

Abstract
In severe myocardial ischemia, histamine 3 (H3) receptor activation affords cardioprotection by preventing excessive norepinephrine release and arrhythmias; pivotal to this action is the inhibition of neuronal Na+/H+ exchanger (NHE). Conversely, angiotensin II, formed locally by mast cell-derived renin, stimulates NHE via angiotensin II type 1 (AT1) receptors, facilitating norepinephrine release and arrhythmias. Thus, ischemic dysfunction may depend on a balance between the NHE-modulating effects of H3 receptors and AT1 receptors. The purpose of this investigation was therefore to elucidate the H3/AT1 receptor interaction in myocardial ischemia/reperfusion. We found that H3 receptor blockade with clobenpropit increased norepinephrine overflow and arrhythmias in Langendorff-perfused guinea pig hearts subjected to ischemia/reperfusion. This coincided with increased neuronal AT1 receptor expression. NHE inhibition with cariporide prevented both increases in norepinephrine release and AT1 receptor expression. Moreover, norepinephrine release and AT1 receptor expression were increased by the nitric oxide (NO) synthase inhibitor NG-methyl-l-arginine and the protein kinase C activator phorbol myristate acetate. H3 receptor activation in differentiated sympathetic neuron-like PC12 cells permanently transfected with H3 receptor cDNA caused a decrease in protein kinase C activity and AT1 receptor protein abundance. Collectively, our findings suggest that neuronal H3 receptor activation inhibits NHE by diminishing protein kinase C activity. Reduced NHE activity sequentially causes intracellular acidification, increased NO synthesis, and diminished AT1 receptor expression. Thus, H3 receptor-mediated NHE inhibition in ischemia/reperfusion not only opposes the angiotensin II-induced stimulation of NHE in cardiac sympathetic neurons, but also down-regulates AT1 receptor expression. Cardioprotection ultimately results from the combined attenuation of angiotensin II and norepinephrine effects and alleviation of arrhythmias.
Introduction
In severe myocardial ischemia, norepinephrine (NE) is abundantly carried out of sympathetic nerve terminals by the NE transporter functioning in a reversed outward mode; this nonvesicular NE release process is known as “carrier-mediated” (Schömig, 1990; Levi and Smith, 2000). Carrier-mediated NE release is a key arrhythmogenic determinant; pivotal for its occurrence is the activation of neuronal Na+/H+ exchanger (NHE) (Levi and Smith, 2000). Angiotensin II (ANG II), formed locally by mast cell-derived renin in myocardial ischemia (Mackins et al., 2006; Reid et al., 2007), is a major NHE activator via ANG II type 1 receptors (AT1Rs) (Reid et al., 2004) and only a minor, indirect inhibitor via AT2 receptors (Avkiran and Haworth, 2003). ANG II elicits reperfusion arrhythmias by a direct action (Harada et al., 1998; Yahiro et al., 2003; Iravanian and Dudley, 2008), as well as by facilitating NE release (Schömig, 1990; Meredith et al., 1991; Levi and Smith, 2000); both of these actions are AT1R-mediated (Maruyama et al., 1999).
Conversely, histamine, also locally released from mast cells in myocardial ischemia (Imamura et al., 1994; Hatta et al., 1997), inhibits NHE activity via H3 receptor (H3R) activation (Silver et al., 2001), thus attenuating NE release and alleviating arrhythmias (Imamura et al., 1996; Levi and Smith, 2000). Accordingly, we hypothesize that the activation of H3R in myocardial ischemia serves a protective function in opposing the NE-releasing proarrhythmogenic actions of ANG II. Indeed, in ischemia/reperfusion (I/R), H3R-blocked and H3R-deleted mouse hearts release more NE and develop more severe arrhythmias than untreated wild-type hearts (Koyama et al., 2003a,b). Thus, the magnitude of carrier-mediated NE release and associated arrhythmias may depend on a balance between the respective NHE-modulating effects of H3R and AT1R.
The purpose of this investigation was therefore to ascertain whether enhanced ischemic cardiac dysfunction, which is manifest when H3Rs are blocked or deleted, results from an unimpeded AT1R-NHE activation. We report that H3R activation at the level of sympathetic nerve endings in myocardial ischemia opposes the deleterious effects of locally formed ANG II, not only by counteracting the NHE-stimulating effect of ANG II, but also by reducing the expression of AT1R.
Materials and Methods
Perfusion of Guinea Pig Hearts Ex Vivo.
All experiments were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Male Hartley guinea pigs (Charles River Laboratories, Stone Ridge, NY), weighing 300 to 350 g, were anesthetized with CO2 and euthanized by exsanguination while under anesthesia. Hearts were rapidly isolated and perfused at constant pressure (40 cm of H2O) in a Langendorff apparatus with a modified Ringer's solution composed of 154 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 5.6 mM glucose, and 6 mM NaHCO3. The perfusion fluid was equilibrated with 100% O2 at 37°C. After a 20-min stabilization period, normothermic ischemia was induced by complete cessation of coronary perfusion, followed by 30-min reperfusion. Hearts receiving drug treatment were treated for 15 min before the induction of ischemia. All drugs were added to the perfusion solution. The coronary effluent was collected into tubes. In the preischemic and ischemic periods, tubes were replaced every 2 min. The volume of effluent collected for each period was weighed and subsequently analyzed for NE content. NE was assayed in the coronary perfusate by high-pressure liquid chromatography with electro-chemical detection (Seyedi et al., 1999). Surface ECG was obtained from leads attached to the left ventricle and the right atrium and analyzed by using PowerLab/8SP (ADInstruments Inc., Colorado Springs, CO). Onset and duration of reperfusion arrhythmias were recorded and quantified according to the Lambeth Conventions (Walker et al., 1988).
