

Abstract
V-ATPases function as ATP-dependent ion pumps in various membrane systems of living organisms. ATP hydrolysis causes rotation of the central rotor complex, which is composed of the central axis D subunit and a membrane c ring that are connected by F and d subunits. Here we determined the crystal structure of the DF complex of the prokaryotic V-ATPase of Enterococcus hirae at 2.0-Å resolution. The structure of the D subunit comprised a long left-handed coiled coil with a unique short β-hairpin region that is effective in stimulating the ATPase activity of V1-ATPase by twofold. The F subunit is bound to the middle portion of the D subunit. The C-terminal helix of the F subunit, which was believed to function as a regulatory region by extending into the catalytic A3B3 complex, contributes to tight binding to the D subunit by forming a three-helix bundle. Both D and F subunits are necessary to bind the d subunit that links to the c ring. From these findings, we modeled the entire rotor complex (DFdc ring) of V-ATPase.
Ion-transporting rotary ATPases are divided into three types based on their function and taxonomic origin: F-, V-, and A-type ATPases. F-ATPases function as ATP synthases in mitochondria, chloroplasts, and oxidative bacteria (1). V-ATPases function as proton pumps in the membranes of acidic organelles and plasma membranes of eukaryotic cells (2). A-ATPases in archaea function as ATP synthases similar to the F-ATPases (the “A” designation in A-type refers to archaea), although the structure and subunit composition of A-ATPases are more similar to those of V-ATPases (3). These ATPases possess an overall similar structure that is composed of a hydrophilic catalytic portion (F1-, V1-, or A1-ATPase) and a membrane-embedded ion-transporting portion (Fo-, Vo-, or Ao-ATPase), and they have a similar reaction mechanism that occurs through rotation (2).
V-ATPases are found in bacteria, such as Thermus thermophilus and Enterococcus hirae. In T. thermophilus, V-ATPase physiologically functions as an ATP synthase (4). Therefore, it has sometimes been called an A-ATPase, although T. thermophilus is a eubacterium that does not belong to archaea (5). However, the E. hirae V-ATPase acts as a primary ion pump (6), which transports Na+ or Li+ instead of H+ (7). The enzyme is composed of nine subunits (Eh-A, -B, -d, -D, -E, -F, -G, -a, -c; previously designated as NtpA, -B, -C, -D, -E, -G, -F, -I, -K) having amino acid sequences that are homologous to those of the corresponding subunits (A, B, d, D, E, F, G, a, c) of eukaryotic V-ATPases (7) (see Fig. 1). The core of the V1 domain of this ATPase is composed of a hexameric arrangement of alternating A and B subunits responsible for ATP binding and hydrolysis (8). The Vo domain, in which rotational energy is converted to drive Na+ translocation, is composed of oligomers of the 16-kDa c subunits and an a subunit (9, 10). The V1 and Vo domains are connected by a central stalk, which is composed of D, F, and d subunits, and two peripheral stalks, which are composed of E and G subunits of V1 (8, 11) (Fig. 1). ATP hydrolysis induces the rotation of the central stalk (DFd complex) and an attached membrane c ring, which causes ion pumping at the interface between the c ring and a subunit (9). To understand the precise function and rotational mechanism of V-ATPase, the details of these subunit structures and their subunit–subunit interactions should be elucidated.
Fig. 1.

Schematic model of E. hirae V-ATPase. Previous names of the corresponding subunits of E. hirae V-ATPase are in parentheses.
