

Abstract
Activation of the water molecule involved in GTP hydrolysis within the HRas⋅RasGAP system is analyzed using a tailored approach based on hybrid quantum mechanics/molecular mechanics (QM/MM) simulation. A new path emerges: transfer of a proton from the attacking water molecule to a second water molecule, then a different proton is transferred from this second water molecule to the GTP. Gln61 will stabilize the transient OH− and H3O+ molecules thus generated. This newly proposed mechanism was generated by using, for the first time to our knowledge, the entire HRas-RasGAP protein complex in a QM/MM simulation context. It also offers a rational explanation for previous experimental results regarding the decrease of GTPase rate found in the HRas Q61A mutant and the increase exhibited by the HRas Q61E mutant.
Introduction
HRas protein is the most representative member of the Ras subfamily of small GTPases, a form of soluble G-proteins involved in signal transduction (1). G-proteins usually behave as molecular switches; they shift between an active conformation, bound to GTP, and an inactive GDP-bound conformation. In keeping with their function, these proteins present very low intrinsic GTPase activity and low catalytic efficiency (Kcat/Km ratio); these characteristics lengthen the lifetime of the active state (2). In the case of HRas, GTP hydrolysis is modulated by the presence of a second protein, the GTPase activating protein RasGAP. RasGAP stabilizes the HRas conformation and supplies an additional arginine residue that helps to place a water molecule in a position that facilitates catalysis (3), thus leading to GTP hydrolysis and subsequent HRas inactivation.
Despite all the existing data on the structure and functionality of the HRas⋅RasGAP complex, the catalytic mechanism of GTP activation and hydrolysis is still controversial (4). Different mechanisms have been proposed.
One based on the initial crystal structure (5) suggested that the water molecule was activated by the Gln61 residue, which acts as the general base proton acceptor. The position of Gln61 is conserved in the family of guanine nucleotide binding proteins (6) and it is known that mutation of Gln61 by Ala reduces the rate of GTP hydrolysis (7), whereas substitution of Gln61 by Glu, which is considered a better proton acceptor, increases the rate of GTP hydrolysis (8).
Some computational studies have simulated GTP hydrolysis on this basis (9). The conclusions, obtained using an ab initio 6-31 basis set but including only 28 residues in the MM region of the simulation instead of the whole protein, involved an interaction between the amide group of Gln61 and the carboxylate group of Glu63, which increases the proton withdrawal potential of Gln61. This was supported by experiments using an E63K mutant protein (10). The role of Gln61 as a proton acceptor has, however, been questioned by other studies (11) that suggest that the GTP molecule acts as the general base for its own hydrolysis (12, 13). In such a substrate-assisted catalysis mechanism, Gln61 would play a role in the stabilization of the transition state (14).
Finally, more recent reports of the prehydrolysis state of the protein obtained using a cryo-technique suggested that a second water molecule is involved in the catalytic mechanism (15), adding an element to the process that, in addition to the introduction of the whole protein structure in the simulations, can be now used to explore alternative ways.
Using a newly developed approach based on the quantum mechanics/molecular mechanics (QM/MM) method, and applying it to the whole HRas-RasGAP protein complex (Fig. 1), we have modeled the activation of the attacking water molecule—a process leading to GTP protonation and subsequent GTP hydrolysis.
Figure 1.
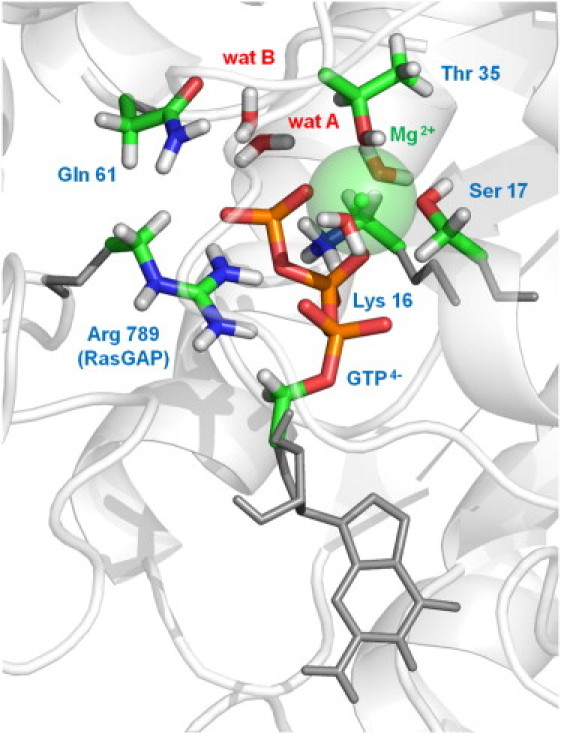
Atoms in the QM/MM system. Atoms in the QM region are represented as thick sticks surrounded by the rest of the molecules in the active center included in the MM region (thin sticks) and part of the protein system also in the MM region (ribbons).
