

Abstract
Evidence for an unusual catalysis of protonation/deprotonation by a reduced flavin mononucleotide cofactor is presented for type-2 isopentenyl diphosphate isomerase (IDI-2), which catalyzes isomerization of the two fundamental building blocks of isoprenoid biosynthesis, isopentenyl diphosphate and dimethylallyl diphosphate. The covalent adducts formed between irreversible mechanism-based inhibitors, 3-methylene-4-penten-1-yl diphosphate or 3-oxiranyl-3-buten-1-yl diphosphate, and the flavin cofactor were investigated by X-ray crystallography and UV-visible spectroscopy. Both the crystal structures of IDI-2 binding the flavin-inhibitor adduct and the UV-visible spectra of the adducts indicate that the covalent bond is formed at C4a of flavin rather than at N5, which had been proposed previously. In addition, the high-resolution crystal structures of IDI-2-substrate complexes and the kinetic studies of new mutants confirmed that only the flavin cofactor can catalyze protonation of the substrates and suggest that N5 of flavin is most likely to be involved in proton transfer. These data provide support for a mechanism where the reduced flavin cofactor acts as a general acid/base catalyst and helps stabilize the carbocationic intermediate formed by protonation.
Keywords: enzyme structure, mechanism of action
Isopentenyl diphosphate isomerase (IDI) catalyzes the interconversion between isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) (1). IDI activity is essential in those organisms that synthesize isoprenoid compounds from mevalonic acid and, although not essential, is found in those that synthesize isoprenoid metabolites from methylerythritol phosphate. Two convergently evolved forms of IDI, which share no sequence or structural homology, are known. Type-1 IDI (IDI-1) is found in eukarya and some bacteria, whereas type-2 IDI (IDI-2) occurs in archaea and other bacteria. There is no apparent correlation between the occurrence of the IDI isoforms and the use of mevalonate or methylerythritol phosphate in isoprenoid biosynthesis.
IDI-1 catalyzes isomerization between IPP and DMAPP by a protonation-deprotonation reaction through a 3° carbocationic intermediate (1). The mechanism of the reaction and identification of essential active cysteine and glutamate residues were confirmed by inhibition studies using active-site-directed irreversible inhibitors (2–5). In contrast, IDI-2 is a flavoprotein that requires reduced FMN for activity (6, 7). UV-visible spectroscopic studies indicate that the anionic flavin in 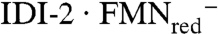 is protonated upon substrate binding to give a catalytically competent IDI-2·FMNred·IPP complex (8). Recent studies indicated that the mechanism for the IDI-2 is similar to the protonation-deprotonation sequence used by IDI-1. These include measurement of deuterium kinetic isotopic effects in D2O or with (R)-[2-2H]-IPP (9), linear free energy studies with seven- and eight-substituted FMN analogues (10), and studies with alkyne and allene analogues of IPP and DMAPP (11). In contrast, attempts to establish radical intermediates in the reaction were unsuccessful. Electron paramagnetic resonance spectra of active IDI-2·FMNred·IPP did not detect signals for putative radical intermediates (9). Attempts to inhibit the enzyme by incubation with a cyclopropyl analogue of IPP, designed to rearrange to a homoallylic structure by a radical clock mechanism, were unsuccessful (12, 13). Instead, the analogue was an alternate substrate that isomerized to the corresponding DMAPP derivative without a cyclopropylcarbinyl-homoallyl radical rearrangement.
is protonated upon substrate binding to give a catalytically competent IDI-2·FMNred·IPP complex (8). Recent studies indicated that the mechanism for the IDI-2 is similar to the protonation-deprotonation sequence used by IDI-1. These include measurement of deuterium kinetic isotopic effects in D2O or with (R)-[2-2H]-IPP (9), linear free energy studies with seven- and eight-substituted FMN analogues (10), and studies with alkyne and allene analogues of IPP and DMAPP (11). In contrast, attempts to establish radical intermediates in the reaction were unsuccessful. Electron paramagnetic resonance spectra of active IDI-2·FMNred·IPP did not detect signals for putative radical intermediates (9). Attempts to inhibit the enzyme by incubation with a cyclopropyl analogue of IPP, designed to rearrange to a homoallylic structure by a radical clock mechanism, were unsuccessful (12, 13). Instead, the analogue was an alternate substrate that isomerized to the corresponding DMAPP derivative without a cyclopropylcarbinyl-homoallyl radical rearrangement.