The crystal structures of several subunits (C and F) and subcomplexes (A3B3 and EG) of T. thermophilus V-ATPase have been obtained at high resolution (5, 12–14). The crystal structure of the Tt-A3B3DF complex of the ATP synthase was recently obtained, although the structure was built as a polyalanine model at 4.5-Å resolution, and the foot portion (residues 56–131) of the axial D subunit was missing (15). Thus, the atomic structure of the D subunit of V-ATPase has not yet been obtained at high resolution, and the detailed functional and structural relationships of the central stalk subunits have not yet been elucidated. Here we report the crystal structure of the DF complex of E. hirae V-ATPase at 2.0-Å resolution and the biochemical properties of the axial complex such as subunit–subunit interactions. The crystal structure revealed a unique β-hairpin region in the D subunit, which was related to the ATPase activity. The structural motif of the left-handed coiled coil of D subunit, which was stabilized by binding to F subunit, was similar to the motifs of other known rotary complexes including that of bacterial flagellum.
Results and Discussion
Sample Preparation and Crystal Structural Analysis.
We previously reported the in vitro reconstitution of the catalytic complex (A3B3DF) of E. hirae V-ATPase from individual subunits, which were expressed using Escherichia coli cell-free protein expression system or were expressed in vivo in E. coli (16). Eh-D was unstable during purification. However, the Eh-DF complex was successfully purified by the coexpression of both the Eh-D and Eh-F, indicating that the binding of Eh-F stabilized Eh-D. The V1 complex that was reconstituted using this Eh-DF showed high ATPase activity equivalent to the native V1-ATPase, which was purified from the cell membrane of E. hirae by EDTA treatment (16). In this study, we crystallized the selenomethionine derivative of the Eh-DF. The V1 complex with the labeled Eh-DF retained a similar level of ATPase activity (Table 1). The initial phases were determined by multiwavelength anomalous diffraction method (MAD) and refined up to 2.0-Å resolution. Data collection and refinement statistics are summarized in Table S1. The final model contains residues 7–194 of Eh-D and 1–101 of Eh-F, 152 water molecules, and a nitrate ion at the crystal contact site.
Table 1.
ATPase activity of the Eh-A3B3 and Eh-A3B3D complexes with excess of the following proteins
| Eh-A3B3 + excess of following protein | ATPase activityμmol Pi/ min per mg A3B3 | Ratio% |
| No addition | 0.6 ± 0.1 | |
| Eh-DF | 7.3 ± 0.5 | 100 |
| SeMet labeled Eh-DF | 7.6 ± 0.7 | 104 |
| Eh-DmF | 3.4 ± 0.3 | 47 |
| EhA3-B3D + excess of following protein | ATPase activityμmol Pi/ min per mg A3B3D | Ratio% |
| No addition | 6.2 ± 0.4 | 100 |
| Eh-F | 7.5 ± 0.6 | 121 |
| Eh-FΔCα | 6.0 ± 0.6 | 97 |
| Eh-F + Eh-d | 7.3 ± 0.4 | 118 |
SeMet, selenomethionine.
Overall Structure of the DF Complex.
The overall structure of the Eh-DF is shown in Fig. 2A. The structure of Eh-D comprises a long pair of α-helices (approximately 100 Å) having N-terminal residues 9–69 and C-terminal residues 127–193, which are twisted into a left-handed coiled coil. The remaining part (residues 70–126) of the Eh-D is composed of a short β-hairpin region formed by residues 89–108, and two flexible loops (residues 82–88 and 114–126) with two short α-helices (residues 74–81 and 111–113), which as a whole connect the two long α-helices (Fig. 2B). Eh-F is bound to the middle portion of Eh-D (Fig. 2A). The N-terminal domain is composed of an α/β-fold with four β-strands (β1, 4–8; β2, 23–25; β3, 45–51; β4, 72–76) and three α-helices (H1, 11–20; H2, 30–42; H4, 59–66). The four β-strands are parallel to each other and separated by the three α-helices (Fig. 2C). The N-terminal domain is connected by a flexible loop (residues 77–84) to a C-terminal α-helix (residues 85–97), which forms a three-helix bundle with the coiled coil of Eh-D (Fig. 2A). The dissociation constant (Kd) of the Eh-A3B3D and Eh-F was estimated to be 3.2 nM by surface plasmon resonance analysis (Fig. S1A). The contact surfaces of the two subunits are about 2,000 Å2 and constituted of hydrophobic interactions on center of the buried surface, 16 hydrogen bonds, and three salt bridges on periphery of the buried surface (Fig. S1B) (except a hydrogen bond between L143 of Eh-D and Y49 of Eh-F which is located inside the buried surface).