Materials and Methods
Molecular dynamics simulations
The system used in our simulations was based on the x-ray structure of the HRas⋅RasGAP complex (Protein DataBank code 1WQ1 (16)). It includes the whole HRas protein and residues 714–1047 of the catalytic domain of the GTPase activating protein RasGAP. Aluminum fluoride and GDP molecules contained in the crystal structure to mimic the geometry of the transition state of the active center were replaced by a GTP molecule, maintaining all common atoms in the same position as in the crystal structure. The distance between the oxygen of the catalytic water molecule (HOH numbered as “230” in PDB structure 1WQ1) and Pγ of GTP was kept at 2–3 Å by an imposed restraint. A second water molecule was placed in the active site according to the crystal structure of the prehydrolysis state obtained by Scheidig et al. (15). K+ ions were added as counterions to neutralize the negative charge of the system and placed in a shell around the system using a Coulombic potential in a grid. The neutralized complexes were then immersed in a rectangular parallelepiped solvent box and a distance of 12 Å was maintained between the wall of the box and the closest atom of the solute.
The counterions and the solvent molecules were added using the LEaP module of AMBER (17, 18). Before the QM/MM simulation itself, 10 ns of unrestricted molecular dynamics (MD) were applied to the system to obtain an equilibrated initial structure, using the PMEMD algorithm of AMBER10 (17, 18) and the parm99 parameter set (17). Initial relaxation of the system was achieved by performing 10,000 steps of energy minimization using a cutoff of 10.0 Å. Subsequently, and to start the MD simulations, the temperature was raised from 0 to 298 K in a 500-ps heating phase, and velocities were reassigned at each new temperature according to a Maxwell-Boltzmann distribution. During this period, the dihedral of the Cα trace were restrained with a force constant of 500 kcal mol−1 rad−2. During the last 200 ps of the equilibration phase of the MD, the force constant was reduced stepwise to 0. The SHAKE algorithm was used throughout to constrain all the hydrogen bonds to their equilibrium values so that an integration time step of 2 fs could be employed. The list of nonbonded pairs was updated every 25 steps, and coordinates were saved every 2 ps. Periodic boundary conditions were applied and electrostatic interactions were represented using the smooth particle mesh Ewald method with a grid spacing of ∼1 Å.
Quantum mechanics/molecular mechanics simulations
After initial relaxation using MD procedures, as described above, quantum mechanics/molecular mechanics simulations (QM/MM) simulations were performed using the Sander module of AMBER10 (17, 18). The hybrid QM/MM approach is a suitable method for simulating processes, such as enzymatic reactions, in which chemical bonds are formed and broken. The method requires the partitioning of the system in two regions: quantum mechanics (QM) and molecular mechanics (MM). Calculations involving the atoms belonging to the QM region were performed using the PM3 semiempirical Hamiltonian. The atoms in the system that were not part of the QM region (the MM region) were treated in a classical MM way. In our system, the QM region included the two water molecules involved in catalysis and the GTP atoms from the γ-phosphate group up to the C5′-C4′ bond. It also included the Mg2+ ion and all the oxygen atoms involved in its coordination sphere, including the hydroxyl groups of the Ser17 and Thr35 residues and two coordinating water molecules. Side chains of the Lys16 and Gln61 residues of HRas and the chain of Arg789 belonging to the RasGAP protein were also included (atoms in the QM region are represented in Fig. 1).