Preparation of Cardiac Synaptosomes.
Guinea pigs were anesthetized with CO2 and exsanguinated while under anesthesia (see above). The rib cage was rapidly opened, and the heart was dissected away. A cannula was inserted in the aorta, and the heart was perfused at constant pressure (40 cm of H2O) in a Langendorff apparatus with oxygenated Ringer's solution. Hearts receiving drug treatment were treated for 15 min before the induction of ischemia. After a 20-min stabilization period, normothermic ischemia was induced by complete cessation of coronary perfusion for 20 min. After a 20-min normothermic ischemia, hearts were subsequently freed from fat and connective tissue and minced in Tris-buffered saline (TBS; 0.28 M sucrose containing 50 mM Tris and 1 mM EDTA, pH 7.4) containing protease inhibitor cocktail (BioVision Research Products, Mountain View, CA). After low-speed centrifugation (10 min at 120g at 4°C), the supernatants were centrifuged for 20 min at 20,000g at 4°C. This pellet, which contained cardiac synaptosomes (pinched-off sympathetic nerve endings), was resuspended in the same Tris-buffered saline with 0.1% Triton X-100. The concentration of protein in homogenate was determined by using Bio-Rad protein assay solution (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin as a standard.
For Western blot, membrane proteins were loaded and run on standard 10 to 20% gradient NuPAGENovex Bis-Tris Mini Gel (Invitrogen, Carlsbad, CA) in NuPAGE MOPS Running Buffer (Invitrogen). Electrophoresis was carried out at 200 V and 100 mA for 70 min. Proteins were transferred onto polyvinylidine difluoride membranes (Immobilon-P; Millipore Corporation, Billerica, MA) for 90 min at 200 V and 300 mA at 4°C. The membranes were blocked in a blocking buffer (TBS containing 0.1% Tween 20 and 5% nonfat dry milk) at room temperature for 2 h. The membrane was then probed overnight at 4°C with rabbit anti-AT1 receptor polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) (1:1000) in the antibody dilution buffer (TBS containing 0.1% Tween 20 and 5% bovine serum albumin). After the membrane was washed four times in TBS containing 0.1% Tween 20, the membrane was subsequently incubated with anti-rabbit antibody horseradish peroxidase-linked IgG (Cell Signaling Technology, Danvers, MA) (1:3000) in the antibody dilution buffer for 1 h at room temperature. The polyvinylidine difluoride membrane was then washed four times with TBS containing 0.1% Tween 20, and the bound antibodies were detected by using enhanced chemiluminescence (Millipore Corporation) followed by exposure to X-ray film (BioMax MR; Eastman Kodak, Rochester NY). Bands were analyzed by densitometry using Fluorchem8800 (Alpha Innotech, San Leandro CA), and the content of β-actin, which was detected by mouse monoclonal anti-human β-actin IgG-horseradish peroxidase conjugate (1:10,000; Alpha Diagnostic International, San Antonio, TX), was used as a control to ensure that the same amount of protein was loaded in each lane.
Cell Culture.
PC12 cells, an AT1R-expressing cell line derived from a pheochromocytoma of the rat adrenal medulla (Zhou et al., 2006), were transfected with the human H3 receptor (donated by Dr. T. W. Lovenberg, Johnson and Johnson Pharmaceutical Research and Development, LLC) using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol (Morrey et al., 2008). PC12-H3 cell lines were selected and maintained in selection media containing 500 μg/ml G418 sulfate (Mediatech, Herndon, VA) and/or Zeocin (Invitrogen), respectively (Morrey et al., 2008).
To examine H3R-mediated effects on AT1R expression, PC12-H3 cells were first grown on collagen from rat tail (Sigma-Aldrich, St. Louis, MO) and cultured in six-well plates in Dulbecco's modified Eagle's medium for 4 to 5 subsequent days containing 0.5% horse serum, 1% fetal bovine serum, G418 sulfate (Mediatech), streptomycin (10 μg/ml), amphotericin B (250 ng/ml), penicillin (100 U), and 1 ng/ml nerve growth factor (NGF). Cells were exclusively used from passages 15 to 24 and grown at 37°C in a humidified atmosphere of 95% air/5% CO2. To determine H3R effects, cells were stimulated with imetit [S-(2-(4-imidazolyl)ethyl)isothiourea] (100 nM) for 30 min. In further experiments, cells were additionally treated with the H3R antagonist clobenpropit [S-(3-(4(5)-imidazolyl))propyl-N-(4-chlorobenzyl) isothiourea] (50 nM). Vehicle-treated cells served as control. Cell lysis buffer (Cell Signaling Technology) was added to the cells, and the expression of AT1R and protein kinase C (PKC) activity in the lysates were examined by Western blotting.
PKC Assay.