Fig. 2.

Structure of the DF complex of the E. hirae V1-ATPase. (A) Cartoon representation of the Eh-DF complex. Eh-D and Eh-F are shown in green and dark red, respectively. (B) Eh-D is presented in blue to red from N to C terminus. (C) Eh-F is colored in the same manner as in B.
Structure and Function of the D Subunit.
The long coiled-coil helices (residues 1–55, 132–205) of the D subunit (Tt-D) of the T. thermophilus V1-ATPase were modeled as polyalanine in the electron density map at 4.5-Å resolution (15). The twisted structure is similar to that of Eh-D (Fig. 3A). The two structures can be superimposed with an rmsd of 2.35 Å for 79 Cα atoms. The amino acid sequences of these regions are similar to those of V- and A-ATPases (Fig. S2). The middle portion of the coiled coil, in particular, is highly conserved, as shown in Fig. 3B. This region is formed mainly by the hydrophobic residues, A22, G25, L28, L29, K30, K32, L36, I160, T163, R165, R166, V167, N168, A169, E171, and I175. Thus, the coiled-coil structure seems to be conserved in eukaryotic V-ATPases and A-ATPases. The short β-hairpin region (residues 89–108) in the middle portion of Eh-D has not been interpreted in the reported structure of Tt-D (15). The superimposed structure (Fig. 3A) shows that the β-hairpin region seems to make contact with the C-terminal domain of Tt-A and/or Tt-B, implying that this region is important for the ATPase activity of the V1-ATPase.
Fig. 3.

Structural similarity of the D subunit. (A) Superimposed structures of Eh-D (green) and Tt-D (gray) of V1-ATPase from T. thermophilus (PDB ID 3A5D). The adjacent Tt-A is shown in blue. The β-hairpin region (residues 90–108) that was deleted in mutation experiments is shown in the red-dotted box. (B and C) Conserved residues of the D subunit. The figures were generated using ConSurf (35). Residues are colored in accordance with conservation among the aligned amino acid sequences of the D subunit from seven species in Fig. S2.
We found a strong similarity between Eh-D and γ-subunit of bovine F1-ATPase [Protein Data Bank (PDB) ID: 1E79] using the DALI server (17). The left-handed coiled-coil structures are similar to each other, although no apparent sequence similarity between these subunits has been observed, and the remaining parts of these subunits show no structural similarities (18) (Fig. S3A). The coiled-coil structure is also similar to that of FliJ, which is a component of the flagellar type III protein export apparatus (19) (Fig. S3B). Thus, the antiparallel, left-handed coiled-coil structure seems to be conserved in these rotary complexes, but the β-hairpin region of Eh-D is unique and seems to be important for specific functions of V-ATPases.
Function of the β-Hairpin Region of the D Subunit.
To understand the function of the β-hairpin region of Eh-D, we synthesized a deletion mutant that lacked the β-hairpin region (residues 90–108) and replaced two linker residues as “GS” (Fig. 3A and Fig. S2) and purified the mutated Eh-D (Eh-Dm) with Eh-F. We could not purify Eh-Dm without Eh-F, indicating that these two subunits made a stable complex similar to the wild-type Eh-DF complex. We assessed the binding affinity of the Eh-DmF to Eh-A3B3 and its effect on the ATPase activity. The Kd of the Eh-DmF interacting with the Eh-A3B3 was approximately 4.6 nM, whereas that of the wild-type Eh-DF was approximately 0.8 nM (Fig. S4 A and B), indicating that the β-hairpin region was not essential for the binding of the Eh-DF to the Eh-A3B3. The Km for the ATP of the Eh-A3B3DmF complex was similar to that of the wild-type complex (Fig. S4 C and D). However, the maximal velocity of the ATPase activity for the mutant complex decreased to approximately half compared with that of the wild-type V1-ATPase (Table 1 and Fig. S4 C and D). These findings indicate that the β-hairpin region is not essential for ATPase activity, but it stimulates the activity approximately two times.