The QM region contained 75 atoms including six link H-atoms used to maintain the integrity of the covalent bonds sectioned by the QM/MM boundary (17). In the case of the experiment illustrated in Fig. 2, carboxylate atoms of Glu63 were also included in QM region. The conformation obtained after MD procedures (see above) was equilibrated again for 200 ps using this QM/MM approach. During this equilibration, constraints corresponding to all the covalent bonds between the atoms of the QM region were maintained. All the constraints, except those corresponding to the parameters of the reaction and to the maximum allowable distance position of the catalytic water, were removed progressively over the next 100 ps. In all cases, position of the catalytic water molecule was stable under QM/MM simulation without position restrains. The average values of significant geometrical parameters of the QM/MM system are summarized in Table 1. SHAKE was not used for either the MM region or the QM region. Due to the presence of a peptide bond in the side chain of the Gln61 residue, an MM correction to the peptide linkages was used. A cutoff of 8 Å was used to calculate the QM/MM electrostatic interactions. The extra Gaussian terms that were used in the PM3 Hamiltonian to improve the core-core repulsion term in QM-QM interactions were also included for the QM-MM interactions.
Figure 2.
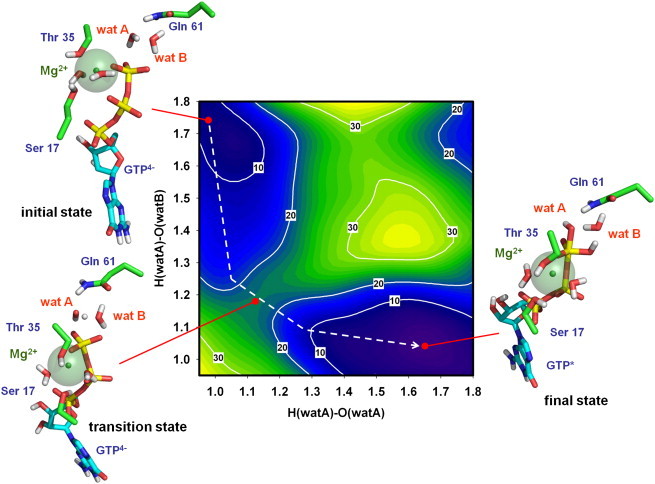
Free energy landscape for the activation of the catalytic water molecule supposing Gln61 as proton acceptor assisted by Glu63. ΔG° values obtained for the whole system are shown for the different states corresponding to the distances: x axis, from proton to oxygen atom in the attacking water molecule (wat A); y axis, from the same proton to the oxygen in ɛ1 position of Gln61. The structure of the active center in the initial state (minimum ΔG°GTP4− value adjusted to 0), transition state (ΔG°GTP4- ≈ 35 kcal·mol−1), and final state (ΔG°GTP4- ≈ 25 kcal·mol−1) is depicted, indicating the relative positions of Ser17, Thr53, Gln61, Glu63, GTP, and the two water molecules (wat A and wat B).
Table 1.
Main geometrical parameters of the QM/MM system
| Distance | Average value mean ± SE |
|---|---|
| Pγ - Ocat | 3.01 ± 0.02 |
| Pγ - O3β | 1.63 ± 0.02 |
| O3β- Pβ | 3.01 ± 0.02 |
| Mg - O1γ | 1.91 ± 0.04 |
| Mg - O2β | 1.90 ± 0.04 |
Average values are mean ± SE of the dynamical fluctuation during the 300-ps stabilization of the QM region.
Energy surface calculations
In the hybrid QM/MM potential, the effective energy of the system is divided into three components, as
where HQM is evaluated using the chosen QM method (in our case PM3 Hamiltonian); EMM is calculated classically from the MM atom positions using the AMBER force field equation and parameters; and HQM/MM is the sum of an electrostatic term and a Lennard-Jones (van der Waals) term, representing the interactions between the atoms of the QM and MM regions. Throughout the trajectories, the EMM term of the effective energy was not taken into account because we considered that the HQM plus HQM/MM terms were sufficiently representative of the influence of the protein moiety on the chemical reaction.