PC12-H3 cells were incubated with phorbol 12-myristate β-acetate (PMA) (30 min) with or without imetit pretreatment (10 min). Other cells were pretreated with clobenpropit (10 min) before imetit and PMA treatment. Cells were then homogenized in buffer (200 μl) (20 mM Tris-HCl, 2 mM EDTA, 10 mM EGTA, 0.25 M sucrose, β-mercaptoethanol, and 1× protease inhibitors cocktail). Cell homogenates were then passed through syringes with a needle (30 gauge) 10 times, and cell lysates were centrifuged at 100,000g for 30 min to collect cytosolic fractions (supernatant). The pellets were resuspended in homogenization buffer (50 μl) with 1% Triton X-100, and then centrifuged at 100,000g for 30 min to collect membrane fractions (supernatant). Translocation of PKC was assessed by using a PKC-specific antibody (Santa Cruz Biotechnology, Inc.; 1:1000 dilution) in Western blot analysis. Methods for Western blot analysis were as described previously (Corti et al., 2011). The ratio of PKC in membrane to that in cytosol was expressed as PKC translocation (i.e., PKC activity).
Drugs.
Imetit was purchased from Tocris Bioscience (Ellisville, MO). Clobenpropit and NG-methyl-l-arginine (l-NMA) were purchased from Sigma-Aldrich (St. Louis, MO). EXP3174 [2-n-butyl-4-chloro-1-((2′-(1H-tetrazol-5-yl) biphenyl-4-yl) methyl) imidazole-5-carboxylic acid] was obtained from Merck Research Laboratories (Rahway, NJ). PMA was purchased from LC Laboratories, now a division of PKC Pharmaceuticals, Inc. (Woburn, MA). HOE642 (cariporide; 4-isopropyl-3-methylsulfonylbenzoyl-guanidine methanesulfonate) was a gift from Hoechst Marion Roussel (Frankfurt, Germany).
Statistical Analysis.
All data were expressed as means ± S.E.M. Analysis of variance (ANOVA) followed by Bonferroni's test or unpaired t test were used to determine statistical significance where appropriate.
Results
H3R-Mediated Cardioprotective Effects Involve NHE Attenuation and AT1R Inhibition.
Langendorff-perfused guinea pig hearts were subjected to global ischemia for 20 min, followed by reperfusion for 30 min (Imamura et al., 1996). Reperfusion was characterized by an increase in NE overflow into the coronary effluent (≅140 pmol/g from an undetectable level in preischemic conditions) (Fig. 1). In hearts perfused with the H3R antagonist clobenpropit (50 nM) NE overflow was ∼2.5-fold greater than in I/R control hearts (Fig. 1). The NHE inhibitor HOE642 (cariporide; 10 μM) reduced the clobenpropit-induced increase in NE overflow by ∼40% (Fig. 1), whereas the AT1R antagonist EXP3174 (100 nM) abolished it (Fig. 1). These results suggested that H3R blockade uncovers a protective H3R-mediated effect in I/R characterized by decreased NE release, an effect probably involving NHE attenuation and AT1R inhibition.
Fig. 1.
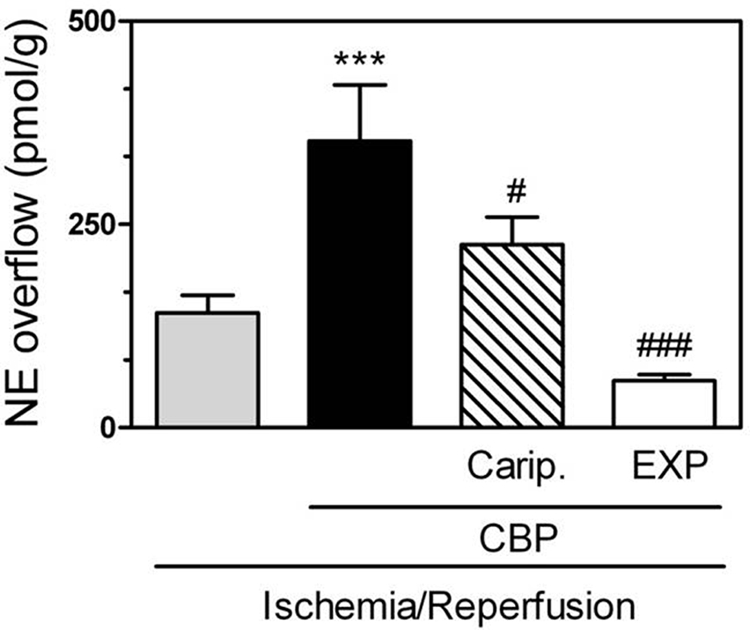
H3R blockade (50 nM CBP) in ex vivo guinea pig hearts subjected to I/R (20-min global ischemia + 30-min reperfusion) uncovers an unopposed NHE-stimulating action of ANG II, resulting in a magnified NE overflow that is markedly diminished by NHE blockade with cariporide (Carip., HOE642; 10 μM) and prevented by AT1R blockade with EXP3174 (EXP; 100 nM). Means ± S.E.M. (n = 5–13). ***, P < 0.001 from I/R; ###, P < 0.001 from I/R + CBP, by ANOVA and Bonferroni's multiple test. #, P < 0.05 from I/R + CBP by unpaired t test.
The enhanced NE spillover in I/R hearts was accompanied by long-lasting reperfusion arrhythmias (i.e., ventricular tachycardia and ventricular fibrillation) whose duration was greatly increased in hearts perfused with the H3R antagonist clobenpropit (50 nM). In contrast, in the presence of the NHE inhibitor cariporide (10 μM) ventricular tachycardia/ventricular fibrillation duration was shortened and abolished in the presence of the AT1R antagonist EXP3174 (100 nM) (data not shown). Thus, the H3R-mediated NHE attenuation and AT1R inhibition in I/R includes both a decrease in NE release and an alleviation of reperfusion arrhythmias.