Mechanism-based inhibitors of IDI-1, which are activated by protonation of epoxide and diene moieties in these compounds, also inactivate IDI-2 (12–15). The inhibitors alkylate FMNred, which indicates that the flavin is in intimate contact with the substrate during isomerization. This arrangement is supported by crystal structures of Sulfolobus shibatae (16) and Thermus thermophilus IDI-2 (17), where the isoalloxazine moiety in FMNred and the side chains of tryptophan, histidine, and glutamine form the substrate binding pocket. Although flavins are not generally regarded as acid-base catalysts in enzyme-catalyzed reactions, site-directed mutagenesis experiments show that none of the active-site amino acids act as the catalyst (16). Furthermore, the isoprene moiety in IPP is orientated over FMNred in the S. shibatae IDI-2·FMNred·IPP complex in a conformation consistent with proton transfers between the substrate and the reduced tricyclic isoalloxazine moiety during the stereoselective reversible isomerization between R-(2-2H)-IPP and unlabeled DMAPP in D2O (18).
We and others initially proposed that mechanism-based inhibitors alkylate FMNred at N5, based on comparisons of UV spectra for the isolated adducts with those reported in the literature for related compounds (14, 15). Because UV spectra for N5 and C4a adducts are highly dependent on solvent and pH (19), we decided to conduct a more thorough study of the structures. In the present work, we describe crystallographic and spectroscopic studies that support modification at C4a by epoxide and diene inhibitors. Consideration of our data leads us to suggest a protonation-deprotonation mechanism for isomerization by IDI-2 in which the flavin cofactor acts as a general acid-base catalyst and helps stabilize the carbocationic intermediate.
Results
Structural Studies of IDI-2·Inhibitor Complexes.
Crystals of S. shibatae IDI-2 were soaked with 3-methylene-4-penten-1-yl diphosphate (vIPP) or 3-oxiranyl-3-buten-1-yl diphosphate (oIPP) (Fig. 1A), NADH, and MgCl2 before analysis. Electron density maps created from X-ray diffraction data indicate that both vIPP and oIPP are bound at the active site of the enzyme. Continuous electron density, which originates on the si face of the isoalloxazine unit between C4a and N5, links FMN and the inhibitors (Table 1, vIPP-IDI and oIPP-IDI, respectively, Fig. 1B, and Fig. S1). Initially, we expected that the inhibitors might be attached to FMN in different structural or conformational isomers and fitted them to the 2Fo - Fc electron density maps (Fig. 1C and SI Methods). Although discernible differences were seen in the levels of fitting for optimized models of the isomeric adducts (SI Methods), it was much more difficult to distinguish between models of C4a and N5 adducts for the same inhibitor from the best fits of map correlation coefficients, which express how well the models fit the electron density map (Fig. 1B). For example, the map correlation coefficients for the vIPP-derived hydrocarbon moieties of the three C4a and N5 adducts shown in Fig. 1C are 0.9435 (C4a E-isomer), 0.9327 (C4a Z-isomer), and 0.9414 (N5 Z-isomer), which indicates that the three models have similar fits (see SI Methods for other isomers of vIPP adducts and those of oIPP adducts). However, the inhibitor-derived carbon atom that is directly attached to FMN fits better into the density map in the C4a adduct models than the N5 model (Fig. 1B). Because this carbon atom in the inhibitor is directly attached to the tricyclic isoalloxazine moiety in FMN, its location in the density map should be especially important for determining the location of the FMN-inhibitor bond. Moreover, a definite negative peak in the noncrystallography symmetry-averaged Fo - Fc map overlaps the carbon in the N5-adduct model (Fig. 1D, red mesh), which also suggests that the model is less reasonable. Similar results were obtained for the oIPP adduct (Fig. S1). We conclude that our crystallographic data favor alkylation of FMN at C4a rather than N5.