To explore the conformational space defined by the reaction coordinates, a new approach, to our knowledge, was developed. In this approach, based on adaptively biased MD (19) and presenting some characteristics of steered MD (20) as well as umbrella sampling (21) procedures, QM/MM trajectories were simulated by restraining both reaction coordinates using harmonic potentials with a flat bottom and parabolic sides. The reaction coordinates used in the calculations were the bond-breaking distance from the proton to the oxygen atom in the attacking water molecule and the bond-forming distance from this same proton to the oxygen in the ɛ1 position of Gln61 (Fig. 2) or to the oxygen of the second water molecule (Figure 3, Figure 4 and see Fig. S1 in the Supporting Material). For each trajectory, the value of one coordinate (x axis) was increased throughout the simulation time, as in steered MD (20), from 0.95 to 1.8 Å in steps of 0.025 Å, whereas the size of the flat-bottomed part of the harmonic potential of the other coordinate (y axis) was kept constant within a narrow range. The next trajectory was generated by adding an increment of 0.025 Å to the constant value of the y axis.
Figure 3.
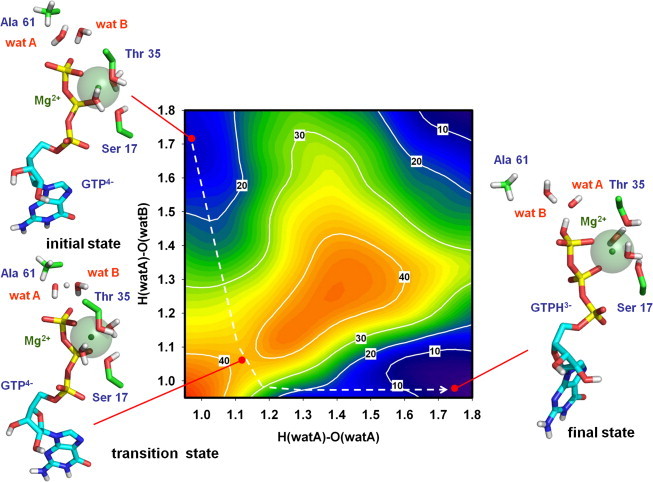
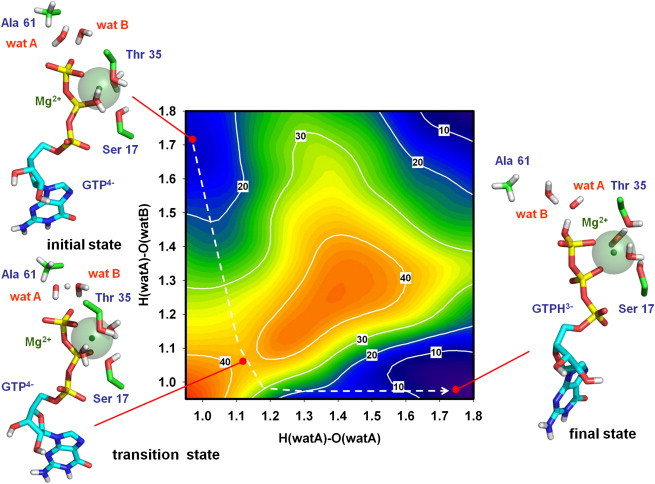
Free energy landscape for the proton transfer between the attacking water molecule (wat A) and the second water molecule (wat B). The axes represent the following distances: x axis, from proton to oxygen atom in the attacking water molecule (wat A); y axis, from the same proton to the oxygen atom in the second water molecule (wat B). The structure of the active center in the initial state (ΔG°GTP4− ≈ 3 kcal·mol−1), transition state (ΔG°GTP4- ≈ 22 kcal·mol−1), and final state (ΔG°GTP∗ value adjusted to 0) is depicted, including the same residues as in Fig. 1. GTP molecule with pentacovalent Pγ in the final state is shown by GTP∗. Note the position of Gln61 in the transition state, where it stabilizes the position of the water molecules during proton transfer.
Figure 4.
Free energy landscape for the Q61A mutant. Axes are defined as in Fig. 3. The structure of the active center in the initial state (ΔG°GTP4- ≈ 11 kcal·mol−1), transition state (ΔG°GTP4- ≈ 38 kcal·mol−1), and final state (ΔG°GTPH3− value adjusted to 0) is depicted, including the side chain of Ala61 residue instead of Gln61.
