with 30 μM FK-506; lanes 5 and 6, treated with 30 μM FK-506 + 20 μM K201. Lanes 1,3, and 5 are supernatant fractions. Lanes 2, 4, and 6 are pellet fractions. Normalized integrated densities are as follows: lane 1, 0.00; lane 2, 1.00; lane 3, 1.02; lane 4, 0.01; lane 5, 1.06; lane 6, 0.00. This transfer was repeated three times with identical findings.
Fig. 6.

K201 inhibition of single-channel activity is independent of FKBP12. A, representative single-channel traces of control after treatment with 30 μM FK-506 and after the addition of K201 at increasing concentrations. B, normalized open probability versus [K201] in the absence (●, n = 7) and presence of 30 μM FK-506 (○, n = 6). Data shown are means ± S.E. Open probability of control (no K201) = 0.039 ± 0.011 (n = 13) and in the presence of FK-506 = 0.154 ± 0.045 (n = 7). Experimental conditions are identical to those described in the legend to Fig. 3.
The specificity of new drugs targeting the RyR is often an important issue in drug development. The nonspecific interactions between K201 and multiple targets have limited its clinical usefulness. In Fig. 7 it is shown that K201 and its dioxole derivative inhibit the Ca2+-ATPase activity of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase type 1 at similar concentrations. The IC50 for K201 = 54.8 ± 12.9 μM, whereas the IC50 for the dioxole derivative of K201 = 48.1 ± 7.0 μM (mean ± S.E.). The enhanced electron donor properties of the dioxole derivative affects the potency of this drug to inhibit the RyR1, but it does not affect the potency of this drug to inhibit Ca2+-ATPase activity. Moreover, as can be seen in Fig. 7, 4-MmC has no effect on ATPase activity at concentrations as high as 0.5 mM.
Fig. 7.

Ca2+-dependent ATPase activity versus concentration of K201 (●), the dioxole derivative (○), and 4-methoxy-3-methyl phenol (▾). Data shown are means ± S.E. (n = 3).
Discussion
In this study we synthesized two new potent RyR1 inhibitors that are derivatives of the known RyR1 modulators, 4-CmC and K201. 4-CmC activates RyR1 at a concentration range from 50 to 400 μM (Herrmann-Frank et al., 1996). By replacing the chloro in the 4 position with a methoxy group, we have converted an electron acceptor (Fig. 1) into an electron donor (Fig. 2) and converted a channel activator into a potent channel inhibitor (Fig. 3). In a similar manner, synthesizing the dioxole derivative of K201 converts a RyR1 inhibitor into a more potent electron donor and significantly increases its effectiveness as a channel inhibitor. This nontraditional approach toward drug design was motivated by our earlier observations that all RyR activators tested were electron acceptors, whereas channel inhibitors were electron donors. Moreover, there was a strong correlation between the effectiveness of these drugs and their potency as either electron donors (inhibitors) or acceptors (activators) (Marinov et al., 2007).
K201 is a strong electron donor (Fig. 2), which inhibits both the skeletal (Almassy et al., 2008) and cardiac muscle RyR (Kohno et al., 2003; Wehrens et al., 2004). It has been shown to have both antiarrhythmic and cardioprotective properties. These beneficial effects on the heart seem to be caused by its ability to decrease the “Ca2+ leak” mediated by the cardiac ryanodine receptor (RyR2). It has also been shown that K201 decreases single-channel activity of the reconstituted RyR1 isolated from rat skeletal muscle SR (IC50 = ∼25 μM) (Almassy et al., 2008). Moreover, subconductance states of RyR1 were observed at low concentrations of K201, which were associated with enhanced Ca2+ fluxes across actively loaded SR vesicles (Almassy et al., 2008). There are several differences between the K201 data described in our study and the previous work carried out by Almassy et al. (2008). All of the bilayer reconstitution work described in this study was done with rabbit fast-twitch muscle SR vesicles that were fused with an artificial membrane as described under Materials and Methods. The SR vesicles were not exposed to detergent. In the article by Almassy et al., SR vesicles were isolated from rat skeletal muscle and then were fractionated on a sucrose gradient in the presence of the nonionic detergent CHAPS before reconstitution in a similar planar bilayer membrane. The IC50 for inhibition of single-channel activity by K201 is ∼6 times lower in the present study, and in none of our studies did we observe K201-induced subconductance states as described by Almassy et al. (2008). The RyR1 is a large multiprotein complex. A number of associated proteins have been shown to functionally regulate activity. It is likely that breaking up of this complex by addition of CHAPS alters the sensitivity to inhibition by K201 and results in subconductance states such as those that occur when FKBP12 dissociates from the RyR1 (Ahern et al., 1997).