Structure and Function of the F Subunit.
The overall structure of the Eh-DF complex is similar to that of the T. thermophilus V1-ATPase at 4.5-Å resolution (rmsd = 2.44 Å for 60 Cα atoms of F subunit) (15). The C-terminal region of Eh-F, which forms a three-helix bundle with the coiled coil of Eh-D, seems to make contact with the C-terminal domain of Tt-B (Fig. 4A), implying that the C-terminal α-helix plays an important role in V1-ATPase activity. The crystal structure of Tt-F has been solved at 2.2-Å resolution, and the structure was interpreted as an artificial domain-swapped dimer in the extended form (13). The C-terminal portion containing the C-terminal α-helices and β4-strand (residues 75–109) of Tt-F was shown to change its conformation into an extended form in the presence of ATP (13). In contrast, the structure of the F subunit (Mm-F) of the A-type ATP synthase of Methanosarcina mazei obtained by NMR indicated that the C-terminal α-helix region (without β4) solely was in an extended form and fluctuating (20). In addition, the low-resolution structure of the F subunit of yeast V-ATPase obtained by small-angle X-ray scattering also indicated that the C-terminal α-helix had an extended conformation (21). We compared these structures of the F subunit with that of the T. thermophilus V1-ATPase (Fig. 4B). The N-terminal portions (α1–3, β1–3) of all of the structures are similar to each other. The C-terminal portion that contains β4 of the extended form of the T. thermophilus F subunit seems to make contact with Tt-B. On the other hand, the shorter C-terminal portion without β4 of the Mm-F seems to make contact with a different Tt-B (Fig. 4B).
Fig. 4.

Structural similarity of the F subunit. (A) Superimposed structures of Eh-F (dark pink) and the F subunit (cyan) of V1-ATPase from T. thermophilus (PDB ID 3A5D). The adjacent Tt-B is shown in violet. (B) Structures of Tt-F (orange) extended form (PDB ID 2D00) and Mm-F (green) are superimposed in A. (C) The sequence alignment of the F subunits from different species (E. hirae, T. thermophilus HB8, Methanosarcina mazei Gö1, Saccharomyces cerevisiae, and Homo sapiens). The secondary structures of Eh-F are shown above the sequence. The deleted regions of the mutations are indicated in the green box.
Function of the C-Terminal α-Helix of the F Subunit.
To elucidate the function of the C-terminal portion of the F subunit in V1- or A1-ATPase, we constructed two deletion mutants that were deleted from the β4-strand (Eh-FΔβ4Cα) and from the C-terminal α-helix (Eh-FΔCα), as shown in Fig. 4C. The shorter F mutant (Eh-FΔβ4Cα) formed an inclusion body and could not be purified. Eh-FΔCα could be expressed and purified. However, when both Eh-FΔCα and Eh-D were expressed, the Eh-D formed an inclusion body and could not be purified as a complex, indicating that the C-terminal helix of Eh-F was important for binding to Eh-D. The binding affinity (Kd = 520 nM; Fig. S5) between Eh-A3B3D and Eh-FΔCα decreased approximately 160 times compared with that in wild-type Eh-F (Kd = 3.2 nM; Fig. S1). The ATPase activity of the Eh-A3B3 was stimulated by approximately 20% on the addition of excess purified Eh-F (Table 1), which was also observed in the studies of the T. thermophilus V1-ATPase (22). However, the addition of excess Eh-FΔCα did not further affect the ATPase activity (Table 1). Thus, the C-terminal α-helix of the F subunit is important for tight binding to the D subunit and for the stimulation of ATPase activity, but it probably does not undergo any conformational changes for the regulation of the activity.