H3R Activation during I/R Decreases AT1R Expression in Cardiac Sympathetic Nerve Terminals: NHE Involvement.
We next questioned whether this H3R-mediated inhibition of AT1R activity might entail a decrease in neuronal AT1R expression. For this, we determined AT1R expression in sympathetic nerve endings (synaptosomes) isolated from guinea pig hearts subjected to I/R. Langendorff-perfused guinea pig hearts were subjected to 20-min global ischemia, followed by 4-min reperfusion. Cardiac synaptosomes were isolated at the end of reperfusion, and expression of AT1R was evaluated by Western blotting. AT1Rs were detected as membrane proteins of 43-kDa molecular mass (Fig. 2, top). In the presence of the H3R antagonist clobenpropit (50 nM) AT1R expression increased by ∼70%; this effect was prevented by NHE inhibition with cariporide (10 μM) and diminished by AT1R blockade with EXP3174 (100 nM) (Fig. 2, bottom). These results suggested that constitutional activation of H3R during I/R decreases the expression of AT1R in cardiac sympathetic nerve terminals and that this effect is likely to involve NHE. Hence, we next investigated the mechanisms mediating the H3R-induced attenuation of AT1R expression.
Fig. 2.

H3R blockade (50 nM CBP) increases AT1R protein abundance in sympathetic nerve endings (cardiac synaptosomes) isolated from Langendorff guinea pig hearts subjected to I/R (20-min global ischemia + 4-min reperfusion), an effect prevented by NHE blockade with cariporide (Carip., HOE642; 10 μM) and diminished by AT1R blockade with EXP3714 (EXP; 100 nM). Top, representative Western blots. Bottom, quantification. Bars are means ± S.E.M. (n = 4–8). **, P < 0.01 from I/R + CBP by ANOVA and Bonferroni's multiple test.
Nitric Oxide Attenuates AT1R Expression in Cardiac Sympathetic Nerve Terminals during I/R.
By inhibiting NHE, H3R activation can lead to intracellular acidification (Silver et al., 2001). In turn, intracellular acidification is known to stimulate nitric oxide (NO) production (Tsutsumi et al., 1999), and NO has been shown to suppress AT1R expression (Ichiki et al., 1998). Hence, we questioned whether NO might be involved in the H3R-induced reduction of AT1R expression. For this, we perfused isolated guinea pig hearts with the NO synthase inhibitor l-NMA (100 μM) and then subjected them to 20-min global ischemia, followed by 4-min reperfusion. At the end of reperfusion, we isolated cardiac synaptosomes and measured the expression of AT1R by Western blotting. The level of AT1R expression in synaptosomes isolated from hearts subjected to I/R in the presence of l-NMA was twice as large as in its absence (Fig. 3), and NE overflow was ∼3-fold greater (Fig. 4). These findings implied that H3R activation during I/R leads to a NO-mediated attenuation of AT1R expression, resulting in a decreased NE release.
Fig. 3.

Inhibition of nitric-oxide synthesis with l-NMA (100 μM) or stimulation of PKC with PMA (300 nM). Each increases AT1R expression in sympathetic nerve endings (cardiac synaptosomes) isolated from ex vivo guinea pig hearts subjected to I/R (20-min global ischemia + 4-min reperfusion). Top, representative Western blots. Bottom, quantification. Bars are means ± S.E.M. (n = 3–7). ***, P < 0.001 from I/R by ANOVA and Bonferroni's multiple test.
Fig. 4.

Inhibition of nitric-oxide synthesis with l-NMA (100 μM), or stimulation of PKC with PMA (300 nM), increases NE overflow from ex vivo guinea pig hearts subjected to I/R (20-min global ischemia + 30-min reperfusion). Bars are means ± S.E.M. of NE overflow collected during the first 6 min of reperfusion (n = 3–13). *, P < 0.05 and **, P < 0.01, from I/R by ANOVA and Bonferroni's multiple test.
An H3R-Mediated PKC/NHE Attenuation Decreases AT1R Expression in Cardiac Sympathetic Nerve Terminals during I/R.
Our findings suggested that NHE is probably involved in the H3R-induced attenuation of AT1R expression in sympathetic nerve terminals isolated from hearts subjected to I/R (see Fig. 2). PKC is known to directly phosphorylate and activate NHE (Wakabayashi et al., 1997; Karmazyn et al., 1999), whereas H3R activation attenuates NHE activity (Silver et al., 2001), possibly by decreasing phosphoinositide turnover (Cherifi et al., 1992), thus inhibiting PKC activation. Hence, we hypothesized that a decrease in PKC activity may play a role in the H3R-induced attenuation of AT1R expression. To verify this hypothesis, we subjected Langendorff-perfused guinea pig hearts to I/R in the presence of the PKC activator PMA (300 nM) and determined NE overflow and AT1R expression in sympathetic nerve endings after 4-min reperfusion. In synaptosomes isolated from I/R hearts perfused with PMA the level of AT1R expression was ∼2.6-fold greater than in the absence of PMA (Fig. 3), whereas NE overflow was ∼3.5-fold greater in the presence than in the absence of PMA (Fig. 4). These data suggested that constitutional H3R activation during I/R exerts cardioprotective effects that may result from a sequential decrease in PKC and NHE activity.
H3R Activation Induces a Decrease in AT1R Protein Expression and PKC Activity in PC12-H3 Cells.