It has been shown by Marks' group (Wehrens et al., 2005) that K201 stabilizes the closed state of both RyR1 and RyR2 by increasing the binding of FKBP12 to RyR1 and FKBP12.6 to RyR2 in a heart failure model. This interpretation has been challenged by the observations that K201 almost completely inhibits the Ca2+ leak from normal dog heart SR under conditions in which the FKBP12.6 has completely dissociated from RyR2 (i.e., in the presence of 30 μM FK-506) (Yano et al., 2003). Moreover, K201 inhibits store overload-induced Ca2+ release in human embryonic kidney 293 cells expressing RyR2 either with or without FKBP12.6 (Hunt et al., 2007). In contrast to the initial hypothesis that K201 stabilizes the RyR by increasing the binding of the FKBP protein, it has recently been shown, using surface plasmon resonance techniques, that K201 in the closed conformation of RyR1/RyR2 reduces the binding affinity of the FKBP12-RyR1 and FKBP12.6-RyR2 complexes (Blayney et al., 2010). It does not strengthen this interaction as originally postulated. Although 30 μM FK-506 increases the channel open probability ∼4- to 8-fold (Fig. 6) and completely displaces FKBP12 from RyR1 (Fig. 5), the potency of K201 as a RyR1 inhibitor is unaffected by the presence or absence of the associated FKBP12 (Fig. 6B). Moreover, RyR1 does not reassociate with FKBP12 in the presence of 20 μM K201, a concentration that significantly inhibits single-channel activity (Fig. 6). This result does not address the issue of whether K201 causes the FKBP12-RyR complex to reassociate in a heart failure model, but it does demonstrate that inhibition of RyR1 by K201 is independent of the presence or absence of FKBP12.
In an alternative model, Yamamoto and Ikemoto (2002) have proposed that the closed state of the RyR is stabilized by interactions between the N-terminal and central domain of the RyR and that mutations to the RyR2 lead to “unzipping” of these two domains, resulting in a Ca2+ leak. The binding of K201 to domain2114–2149 has been proposed to correct defective interdomain interactions within RyR2 and decrease the Ca2+ leak associated with the failing heart (Yamamoto and Ikemoto, 2002). Mutations to Q4020K4021 on RyR1 (corresponding to amino acids 3976–3977 in RyR2) abolishes channel activation by 4-CmC (Fessenden et al., 2006). If 4-MmC binds to the same region of RyR1 as 4-CmC, then the inhibitory action of 4-MmC is remote from the interdomain region and the action of 4-MmC is unlikely to involve the direct stabilization or “zipping” of the RyR1 as proposed for the action of K201. Moreover, it is difficult to understand how activation of channel activity by 4-CmC might result in unzipping of the N-terminal and central core domains, whereas binding of 4-MmC results in domain zipping, unless the domain interactions are sensitive to the electron acceptor/donor properties of these compounds that seem to bind in a region distant from interdomain interaction.
It is well established that thiol-reducing agents inhibit RyR1, whereas thiol-oxidizing agents activate the release channel. Reduction of endogenous disulfides on the RyR1 inhibits the flux of Ca2+ across the SR (Trimm et al., 1986), channel activity, and high-affinity ryanodine binding (Zable et al., 1997). The data presented in this article show that increasing the electron donor properties of drugs targeting the RyR1 results in new potent inhibitors of channel activity. Moreover, the role of electron donors in closing down RyR1 seems to be evident at the level of protein/protein interactions. As shown in Fig. 2, the purified FKBP12 protein is a more potent electron donor than is K201, its dioxole derivative, or 4-MmC. This result suggests that FKBP12 stabilizes RyR1 in its closed configuration by virtue of its electron donor properties. Removal or decreased binding of FKBP12 to RyR1 results in a Ca2+ leak, which can be reversed by addition of any of a number of drugs that act as strong electron donors. The redox activity of drugs and proteins that interact with the Ca2+ release channel is a key factor in modifying function of the SR. The observation that novel drugs with enhanced electron donor properties act as more potent inhibitors of RyR1 supports the role of redox reactions in channel gating and presents a new methodology for designing new, more potent channel inhibitors.
In a previous publication, we proposed that nonthiol electron acceptors or donors form a reversible charge transfer complex upon binding to RyR1 (Marinov et al., 2007). Channel activators (electron acceptors) were shown to shift the redox potential of RyR1 to more negative redox potentials (RyR1 oxidizes more readily), and the thiol/disulfide balance of the receptor shifts to a more oxidized state. In contrast, channel inhibitors (electron donors) shift the redox potential of the RyR to a more positive potential and the thiol/disulfide balance shifts to a more reduced state (the SH/S–S ratio increases) (Marinov et al., 2007). Consistent with the formation of a charge transfer complex, we have observed that the inhibitory effects of the dioxole derivative of K201 at the single-channel level are reversible (not shown). Changing solutions after addition of the dioxole derivative restores channel activity.