Coupling of the DF Complex and the d Subunit.
The crystal structure of a C subunit (corresponding to the d subunit) of the T. thermophilus V1-ATPase has been solved at 1.95-Å resolution (12). Cross-linking experiments revealed that the C subunit (Tt-d) is attached to both the F subunit and the membrane ring. Therefore, this subunit seems to act as a socket for the attachment of the central stalk subunits of V1-ATPase onto the membrane ring of Vo-ATPase (12) (Fig. 1). Here we examined the protein–protein interactions between V1-ATPase and the d subunit by using the Biacore system. We used the Eh-A3B3D as the ligand on the Biacore chip. When Eh-d was passed over this chip, on which the Eh-A3B3D was fixed, no interaction signal was detected (Fig. S6A). However, an interaction signal was obtained after binding Eh-F on the Eh-A3B3D (Fig. S6A), indicating that Eh-d interacted with the Eh-DF. The Kd value was estimated as 82 nM (Fig. S6B). We did not observe any binding interactions between the Eh-F and Eh-d by gel filtration or the Biacore assay, in which Eh-d was fixed onto the chip. Therefore, we concluded that the Eh-d interacts with the Eh-DF complex.
The crystal structure of Tt-d has threefold pseudosymmetry and a cone-like shape (12). The sequence of Eh-d is 22% identical and 55% similar to that of the Tt-d, and it is likely to have a similar structure (Fig. S7A). The Eh-d structure was created by homology modeling according to the template structure of Tt-d (PDB ID 1R5Z) using the LOOPP (Learning, Observing, and Outputting Protein Patterns) server (23) (Fig. 5B and Fig. S7B). The homology model exhibited considerably different electrostatic potentials on the surface from that of the template structure: The surface of Eh-d except the concave face was rich in acidic amino acids (Fig. 5B and Fig. S7 C and D). The electrostatic potential of the bottom of the Eh-DF was also acidic, whereas only small portion in the concave face of the Eh-d was basic (Fig. 5 A and B). Therefore, the acidic bottom of Eh-DF was docked manually in this small basic pit of Eh-d (Fig. 5D). Most of the surfaces of both Eh-DF and Eh-d were acidic, implying that the binding at these surfaces is inhibited by electrostatic repulsion. The binding affinity (Kd = 82 nM) of Eh-d and Eh-DF was lower compared with other subunit–subunit interactions (A3B3-D, Kd = 0.8 nM; A3B3D-F, Kd = 3.2 nM). Previous studies on eukaryotic V-ATPases showed that the enzyme was regulated by the reversible dissociation and reassociation of V1-ATPase (2). The weak interaction between the DF complex and d subunit might be responsible for the unique regulation of V-ATPases.
Fig. 5.

Electrostatic potential of the rotor complex. The electrostatic potential surfaces were calculated using APBS (36) and mapped at contouring levels from -3 kT (blue) to 3 kT (red). (A) Eh-DF (Upper, bottom view; Lower, side view). (B) Homology modeled Eh-d (Upper, top view; Lower, side view). (C) Eh-c ring (Upper, top view; Lower, side view). (D) The model of the entire rotor complex.
We have solved the crystal structure of the membrane Eh-c ring previously (9). The electrostatic potential of the central cavity of the membrane Eh-c ring was basic (Fig. 5C). Therefore, the acidic convex face of the Eh-d is expected to bind to this basic cavity. The Eh-d was docked manually into the Eh-c ring, where the convex face of the Eh-d penetrates deeply into the central cavity of the ring, according to our previous docking model of Tt-d and Eh-c ring (9). In Fig. 5D, we show this current structure model of entire rotor complex of V-ATPase. Recently, an interesting model was reported based on the asymmetric structure of averaged cryo-EM images of T. thermophilus V-ATPase, where the convex face of the Tt-d penetrates only slightly into the Tt-c ring with the narrowest point of Tt-d contacting one edge of the ring (24). This feature seems to be important for the coupling mechanism of V-ATPases. Further investigation for protein–protein interactions between Eh-d and Eh-c ring will be needed to elucidate this aspect.