We directly tested the hypothesis that H3R activation decreases neuronal AT1R expression in a cell line bearing a sympathetic neuron phenotype, i.e., NGF-differentiated rat pheochromocytoma PC12 cells permanently transfected with H3R cDNA (PC12-H3 cells) (Morrey et al., 2008). In the presence of the H3R agonist imetit (100 nM), AT1R expression was decreased by ∼40%; this effect was prevented by the H3R antagonist clobenpropit (50 nM). In the absence of H3R activation, clobenpropit had no effect on AT1R expression (Fig. 5).
Fig. 5.

H3R activation with imetit (100 nM; 30 min) attenuates the expression of AT1R in PC12-H3 cells. Prevention by H3R blockade with CBP (50 nM) is shown. Top, representative Western blot analysis. Bottom, quantification. Bars are means ± S.E.M. (n = 3–6). *, P < 0.05 from control; ##, P < 0.01 from imetit + CBP by ANOVA and Bonferroni's multiple test.
We next directly tested the postulate that H3R activation decreases neuronal PKC activity. As evidence of PKC activation, we measured cytosol-to-membrane translocation of PKC in NGF-differentiated PC12-H3 cells. We found that H3R activation with imetit (100 nM) attenuated the increase in PKC translocation induced by PMA (300 nM), an effect that was prevented by the H3R antagonist clobenpropit (50 nM) (Fig. 6). These results suggested that H3R activation induces a decrease in neuronal AT1R protein expression that involves a reduction in PKC activity.
Fig. 6.

H3R activation attenuates the PMA-induced PKC activation (i.e., translocation from cytosol to membrane) in PC12-H3 cells. Top, representative blots. Cells were treated with the PKC activator PMA (300 nM; 20 min), the H3R agonist imetit (100 nM; 10 min), and the H3R antagonist CBP (50 nM; 10 min). PKC expression in cytosolic (C) and membrane (M) fractions was determined by Western immunoblotting. Bottom, PKC translocation expressed as the ratio of PKC abundance between membrane and cytosolic fractions. Bars are means (± S.E.M.; n = 3) normalized relative to β-actin. *, P < 0.05 from control by ANOVA and Bonferroni's multiple test; ##, P < 0.01 from PMA by unpaired t test; †, P < 0.05 from PMA + imetit + CBP by ANOVA and Bonferroni.
Discussion
The goal of our study was to determine whether H3Rs afford cardioprotection in I/R by counteracting the AT1R-mediated stimulation of NHE. We found that activation of neuronal H3R opposes the deleterious effects of locally formed ANG II, not only by inhibiting NHE, but also by reducing the expression of AT1R. This novel phenomenon broadens the known cardioprotective effects of H3R activation (Levi and Smith, 2000; Mackins and Levi, 2000).
We had reported previously that H3Rs are activated in I/R and limit the release of pathological amounts of NE and associated reperfusion arrhythmias by decreasing NHE activation, thus counteracting a pivotal mechanism of nonvesicular, carrier-mediated NE release (Levi and Smith, 2000). Because ANG II is formed locally in the heart during I/R (Mackins et al., 2006) and ANG II is a major NHE activator (Reid et al., 2004), we assumed that in I/R the activation of H3R could counterbalance the NHE-stimulating effect of ANG II. Indeed, we found that pharmacological blockade of H3R in I/R enhanced the magnitude of NE overflow and the severity of arrhythmias, and this effect was prevented by NHE and AT1R blockade. This clearly indicated that in protracted I/R H3R activation serves to counteract the NHE-stimulating effect of intraneuronal anaerobic acidification and the AT1R-mediated effects of ANG II.
The first hint that H3R might do more than just antagonize ANG II at the NHE level came from the finding that AT1R protein abundance in synaptosomes isolated from I/R hearts increased when H3Rs were blocked. This, in fact, suggested that H3Rs may influence neuronal AT1R expression in hearts subjected to I/R. Inasmuch as cardiac synaptosomes probably include cholinergic, purinergic, and sensory C-fiber terminals (Seyedi et al., 1999; Morrey et al., 2010), we tested whether H3R activation would influence AT1R expression in PC12-H3 cells, which once differentiated with NGF express a typical sympathetic neuron phenotype (Taupenot, 2007; Morrey et al., 2008; Corti et al., 2011). Indeed, we found that activation of H3R with imetit markedly diminished AT1R protein abundance in these cells, an action that was abolished by the H3R antagonist clobenpropit. It is noteworthy that, although the down-regulation of AT1R protein expression by H3R activation seemed to be fairly rapid, the time of exposure to the H3R agonist (i.e., 30 min in PC12-H3 cells) was comparable with that reported for the down-regulation of AT1R by dopamine D5 receptor activation in renal proximal tubule cells (i.e., half-life 0.47 ± 0.18 h) (Gildea et al., 2008).
It is noteworthy that the CBP-induced increase in NE overflow was completely suppressed by the AT1R blocker EXP3174, but partially suppressed by the NHE blocker cariporide, whereas cariporide completely blocked the CBP-induced AT1R up-regulation. These apparent discrepancies probably result from the fact that ANG II elicits NE release by two mechanisms, exocytotic and carrier-mediated (Reid et al., 2004), which are both mediated by AT1R, but are Ca2+- and NHE-dependent, respectively. Accordingly, AT1R blockade by EXP3174 is likely to be more effective in preventing NE release because it blocks both exocytosis and carrier-mediated release. In contrast, cariporide only blocks NHE without affecting exocytosis (Levi and Smith, 2000). On the other hand, cariporide completely blocks the CBP-induced increase in AT1R expression because this H3R-mediated phenomenon seems to be solely NHE-dependent.