Fig. 1.

Structural information of the flavin adducts formed in S. shibatae IDI-2 by treatment with the mechanism-based irreversible inhibitors. (A) Chemical structures of vIPP and oIPP. OPP designates the diphosphate group. (B) C4a and N5 adduct model structures for vIPP-IDI with a 2Fo - Fc electron density map. The C4a and N5 models are shown in blue and pink, respectively. Mg2+ ions are indicated in spheres. The electron map (blue) is contoured at 1.5σ. For the construction of the models shown here, the structures of vIPP adduct with the highest map coefficients (C) were used. C5H10O4P and OPP in the structures mean ribitol phosphate and diphosphate groups, respectively. Other possible chemical structures are shown in SI Methods. (D) 2Fo - Fc and noncrystallography symmetry-averaged Fo - Fc maps overlapping the N5-model structure of vIPP-IDI. The electron maps of 2Fo - Fc (blue) and Fo - Fc (red) are contoured at 1.5σ and -3.5σ, respectively. Carbon, oxygen, and nitrogen atoms are colored yellow, red, and blue, respectively.
Table 1.
Data collection and refinement statistics
| Crystal type | vIPP-IDI | oIPP-IDI | IPP-IDI | DMAPP-IDI |
| Data collection and processing statistics | ||||
| Beamline | PF-NE3A | PF-NE3A | PF-BL5A | PF-BL5A |
| Space group | P43212 | P43212 | P43212 | P43212 |
| Unit cell dimension, Å | ||||
| a = b, Å | 101.197 | 101.234 | 101.617 | 100.842 |
| c, Å | 336.837 | 337.42 | 333.078 | 334.499 |
| α = β = γ, ° | 90.000 | 90.000 | 90.000 | 90.000 |
| Wavelength, Å | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Resolution, Å | 50.00–2.20 | 50.00–2.30 | 50.0–2.20 | 50.0–2.29 |
| (2.28–2.20) | (2.38–2.30) | (2.28–2.20) | (2.34–2.30) | |
| Measured | 459,056 | 512,211 | 619,459 | 439,989 |
| I/σI | 17.1 (3.0) | 21.3 (2.0) | 22.8 (4.7) | 22.5 (5.7) |
| Redundancy | 5.3 (2.2) | 6.7 (2.7) | 7.0 (7.0) | 6.0 (6.5) |
| Completeness, % | 95.9 (78.6) | 96.1 (71.1) | 98.1 (99.4) | 93.8 (98.8) |
| Rmerge,* % | 9.2 (26.2) | 8.6 (27.6) | 6.9 (39.8) | 11.3 (35.5) |
| Refinement statistics | ||||
| Resolution | 46.13-2.20 | 48.48-2.30 | 31.62-2.20 | 37.39-2.29 |
| Protein atoms | 11,244 | 11,184 | 11,232 | 11,232 |
| Ligand atoms | 187 | 192 | 184 | 184 |
| Water molecule | 420 | 452 | 467 | 294 |
| Rwork/Rfree, % | 20.1/23.1 | 18.6/22.0 | 20.0/24.2 | 20.4/23.9 |
| Root mean square deviations | ||||
| Bond lengths, Å | 0.016 | 0.023 | 0.022 | 0.023 |
| Bond angles, ° | 1.635 | 1.935 | 1.945 | 1.975 |
Numbers in parentheses are for the highest shell.
*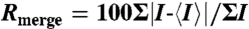 , where I is the observed intensity and
, where I is the observed intensity and 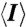 is the average intensity of multiple observations of symmetry-related reflections.
is the average intensity of multiple observations of symmetry-related reflections.