Materials and Methods
Protein Preparation.
An E. coli cell-free protein expression system, as described elsewhere (25), was used to synthesize the Eh-d, -F, and -DF polypeptides using plasmids harboring the corresponding genes or a mixture of plasmids for two genes. We used selenomethionine to synthesize the Eh-DF to facilitate X-ray crystallographic analysis. More than 15 mg of the complex was synthesized with this system, using 27 mL of the reaction solution in the presence of 0.1 mg of plasmids (25). The expressed protein was purified as previously described (11). The mutagenesis of Eh-D and -F were performed using the QuikChange site-directed mutagenesis kit (Agilent Technologies). We expressed Eh-A, -B, and -D in E. coli and purified according to a previous report (16). Eh-D was unstable during purification, and we could not concentrate it to more than 0.1 mg of protein per mL. Therefore, a low concentration of purified Eh-D was immediately reconstituted with Eh-A3B3 to form the Eh-A3B3D complex. The Eh-A3B3DF complex was reconstituted according to a previous report (16).
Binding Assay by Surface Plasmon Resonance.
The Biacore T100 protein interaction analysis system and sensor chips were obtained from GE Healthcare. Eh-B was purified without treatment with tobacco etch virus (TEV) protease, thereby retaining the His6-tag. Using this His-tagged B subunit, the Eh-A3B3 (or Eh-A3B3D) was reconstituted in the presence of adenosine 5’-[β,γ-imido]triphosphate, as reported previously (16), and it was immobilized as a ligand on a sensor chip to bind His-tagged proteins (Series S sensor chip NTA; GE Healthcare). Then, several subunits or complexes made contact with the chip as analytes to detect any interaction with the proteins on the chip. Proteins were used at concentrations ranging from 2 nM to 5.5 μM in a buffer (pH 6.5) composed of 20 mM MES-Tris, 150 mM NaCl, 50 μM EDTA, and 0.005% Tween 20. Measurements were performed according to manufacturer’s instructions. Kd was determined using the BIAevaluation software (version 1.1), which employs the Langmuir isotherm model that assumes a 1∶1 binding stoichiometry.
Crystallization of the Eh-DF Complex and Determination of Its Structure.
The selenomethionine-labeled crystals of the Eh-DF complex were obtained at 293 K by the sitting drop vapor diffusion method, which was achieved by mixing 0.5 μL of protein solution [10 mg/mL protein solution in 20 mM Tris·HCl (pH 8.0), 150 mM NaCl, and 2 mM DTT] with 0.5 μL reservoir solution [0.1 M MES-Tris (pH 6.5), 18% PEG-3350, and 0.2 M sodium nitrate]. X-ray diffraction datasets (wavelengths 0.9791, 0.9794, and 1.000 Å) were obtained using an MX225HE X-ray CCD detector (Rayonix) at a cryogenic temperature (100 K) on the beamline BL41XU at SPring-8 (Harima, Japan). The collected datasets were indexed and integrated using Mosflm (ver. 7.0.4) and scaled using Scala from CCP4 packages (26). The crystal belonged to the space group C2 with the following unit cell parameters: a = 105.79, b = 68.43, and c = 51.15 Å, and β = 114.99°. A heterodimer was contained in the crystallographic asymmetric unit (Vm = 2.34 Å3/Da, 47.4% solvent content). The crystal structure of the Eh-DF complex was determined by MAD method. Six out of 11 Se atoms were found using SOLVE/RESOLVE (27). The overall figure of merit [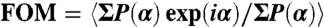 , where P(α) is the probability distribution for the phase (α)] was 0.42 after MAD phasing and 0.60 after density modification. The initial model building was performed automatically using ARP/wARP (28). The three-dimensional model was manually modified using Coot (29) and iteratively refined using CNS (Crystallography and NMR System) (30) and REFMAC5 (31). The Ramachandran plot produced using RAMPAGE (32) revealed that 97.5% residues were in the favored region, 2.5% in the allowed region, and 0.0% in the outlier region. All molecular graphics were generated using PyMOL (http://www.pymol.org/).