The finding that NHE blockade in I/R prevented the increase in AT1R expression in cardiac synaptosomes, whereas PKC stimulation with PMA increased it, implied an involvement of both NHE and PKC. First, we tested whether PKC could be involved in the H3R-mediated inhibition of NHE and associated reduction of AT1R expression. Given that PKC is translocated/activated in I/R (Strasser et al., 1992) and this stimulates NHE (Karmazyn et al., 1999), we thought it plausible that the H3R-induced inhibition of NHE may involve a decrease in PKC translocation/activation. Indeed, we found that the PMA-induced activation of PKC in PC12-H3 cells, similar to that occurring in myocardial ischemia (Prasad and Jones, 1992), was abolished by H3R activation. It is conceivable that this H3R-mediated decrease in PKC activation stems from a decreased phosphoinositide turnover, because H3Rs have been found previously to be negatively coupled to it (Cherifi et al., 1992). Although we did not examine which PKC isoform may be specifically involved in the down-regulation of AT1R in sympathetic nerve endings, if PKCε were involved (Koda et al., 2010) it could be plausibly activated by NO via a positive feedback mechanism (Ping et al., 1999). If so, this could counteract in part the H3R-mediated decrease in PKC activity.
An H3R-mediated decrease in NHE activity is expected to lead to acidification of cardiac sympathetic nerve endings (Levi and Smith, 2000), and intracellular acidification was shown to stimulate NO production (Tsutsumi et al., 1999). Moreover, NO production increases during I/R in the mammalian heart (Zweier et al., 1995a; Lecour et al., 2001), and NO has been shown to suppress AT1R expression (Ichiki et al., 1998). It thus became apparent that NO might mediate the decreased AT1R protein abundance in sympathetic nerve endings initiated by the H3R-mediated decrease in NHE activity. The finding that inhibition of NO synthase enhanced AT1R expression in I/R synaptosomes, and markedly increased NE release, suggests that H3R activation increases NO production, ultimately reducing AT1R protein abundance in sympathetic nerve endings. Although we did not measure NO synthesis, our results with l-NMA indicate an involvement of NO and NO synthase. Yet, it is possible that NO was also produced by an enzyme-independent mechanism (e.g., by direct reduction of nitrite to NO) as demonstrated by Zweier et al. (1995b).
It is conceivable that additional NO might be generated by the activation of Mas, an orphan G protein-coupled receptor that is a functional ligand site for angiotensins 1 to 7 (ANG-1–7) (Santos et al., 2003). ANG-1–7 are produced most efficiently from ANG II by ACE2, a newly discovered carboxyl-peptidase (Raizada and Ferreira, 2007; Ferrario, 2010). Yet, although ACE2 is widely distributed throughout the brain (Doobay et al., 2007), its presence in peripheral sympathetic nerves has yet to be demonstrated. Thus, the contribution of ACE2 and ANG-1–7 to the formation of NO in cardiac sympathetic nerves and their possible roles in the down-regulation of AT1R remain to be determined.
Although imetit and clobenpropit have long been considered to be a selective H3R agonist and antagonist, respectively (Garbarg et al., 1992; Koyama et al., 2003b), they have also been shown to activate H4R (Lim et al., 2005). Similar to H3Rs, H4Rs are Gi/o-coupled; hence, it is conceivable that H4Rs may share some of the characteristic effects of H3Rs, such as NHE inhibition and thus, might contribute to the H3R-mediated attenuation of AT1R expression. Yet, although H4Rs have been described in the central nervous system (Connelly et al., 2009), their presence in cardiac sympathetic neurons has not been reported. Thus, whether the H4R-agonistic effects of clobenpropit might have contributed to the increase in synaptosomal AT1R expression (see Fig. 2) cannot be confirmed at this time.
In conclusion, we have uncovered a novel cardioprotective action resulting from activation of neuronal H3R in mammalian heart (see Fig. 7). Binding of an endogenous ligand to H3R, most likely histamine, released from local mast cells (Imamura et al., 1994; Hatta et al., 1997) by the action of reactive oxygen species produced during I/R (Koda et al., 2010), probably causes a decrease in phosphoinositide turnover (Cherifi et al., 1992). This is expected to reduce the formation of diacylglycerol and thus diminish PKC activity, as we have demonstrated here (see Fig. 6). This, in turn, decreases NHE activity (Karmazyn et al., 1999), so that H+ ions accumulate intraneuronally. Intracellular acidification, as it occurs in myocardial ischemia (Poole-Wilson, 1989; Levi and Smith, 2000), stimulates the production of NO (Tsutsumi et al., 1999), which has been shown to suppress AT1R expression (Ichiki et al., 1998). Hence, the H3R-induced decrease in NHE activity leading to an increased NO synthesis may well be responsible for the ultimate decrease in AT1R protein abundance (Fig. 7). We propose that our findings of a down-regulation of AT1R signaling and attenuation of NE release by activation of neuronal H3R are both crucial and plausibly translatable mechanisms of cardioprotection, not only in myocardial ischemia but also in other cardiac dysfunctions in which ANG II plays a major role, such as heart failure.
Fig. 7.