A certain degree of overlapping of points was observed from one trajectory to the next, as occurs in umbrella sampling procedures (21), ensuring a better coverage of the whole surface. The generation of a large number of trajectories made it possible to explore in detail the conformational space defined by the reaction coordinates. We sampled the conformational space of the reaction with ∼12,000 homogeneously distributed points on a surface of 0.85 × 0.85 Å. Three-dimensional smoothing of the data was applied using the local smoothing technique with tricube weighting and polynomial regression. Final ΔG° values in the energy maps were normalized by subtracting from all of them the lowest value obtained in each experiment. Minimum values that were adjusted to 0 in each map were those corresponding to the initial state (Fig. 2 and see Fig. S1) or to the final state (Figure 3, Figure 4).
Results and Discussion
To evaluate the different mechanisms proposed for the activation of the water molecule involved in catalysis, as well as to identify the proton acceptor and the role of Gln61 in each pathway, a specific simulation method was devised based on the hybrid QM/MM potential (22) implemented in the AMBER10 package (17, 18, 23). The approach, which shares some characteristics with steered MD and also with umbrella sampling procedures, uses the adaptively biased MD (19) method to obtain a free energy surface in the conformational space defined by the reaction coordinates (see Materials and Methods). Analysis of the surface characteristics provided us with variations in the free energy that helped to describe suitable reaction mechanisms.
To begin, and as a previous validation test, the approach was used to study a well-known mechanism: the ionization of two water molecules resulting in a molecule of OH− plus a molecule of H3O+. The free energy landscape obtained (see Fig. S1) gave a ΔG° of ∼19 kcal⋅mol−1 between the initial and the final states, in complete agreement with the bulk water value (ΔG = −RT ln K = −19.1 kcal⋅mol−1 at 298.15°K and pH = 7), and thus supporting the accuracy of the method.
Using the same approach, and including in the QM/MM system the whole crystallized structure of the HRas⋅RasGAP complex (16), two alternative hypotheses for the activation of the attacking water molecule were tested. As indicated under Materials and Methods, the complete simulation system comprises not only the atoms in the active center but, for the first time to our knowledge, the whole protein complex, all included in a solvent box (total number of atoms >53,000). The first mechanism analyzed included the assumption that Gln61 acts as a proton acceptor assisted by Glu63. The free energy landscape obtained for the activation of the catalytic water molecule (Fig. 2) gave a ΔG° of ∼25 kcal⋅mol−1 between the initial and the final states; even higher than for the ion product of water in a polar environment (see above). This result ruled out the possibility of Gln61 acting as a general base, in agreement with previous equilibrium studies of proton transfer from the catalytic water molecule to this residue (11). To provide further support to this statement, an unconstrained simulation of the reaction once the final state was reached, was performed (see Fig. S2). As expected, in absence of restrictions, the system reverted spontaneously from these situation to the initial reaction substrates (unprotonated Gln61 plus two water molecules), indicating that the products were not stable in the simulated conditions.
Once Gln61 was ruled out as the proton acceptor, an alternative mechanism was tested. According to the arrangement of the side chain of residues and water molecules at the catalytic site of the prehydrolysis state of HRas (15), the proton transfer may occur between an attacking water molecule (wat A) and a second water molecule (wat B) acting as the proton acceptor. The free energy landscape obtained in these conditions (Fig. 3) indicated that ΔG° between the initial and the final states is ∼−3 kcal⋅mol−1. Detailed analysis of the structure that corresponds to the final state (Fig. 3, bottom right) showed that the GTP molecule is protonated, suggesting that GTPH3− is more stable than GTP4− in the environment of the catalytic site. In addition, and unexpectedly, the results indicated that the proton bound to the GTP molecule is initially not part of wat A but of wat B, which acted as the initial proton acceptor.
To analyze the behavior of the protons in detail throughout the process, the reaction along the minimum energy path between the initial and the final states was simulated (dashed line in Fig. 3). Tracing the proton movements (a video of the entire process is provided as Movie S1 in the Supporting Material) revealed that a proton from wat A is initially transferred to wat B, thus forming a hydronium ion. Then, toward the end of the trajectory, a different proton from wat B is transferred to GTP. In addition, at this precise moment, the free OH− molecule approaches the GTP γ-phosphate forcing it to adopt the trigonal bipyramidal geometry characteristic of the pentacovalent state, suggesting a SN2 reaction mechanism (24). Although, in our conditions, a SN2 mechanism appeared as favorable, further studies are still needed to fully unravel the complete mechanism of the GTP hydrolysis as we cannot completely rule out alternative mechanisms, i.e., SN1 as it has been proposed for Hsc70 ATPase (25).