The approach taken to design new drugs with enhanced electron donor properties has resulted in the development of new, more potent inhibitors of the sarcoplasmic reticulum Ca2+ release channel. It is not yet known whether a similar approach can be applied to other protein targets. Previous studies have shown that inhibitors of the L-type Ca2+ channel (i.e., nifedipine, verapamil, and diltiazem) are electron donors (Marinov and Saxon, 1985), whereas local anesthetics, antiarrhythmic agents, and some anticonvulsants, which inhibit Na+ channels, are also electron donors (Marinov, 1985). In contrast, both K201 and its dioxole derivative, with enhanced electron donor properties, show similar potency for inhibiting ATPase activity of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase type 1.
Acknowledgments
The expression vector for the FKBP12 protein was a generous gift of Professor Susan L. Hamilton (Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX). We thank Professor Dirk Iwata-Reuyl and Dr. Vimbai Masiyanise (Department of Chemistry, Portland State University, Portland, OR) for assistance in expressing the FKBP12 protein.
This work was supported by the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant R01-AR48911] (to J.J.A.); the National Institutes of Health National Institute of Biomedical Imaging and Bioengineering [Grant R01-EB002044] (to R.M.S.); the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM082940] (to J.D.S.); and the Portland State University Faculty development award and University Venture Development Fund; Office of Naval Research–Oregon Nanoscience and Microtechnologies Institute [Grant N00014-11-1-0193];.
J.J.A. and R.M.S. are founding members of ELEX Biotech LLC, a start-up company that is developing drugs targeting RyR1 and RyR2.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.074740.
- SR
- sarcoplasmic reticulum
- 4-CmC
- 4-chloro-3-methyl phenol
- RyR1
- ryanodine receptor type 1
- K201
- (4-[-3{1-(4-benzyl) piperidinyl}propionyl]-7-methoxy-2,3,4,5-tetrahydro-1,4-benzothiazepine, JTV519
- S107
- 7-methoxy-4-methyl-2,3,4,5-tetrahydro-1,4-benzothiazepine
- RyR2
- ryanodine receptor type 2
- FKBP12
- 12-kDa FK-506 binding protein
- FK-506
- hexadecahydro-5,19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone
- XTT
- 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide sodium salt
- Pipes
- piperazine-N,N′-bis (2-ethanesulfonic acid)
- TBST
- Tris-buffered saline-Tween 20
- 4-MmC
- 4-methoxy-3-methyl-phenol
- CHAPS
- 3-[(3-cholamido-propyl) dimethylammonio]-1-propanesulfonate.
Authorship Contributions
Participated in research design: Ye, Yaeger, Owen, Escobedo, Wang, Singer, Strongin, and Abramson.
Conducted experiments: Ye, Yaeger, Owen, Escobedo, Wang, Singer, Strongin, and Abramson.
Contributed new reagents or analytic tools: Escobedo, Wang, Singer, and Strongin.
Performed data analysis: Ye, Yaeger, Owen, Escobedo, Wang, Singer, Strongin, and Abramson.
Wrote or contributed to the writing of the manuscript: Ye, Owen, Escobedo, Singer, Strongin, and Abramson.
References
- Ahern GP, Junankar PR, Dulhunty AF. (1997) Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys J 72:146–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mousa F, Michelangeli F. (2009) Commonly used ryanodine receptor activator, 4-chloro-m-cresol (4CmC), is also an inhibitor of SERCA Ca2+ pumps. Pharmacol Rep 61:838–842 [DOI] [PubMed] [Google Scholar]
- Almassy J, Sztretye M, Lukacs B, Dienes B, Szabo L, Szentesi P, Vassort G, Csernoch L, Jona I. (2008) Effects of K-201 on the calcium pump and calcium release channel of rat skeletal muscle. Pflugers Arch 457:171–183 [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, et al. (2008) Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA 105:2198–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blayney LM, Jones JL, Griffiths J, Lai FA. (2010) A mechanism of ryanodine receptor modulation by FKBP12/12.6, protein kinase A, and K201. Cardiovasc Res 85:68–78 [DOI] [PubMed] [Google Scholar]
- Favero TG, Zable AC, Abramson JJ. (1995) Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J Biol Chem 270:25557–25563 [DOI] [PubMed] [Google Scholar]
- Fessenden JD, Feng W, Pessah IN, Allen PD. (2006) Amino acid residues Gln4020 and Lys4021 of the ryanodine receptor type 1 are required for activation by 4-chloro-m-cresol. J Biol Chem 281:21022–21031 [DOI] [PubMed] [Google Scholar]
- Herrmann-Frank A, Richter M, Lehmann-Horn F. (1996) 4-Chloro-m-cresol: a specific tool to distinguish between malignant hyperthermia-susceptible and normal muscle. Biochem Pharmacol 52:149–155 [DOI] [PubMed] [Google Scholar]