Proposed mechanisms for the H3R-induced reduction of neuronal AT1R expression. DAG, diacylglycerol; PI, phosphoinositide; PIP, phosphatidylinositol phosphate; PIP2, phosphatidylinositol-4,5-biphosphate; PLC, phospholipase C.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL034215]; and a Pharmaceutical Research Manufacturers of America Foundation predoctoral fellowship.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- NE
- norepinephrine
- ANG
- angiotensin
- AT1R
- ANG II type 1 receptor
- H3R
- histamine 3 receptor
- H4R
- histamine 4 receptor
- I/R
- ischemia/reperfusion
- l-NMA
- NG-methyl-l-arginine
- NHE
- Na+/H+ exchanger
- NO
- nitric oxide
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate β-acetate
- TBS
- Tris-buffered saline
- NGF
- nerve growth factor
- ANOVA
- analysis of variance
- ACE2
- angiotensin I converting enzyme 2
- CBP
- clobenpropit
- EXP3174
- 2-n-butyl-4-chloro-1-((2′-(1H-tetrazol-5-yl) biphenyl-4-yl) methyl) imidazole-5-carboxylic acid
- HOE642
- 4-isopropyl-3-methylsulfonylbenzoyl-guanidine methanesulfonate.
Authorship Contributions
Participated in research design: Hashikawa-Hobara, Chan, and Levi.
Conducted experiments: Hashikawa-Hobara and Chan.
Performed data analysis: Hashikawa-Hobara and Chan.
Wrote or contributed to the writing of the manuscript: Hashikawa-Hobara, Chan, and Levi.
References
- Avkiran M, Haworth RS. (2003) Regulatory effects of G protein-coupled receptors on cardiac sarcolemmal Na+/H+ exchanger activity: signalling and significance. Cardiovasc Res 57:942–952 [DOI] [PubMed] [Google Scholar]
- Cherifi Y, Pigeon C, Le Romancer M, Bado A, Reyl-Desmars F, Lewin MJ. (1992) Purification of a histamine H3 receptor negatively coupled to phosphoinositide turnover in the human gastric cell line HGT1. J Biol Chem 267:25315–25320 [PubMed] [Google Scholar]
- Connelly WM, Shenton FC, Lethbridge N, Leurs R, Waldvogel HJ, Faull RL, Lees G, Chazot PL. (2009) The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br J Pharmacol 157:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti F, Olson KE, Marcus AJ, Levi R. (2011) The expression level of ecto-NTP diphosphohydrolase1/CD39 modulates exocytotic and ischemic release of neurotransmitters in a cellular model of sympathetic neurons. J Pharmacol Exp Ther 337:524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. (2007) Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 292:R373–R381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CM. (2010) New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 55:445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarg M, Arrang JM, Rouleau A, Ligneau X, Tuong MD, Schwartz JC, Ganellin CR. (1992) S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist. J Pharmacol Exp Ther 263:304–310 [PubMed] [Google Scholar]
- Gildea JJ, Wang X, Jose PA, Felder RA. (2008) Differential D1 and D5 receptor regulation and degradation of the angiotensin type 1 receptor. Hypertension 51:360–366 [DOI] [PubMed] [Google Scholar]
- Harada K, Komuro I, Hayashi D, Sugaya T, Murakami K, Yazaki Y. (1998) Angiotensin II type 1a receptor is involved in the occurrence of reperfusion arrhythmias. Circulation 97:315–317 [DOI] [PubMed] [Google Scholar]
- Hatta E, Yasuda K, Levi R. (1997) Activation of histamine H3 receptors inhibits carrier-mediated norepinephrine release in a human model of protracted myocardial ischemia. J Pharmacol Exp Ther 283:494–500 [PubMed] [Google Scholar]
- Ichiki T, Usui M, Kato M, Funakoshi Y, Ito K, Egashira K, Takeshita A. (1998) Down-regulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 31:342–348 [DOI] [PubMed] [Google Scholar]
- Imamura M, Lander HM, Levi R. (1996) Activation of histamine H3 receptors inhibits carrier-mediated norepinephrine release during protracted myocardial ischemia. Comparison with adenosine A1 receptors and α2-adrenoceptors. Circ Res 78:475–481 [DOI] [PubMed] [Google Scholar]
- Imamura M, Poli E, Omoniyi AT, Levi R. (1994) Unmasking of activated histamine H3 receptors in myocardial ischemia: their role as regulators of exocytotic norepinephrine release. J Pharmacol Exp Ther 271:1259–1266 [PubMed] [Google Scholar]
- Iravanian S, Dudley SC., Jr. (2008) The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm 5:S12–S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmazyn M, Gan XT, Humphreys RA, Yoshida H, Kusumoto K. (1999) The myocardial Na+-H+ exchange: structure, regulation, and its role in heart disease. Circ Res 85:777–786 [DOI] [PubMed] [Google Scholar]
- Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly-Rosen D, Levi R. (2010) Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 122:771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Heerdt PM, Levi R. (2003a) Increased severity of reperfusion arrhythmias in mouse hearts lacking histamine H3 receptors. Biochem Biophys Res Commun 306:792–796 [DOI] [PubMed] [Google Scholar]
- Koyama M, Seyedi N, Fung-Leung WP, Lovenberg TW, Levi R. (2003b) Norepinephrine release from the ischemic heart is greatly enhanced in mice lacking histamine H3 receptors. Mol Pharmacol 63:378–382 [DOI] [PubMed] [Google Scholar]
- Lecour S, Maupoil V, Zeller M, Laubriet A, Briot T, Rochette L. (2001) Levels of nitric oxide in the heart after experimental myocardial ischemia. J Cardiovasc Pharmacol 37:55–63 [DOI] [PubMed] [Google Scholar]