In our model, the atomic rearrangement at the end of the trajectory suggests that, in the absence of the simulation restraints used to obtain the energy surface, the hydrolysis of GTP can eventually occur spontaneously from the pentacovalent structural state. Additionally, it suggests that the rate-limiting step of the reaction catalyzed by HRas in the presence of RasGAP would be the initial activation of wat A.
Under this hypothesis, the value of the energy barrier located between the initial and final states and corresponding to the transition state, is ∼22 kcal⋅mol−1, lower than the 28 kcal⋅mol−1 of the barrier for the ionization of water in a polar environment (see Fig. S1). Analysis of the structure of the transition state at the saddle point (Fig. 3, bottom left) showed a strong interaction between the amide group of Gln61 and the OH− formed from wat A. An interaction between a carbonyl atom in the side chain of Gln61 and the hydronium ion formed by wat B also contributes to the stabilization of the transition state. These results agree with the previously proposed mechanism of substrate-assisted catalysis (12, 13), although, for the first time to our knowledge, they indicate the essential role of wat B in the process: to mediate the transfer of the proton from wat A to GTP. The role of Gln61 in this new scenario consists of stabilizing the OH− and H3O+ molecules that are transiently generated during proton transfer.
In the same free energy landscape, a second local minimum can be observed (Fig. 3, top right-hand corner). The structure of the active center in this region corresponds to protonated GTP. Nevertheless, the origin of the proton bound to GTP in this case is not wat B but wat A, without the mediation of wat B. Although this alternative pathway cannot be completely ruled out, its higher energy barrier (28 kcal⋅mol−1 vs. 22 kcal⋅mol−1) make it less favorable.
To obtain additional results to support the proposed role of Gln61, a second simulation using the same QM/MM approach was performed. In this case, the Gln61 residue was replaced by Ala to mimic experimental conditions in which reduced GTPase activity of HRas was previously measured in vitro (7). The free energy landscape obtained for the same proton transfer path is shown in Fig. 4. The value of the new energy barrier is clearly higher (∼38 kcal⋅mol−1). In fact, in the absence of the Gln61 side chain that stabilizes the transient OH− and H3O+ molecules, the alternative path for direct protonation of GTP appeared to be more favorable, in direct opposition to the situation observed for the wild-type protein. These results support the role of Gln61 in the stabilization of the proton transfer path from wat A to wat B and then to GTP. In addition, they also explain the decrease of the GTPase rate found in vitro for the Q61A mutant (7). Moreover, the proposed mechanism could also offer a rational explanation for the increased rate of GTP hydrolysis found in the Q61E mutant (8), as it is conceivable that a negatively charged glutamic residue would stabilize the transient hydronium ion better than glutamine.
Acknowledgments
We thank Dr. Galo Ramírez for his continued scientific advice and encouragement. We are grateful to Toffa Evans for valuable assistance in the preparation of the manuscript.
This work was supported by fellowships and grants from the Spanish Ministerio de Ciencia e Innovación (SAF2007-61926), the Madrid Regional Authority (S-BIO-0260/2006-COMBACT), and the European Commission (FP7 HEALTH-F3-2009-223431). Biomol-Informatics was financed by the European Social Fund. Support from the “Fundación Ramón Areces” is acknowledged. We also thank the Centro de Computación Científica-Universidad Autónoma de Madrid for computational support.
Editor: Carmen Domene.
Footnotes
Two figures and one movie are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(11)05356-2.