- Higgins L, Korzekwa KR, Rao S, Shou M, Jones JP. (2001) An assessment of the reaction energetics for cytochrome P450-mediated reactions. Arch Biochem Biophys 385:220–230 [DOI] [PubMed] [Google Scholar]
- Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. (2010) Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol 48:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, Shimoni Y, Chen SR. (2007) K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J 404:431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson AR, Moe ST, Allen PD, Fessenden JD. (2006) Structural determinants of 4-chloro-m-cresol required for activation of ryanodine receptor type 1. Mol Pharmacol 70:259–266 [DOI] [PubMed] [Google Scholar]
- Kaneko N, Ago H, Matsuda R, Inagaki E, Miyano M. (1997) Crystal structure of annexin V with its ligand K-201 as a calcium channel activity inhibitor. J Mol Biol 274:16–20 [DOI] [PubMed] [Google Scholar]
- Kiriyama K, Kiyosue T, Wang JC, Dohi K, Arita M. (2000) Effects of JTV-519, a novel anti-ischaemic drug, on the delayed rectifier K+ current in guinea-pig ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol 361:646–653 [DOI] [PubMed] [Google Scholar]
- Kohno M, Yano M, Kobayashi S, Doi M, Oda T, Tokuhisa T, Okuda S, Ohkusa T, Kohno M, Matsuzaki M. (2003) A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure 1. Am J Physiol Heart Circ Physiol 284:H1035–H1042 [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, et al. (2008) Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest 118:2230–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisy O, Burnett JC., Jr (2006) New cardioprotective agent K201 is natriuretic and glomerular filtration rate enhancing. Circulation 113:246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan DH. (1970) Purification and properties of an adenosine triphosphatase from sarcoplasmic reticulum. J Biol Chem 245:4508–4518 [PubMed] [Google Scholar]
- Marinov BS. (1985) Na+ channel antagonists act as electron donors while agonists act as electron acceptors with free radicals. FEBS Lett 191:159–162 [Google Scholar]
- Marinov BS, Olojo RO, Xia R, Abramson JJ. (2007) Non-thiol reagents regulate ryanodine receptor function by redox interactions that modify reactive thiols 1. Antioxid Redox Signal 9:609–621 [DOI] [PubMed] [Google Scholar]
- Marinov BS, Saxon ME. (1985) Dihydropyridine Ca2+ agonists and channel blockers interact in the opposite manner with photogenerated unpaired electrons. FEBS Lett 186:251–254 [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376 [DOI] [PubMed] [Google Scholar]
- Olojo RO, Xia RH, Abramson JJ. (2005) Spectrophotometric and fluorometric assay of superoxide ion using 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole. Anal Biochem 339:338–344 [DOI] [PubMed] [Google Scholar]
- Salama G, Abramson J. (1984) Silver ions trigger Ca2+ release by acting at the apparent physiological release site in sarcoplasmic reticulum. J Biol Chem 259:13363–13369 [PubMed] [Google Scholar]
- Sutherland MW, Learmonth BA. (1997) The tetrazolium dyes MTS and XTT provide new quantitative assays for superoxide and superoxide dismutase. Free Radic Res 27:283–289 [DOI] [PubMed] [Google Scholar]
- Trimm JL, Salama G, Abramson JJ. (1986) Sulfhydryl oxidation induces rapid calcium release from sarcoplasmic reticulum vesicles. J Biol Chem 261:16092–16098 [PubMed] [Google Scholar]
- Van Lanen SG, Reader JS, Swairjo MA, de Crécy-Lagard V, Lee B, Iwata-Reuyl D. (2005) From cyclohydrolase to oxidoreductase: discovery of nitrile reductase activity in a common fold. Proc Natl Acad Sci USA 102:4264–4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. (2009) Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15:380–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. (2004) Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304:292–296 [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, et al. (2005) Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA 102:9607–9612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Ikemoto N. (2002) Peptide probe study of the critical regulatory domain of the cardiac ryanodine receptor. Biochem Biophys Res Commun 291:1102–1108 [DOI] [PubMed] [Google Scholar]
- Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, et al. (2003) FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 107:477–484 [DOI] [PubMed] [Google Scholar]
- Zable AC, Favero TG, Abramson JJ. (1997) Glutathione modulates ryanodine receptor from skeletal muscle sarcoplasmic reticulum. Evidence for redox regulation of the Ca2+ release mechanism. J Biol Chem 272:7069–7077 [DOI] [PubMed] [Google Scholar]