- Levi R, Smith NC. (2000) Histamine H3 receptors: a new frontier in myocardial ischemia. J Pharmacol Exp Ther 292:825–830 [PubMed] [Google Scholar]
- Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. (2005) Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective histamine H4 receptor agonist. J Pharmacol Exp Ther 314:1310–1321 [DOI] [PubMed] [Google Scholar]
- Mackins CJ, Kano S, Seyedi N, Schäfer U, Reid AC, Machida T, Silver RB, Levi R. (2006) Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 116:1063–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackins CJ, Levi R. (2000) Therapeutic potential of H3 receptor agonists in myocardial infarction. Expert Opin Investig Drugs 9:2537–2542 [DOI] [PubMed] [Google Scholar]
- Maruyama R, Hatta E, Levi R. (1999) Norepinephrine release and ventricular fibrillation in myocardial ischemia/reperfusion: roles of angiotensin and bradykinin. J Cardiovasc Pharmacol 34:913–915 [DOI] [PubMed] [Google Scholar]
- Meredith IT, Broughton A, Jennings GL, Esler MD. (1991) Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med 325:618–624 [DOI] [PubMed] [Google Scholar]
- Morrey C, Brazin J, Seyedi N, Corti F, Silver RB, Levi R. (2010) Interaction between sensory C-fibers and cardiac mast cells in ischemia/reperfusion: activation of a local renin-angiotensin system culminating in severe arrhythmic dysfunction. J Pharmacol Exp Ther 335:76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey C, Estephan R, Abbott GW, Levi R. (2008) Cardioprotective effect of histamine H3 receptor activation: pivotal role of Gβγ-dependent inhibition of voltage-operated Ca2+ channels. J Pharmacol Exp Ther 326:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. (1999) Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res 84:587–604 [DOI] [PubMed] [Google Scholar]
- Poole-Wilson PA. (1989) Regulation of intracellular pH in the myocardium; relevance to pathology. Mol Cell Biochem 89:151–155 [DOI] [PubMed] [Google Scholar]
- Prasad MR, Jones RM. (1992) Enhanced membrane protein kinase C activity in myocardial ischemia. Basic Res Cardiol 87:19–26 [DOI] [PubMed] [Google Scholar]
- Raizada MK, Ferreira AJ. (2007) ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol 50:112–119 [DOI] [PubMed] [Google Scholar]
- Reid AC, Mackins CJ, Seyedi N, Levi R, Silver RB. (2004) Coupling of angiotensin II AT1 receptors to neuronal NHE activity and carrier-mediated norepinephrine release in myocardial ischemia. Am J Physiol Heart Circ Physiol 286:H1448–H1454 [DOI] [PubMed] [Google Scholar]
- Reid AC, Silver RB, Levi R. (2007) Renin: at the heart of the mast cell. Immunol Rev 217:123–140 [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, et al. (2003) Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 100:8258–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schömig A. (1990) Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation 82:II13–II22 [PubMed] [Google Scholar]
- Seyedi N, Maruyama R, Levi R. (1999) Bradykinin activates a cross-signaling pathway between sensory and adrenergic nerve endings in the heart: a novel mechanism of ischemic norepinephrine release? J Pharmacol Exp Ther 290:656–663 [PubMed] [Google Scholar]
- Silver RB, Mackins CJ, Smith NC, Koritchneva IL, Lefkowitz K, Lovenberg TW, Levi R. (2001) Coupling of histamine H3 receptors to neuronal Na+/H+ exchange: a novel protective mechanism in myocardial ischemia. Proc Natl Acad Sci U S A 98:2855–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser RH, Braun-Dullaeus R, Walendzik H, Marquetant R. (1992) α1 Receptor-independent activation of protein kinase C in acute myocardial ischemia. Mechanisms for sensitization of the adenylyl cyclase system. Circ Res 70:1304–1312 [DOI] [PubMed] [Google Scholar]
- Taupenot L. (2007) Analysis of regulated secretion using PC12 cells. Curr Protoc Cell Biol Chapter 15, Unit 15.12 [DOI] [PubMed] [Google Scholar]
- Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, et al. (1999) Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest 104:925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi S, Shigekawa M, Pouyssegur J. (1997) Molecular physiology of vertebrate Na+/H+ exchangers. Physiol Rev 77:51–74 [DOI] [PubMed] [Google Scholar]
- Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DW. (1988) The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction and reperfusion. Cardiovasc Res 22:447–455 [DOI] [PubMed] [Google Scholar]
- Yahiro E, Ideishi M, Wang LX, Urata H, Kumagai K, Arakawa K, Saku K. (2003) Reperfusion-induced arrhythmias are suppressed by inhibition of the angiotensin II type 1 receptor. Cardiology 99:61–67 [DOI] [PubMed] [Google Scholar]
- Zhou J, Pavel J, Macova M, Yu ZX, Imboden H, Ge L, Nishioku T, Dou J, Delgiacco E, Saavedra JM. (2006) AT1 receptor blockade regulates the local angiotensin II system in cerebral microvessels from spontaneously hypertensive rats. Stroke 37:1271–1276 [DOI] [PubMed] [Google Scholar]
- Zweier JL, Wang P, Kuppusamy P. (1995a) Direct measurement of nitric oxide generation in the ischemic heart using electron paramagnetic resonance spectroscopy. J Biol Chem 270:304–307 [DOI] [PubMed] [Google Scholar]
- Zweier JL, Wang P, Samouilov A, Kuppusamy P. (1995b) Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1:804–809 [DOI] [PubMed] [Google Scholar]