Supporting Material
References
- 1.Neves S.R., Ram P.T., Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 2.Kosloff M., Selinger Z. Substrate assisted catalysis—application to G proteins. Trends Biochem. Sci. 2001;26:161–166. doi: 10.1016/s0968-0004(00)01748-5. [DOI] [PubMed] [Google Scholar]
- 3.Scheffzek K., Ahmadian M.R., Wittinghofer A. GTPase-activating proteins: helping hands to complement an active site. Trends Biochem. Sci. 1998;23:257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 4.Wittinghofer A. Phosphoryl transfer in Ras proteins, conclusive or elusive? Trends Biochem. Sci. 2006;31:20–23. doi: 10.1016/j.tibs.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Pai E.F., Krengel U., et al. Wittinghofer A. Refined crystal structure of the triphosphate conformation of H-Ras p21 at 1.35 Å resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitayama H., Sugimoto Y., et al. Noda M. A Ras-related gene with transformation suppressor activity. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- 7.Der C.J., Finkel T., Cooper G.M. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell. 1986;44:167–176. doi: 10.1016/0092-8674(86)90495-2. [DOI] [PubMed] [Google Scholar]
- 8.Frech M., Darden T.A., et al. Wittinghofer A. Role of glutamine-61 in the hydrolysis of GTP by p21H-Ras: an experimental and theoretical study. Biochemistry. 1994;33:3237–3244. doi: 10.1021/bi00177a014. [DOI] [PubMed] [Google Scholar]
- 9.Grigorenko B.L., Nemukhin A.V., et al. Burt S.K. QM/MM modeling the Ras-GAP catalyzed hydrolysis of guanosine triphosphate. Proteins. 2005;60:495–503. doi: 10.1002/prot.20472. [DOI] [PubMed] [Google Scholar]
- 10.Fasano O., Aldrich T., et al. Wigler M. Analysis of the transforming potential of the human H-Ras gene by random mutagenesis. Proc. Natl. Acad. Sci. USA. 1984;81:4008–4012. doi: 10.1073/pnas.81.13.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maegley K.A., Admiraal S.J., Herschlag D. Ras-catalyzed hydrolysis of GTP: a new perspective from model studies. Proc. Natl. Acad. Sci. USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweins T., Geyer M., et al. Wittinghofer A. Substrate-assisted catalysis as a mechanism for GTP hydrolysis of p21Ras and other GTP-binding proteins. Nat. Struct. Biol. 1995;2:36–44. doi: 10.1038/nsb0195-36. [DOI] [PubMed] [Google Scholar]
- 13.Schweins T., Langen R., Warshel A. Why have mutagenesis studies not located the general base in Ras p21. Nat. Struct. Biol. 1994;1:476–484. doi: 10.1038/nsb0794-476. [DOI] [PubMed] [Google Scholar]
- 14.Privé G.G., Milburn M.V., et al. Kim S.H. X-ray crystal structures of transforming p21 Ras mutants suggest a transition-state stabilization mechanism for GTP hydrolysis. Proc. Natl. Acad. Sci. USA. 1992;89:3649–3653. doi: 10.1073/pnas.89.8.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheidig A.J., Burmester C., Goody R.S. The pre-hydrolysis state of p21(Ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of Ras-like proteins. Structure. 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
- 16.Scheffzek K., Ahmadian M.R., et al. Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 17.Case, D.A., T.A. Darden, …, P.A. Kollman. 2010. AMBER 10 User's Manual. http://www.ambermd.org.
- 18.Case D.A., Cheatham T.E., 3rd, et al. Woods R.J. The AMBER biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babin V., Roland C., Sagui C. Adaptively biased molecular dynamics for free energy calculations. J. Chem. Phys. 2008;128:134101. doi: 10.1063/1.2844595. [DOI] [PubMed] [Google Scholar]
- 20.Park S., Khalili-Araghi F., et al. Schulten K. Free energy calculation from steered molecular dynamics simulations using Jarzynski's equality. J. Chem. Phys. 2003;119:3559–3566. [Google Scholar]
- 21.Kästner J., Thiel W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “umbrella integration”. J. Chem. Phys. 2005;123:144104. doi: 10.1063/1.2052648. [DOI] [PubMed] [Google Scholar]
- 22.Kamerlin S.C., Haranczyk M., Warshel A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B. 2009;113:1253–1272. doi: 10.1021/jp8071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker R.C., Crowley M.F., Case D.A. The implementation of a fast and accurate QM/MM potential method in AMBER. J. Comput. Chem. 2008;29:1019–1031. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 24.Lahiri S.D., Zhang G., et al. Allen K.N. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 25.Boero M., Ikeda T., et al. Terakura K. Hsc70 ATPase: an insight into water dissociation and joint catalytic role of K+ and Mg2+ metal cations in the hydrolysis reaction. J. Am. Chem. Soc. 2006;128:16798–16807. doi: 10.1021/ja064117k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.