, where P(α) is the probability distribution for the phase (α)] was 0.42 after MAD phasing and 0.60 after density modification. The initial model building was performed automatically using ARP/wARP (28). The three-dimensional model was manually modified using Coot (29) and iteratively refined using CNS (Crystallography and NMR System) (30) and REFMAC5 (31). The Ramachandran plot produced using RAMPAGE (32) revealed that 97.5% residues were in the favored region, 2.5% in the allowed region, and 0.0% in the outlier region. All molecular graphics were generated using PyMOL (http://www.pymol.org/).
Other.
ATPase activity was measured according to a previous report using the ATP regeneration system (33). Protein concentrations were determined using the DC Protein Assay Kit (Bio-Rad Laboratories) with bovine serum albumin used as the standard. SDS-PAGE was performed according to Laemmli (34) and stained with Coomassie brilliant blue. All other chemicals were obtained from Sigma-Aldrich or Wako Pure Chemical Industries.
Supplementary Material
Acknowledgments.
We thank Mio Inoue and Mari Aoki for technical support of DNA construction. The synchrotron radiation experiments were performed at SPring-8 and Photon Factory (Proposals 2008S2-001 and 2009G660). We also thank the beamline staff at BL41XU of SPring-8 (Harima, Japan) and NE3A of Photon Factory Advanced Ring (PF-AR; Tsukuba, Japan) for help during data collection. This work was supported by Targeted Proteins Research Program, grants-in-aid (18074003, 23118705, 23370047), Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science, and Technology of Japanese Government, and Hamaguchi Foundation for the Advancement of Biochemistry.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and the structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3AON).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108810108/-/DCSupplemental.
References
- 1.Walker JE. ATP synthesis by rotary catalysis (Nobel Lecture) Angew Chem Int Ed Engl. 1998;37:2308–2319. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2308::AID-ANIE2308>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 2.Forgac M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 3.Schäfer G, Engelhard M, Müller V. Bioenergetics of the archaea. Microbiol Mol Biol Rev. 1999;63:570–620. doi: 10.1128/mmbr.63.3.570-620.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toei M, et al. Dodecamer rotor ring defines H+/ATP ratio for ATP synthesis of prokaryotic V-ATPase from Thermus thermophilus. Proc Natl Acad Sci USA. 2007;104:20256–20261. doi: 10.1073/pnas.0706914105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee LK, Stewart AG, Donohoe M, Bernal RA, Stock D. The structure of the peripheral stalk of Thermus thermophilus H+-ATPase/synthase. Nat Struct Mol Biol. 2010;17:373–378. doi: 10.1038/nsmb.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heefner DL, Harold FM. ATP-driven sodium pump in Streptococcus faecalis. Proc Natl Acad Sci USA. 1982;79:2798–2802. doi: 10.1073/pnas.79.9.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murata T, Igarashi K, Kakinuma Y, Yamato I. Na+ binding of V-type Na+-ATPase in Enterococcus hirae. J Biol Chem. 2000;275:13415–13419. doi: 10.1074/jbc.275.18.13415. [DOI] [PubMed] [Google Scholar]
- 8.Murata T, Takase K, Yamato I, Igarashi K, Kakinuma Y. Properties of the V0V1 Na+-ATPase from Enterococcus hirae and its V0 moiety. J Biochem. 1999;125:414–421. doi: 10.1093/oxfordjournals.jbchem.a022302. [DOI] [PubMed] [Google Scholar]
- 9.Murata T, Yamato I, Kakinuma Y, Leslie AGW, Walker JE. Structure of the rotor of the V-Type Na+-ATPase from Enterococcus hirae. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- 10.Murata T, et al. Ion binding and selectivity of the rotor ring of the Na+-transporting V-ATPase. Proc Natl Acad Sci USA. 2008;105:8607–8612. doi: 10.1073/pnas.0800992105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto M, et al. Interaction and stoichiometry of the peripheral stalk subunits NtpE and NtpF and the N-terminal hydrophilic domain of NtpI of Enterococcus hirae V-ATPase. J Biol Chem. 2008;283:19422–19431. doi: 10.1074/jbc.M801772200. [DOI] [PubMed] [Google Scholar]
- 12.Iwata M, et al. Crystal structure of a central stalk subunit C and reversible association/dissociation of vacuole-type ATPase. Proc Natl Acad Sci USA. 2004;101:59–64. doi: 10.1073/pnas.0305165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makyio H, et al. Structure of a central stalk subunit F of prokaryotic V-type ATPase/synthase from Thermus thermophilus. EMBO J. 2005;24:3974–3983. doi: 10.1038/sj.emboj.7600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher MJ, et al. Crystal structure of A3B3 complex of V-ATPase from Thermus thermophilus. EMBO J. 2009;28:3771–3779. doi: 10.1038/emboj.2009.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Numoto N, Hasegawa Y, Takeda K, Miki K. Inter subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase. EMBO Rep. 2009;10:1228–1234. doi: 10.1038/embor.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arai S, et al. Reconstitution in vitro of the catalytic portion (NtpA3-B3-D-G complex) of Enterococcus hirae V-type Na+-ATPase. Biochem Biophys Res Commun. 2009;390:698–702. doi: 10.1016/j.bbrc.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Kääriäinen S, Rosenström P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbons C, Montgomery MG, Leslie AG, Walker JE. The structure of the central stalk in bovine F1-ATPase at 24 Å resolution. Nat Struct Biol. 2000;7:1055–1061. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 19.Ibuki T, et al. Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat Struct Mol Biol. 2011;18:277–282. doi: 10.1038/nsmb.1977. [DOI] [PubMed] [Google Scholar]
- 20.Gayen S, Vivekanandan S, Biuković G, Grüber G, Yoon HS. NMR solution structure of subunit F of the methanogenic A1AO adenosine triphosphate synthase and its interaction with the nucleotide-binding subunit B. Biochemistry. 2007;46:11684–11694. doi: 10.1021/bi701102n. [DOI] [PubMed] [Google Scholar]
- 21.Basak S, et al. Solution structure of subunit F (Vma7p) of the eukaryotic V1VO ATPase from Saccharomyces cerevisiae derived from SAXS and NMR spectroscopy. Biochim Biophys Acta. 2011;1808:360–368. doi: 10.1016/j.bbamem.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Imamura H, Funamoto S, Yoshida M, Yokoyama K. Reconstitution in vitro of V1 complex of Thermus thermophilus V-ATPase revealed that ATP binding to the A subunit is crucial for V1 formation. J Biol Chem. 2006;281:38582–38591. doi: 10.1074/jbc.M608253200. [DOI] [PubMed] [Google Scholar]
- 23.Vallat BK, et al. Building and assessing atomic models of proteins from structural templates: Learning and benchmarks. Proteins. 2009;76:930–945. doi: 10.1002/prot.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau WC, Rubinstein JL. Structure of intact Thermus thermophilus V-ATPase by cryo-EM reveals organization of the membrane-bound V(O) motor. Proc Natl Acad Sci USA. 2010;107:1367–1372. doi: 10.1073/pnas.0911085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kigawa T, et al. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J Struct Funct Genomics. 2004;5:63–68. doi: 10.1023/B:JSFG.0000029204.57846.7d. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative Computational Project N. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Lovell SC, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 33.Murata T, Kakinuma Y, Yamato I. ATP-dependent affinity change of Na+-binding sites of V-ATPase. J Biol Chem. 2001;276:48337–48340. doi: 10.1074/jbc.M106821200. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.