Formation of the IDI-2-vIPP adduct most likely results from a mechanism-based alkylation of the flavin cofactor in analogy to inhibition of IDI-1 by the same compound (15). In this scenario, protonation of the diene moiety gives an allylic cation, which then alkylates the cofactor. Four different isomeric allylic cations can be generated from vIPP, resulting from protonation of the diene at either end of the s-cis and s-trans conformers (see Fig. 2). Once formed, the allylic cations should be stable with regard to E-Z–isomerization of the allylic moiety before reacting with the flavin nucleophile (20). The three most probable structures for the vIPP-flavin adduct as determined from map correlation coefficients (Fig. 1C, designated in Fig. S2 as structures 3 and 4 for C4a adducts and 5 for N5 adduct) are generated from allylic cations D, B, and C, respectively, by a net 1,4 addition of the proton and flavin to the diene. Formation of the 1,4 product requires movement of the allylic cation prior to alkylation of flavin. Although the results of our UV-visible studies (see below) make the N5 adduct depicted by structure 5 unlikely, we cannot unambiguously distinguish between C4a adducts 3 and 4.
Fig. 2.

Isomeric allylic cations formed by protonation of the s-cis and s-trans conformers of vIPP.
We have also obtained structural data for substrate complexes of S. shibatae IDI-2 in the reduced state (Table 1, IPP-IDI and DMAPP-IDI). These structures diffract to 2.20 and 2.29 Å for IPP-IDI and DMAPP-IDI, respectively, which are higher than previously reported values of 2.64 and 2.90 Å, respectively (16). It is noteworthy that the IDI-2·substrate crystals are in the active reduced state and probably represent an equilibrium mixture of the IDI-2·IPP and IDI-2·DMAPP forms. This mixture cannot be discerned in the crystal, because the substrates can adopt conformations where the corresponding atoms of each locate at almost the same position in the active sites.
Spectroscopic Studies on Covalent Adducts.
S. shibatae IDI-2 was incubated with oIPP and vIPP in buffer containing NADH. Small molecules were removed by ultrafiltration under aerobic conditions, and UV-visible spectra of the retained protein were recorded. The spectrum of the S. shibatae IDI-2-vIPP adduct has a peak at approximately 400 nm (Fig. 3A) and resembles the spectrum previously reported for the corresponding T. thermophilus adduct generated under similar conditions (15). The spectrum of S. shibatae IDI-2-oIPP, which also has a peak at approximately 400 nm (Fig. 3A), is different from those reported for T. thermophilus IDI-2-oIPP, which have peaks at approximately 350 and approximately 410 nm when formed under anaerobic conditions (15) and at approximately 350 and approximately 430 nm after aerobic washes (12). When the flavin-inhibitor adducts are removed from S. shibatae IDI-2, both of their UV-visible spectra have a peak at approximately 370 nm and a characteristic shoulder at approximately 305 nm (Fig. 3B). These spectra are similar to those reported for the corresponding flavin-inhibitor adducts isolated from T. thermophilus IDI-2 (12, 15). Negative ion mass spectra of the S. shibatae adducts gave characteristic peaks for the [M-H]- ions at m/z = 731.0 for oIPP-adduct and m/z = 714.9 for vIPP-adduct (Fig. S3) in agreement with [M-H]- ions seen for the adducts formed by T. thermophilus IDI-2 (12, 15). Thus, both enzymes appear to give the same inhibitor-flavin adducts when incubated with vIPP and oIPP. The differences seen in the UV-visible spectra of S. shibatae and T. thermophilus IDI-2-oIPP probably result from differences in the local environment within the catalytic site of the two IDIs.
Fig. 3.

UV-visible spectra of flavin adducts. (A) UV-visible spectra of S. shibatae IDI-2 treated with vIPP (red), oIPP (blue), and IPP (purple). (B) UV-visible spectra of free vIPP-FMNred (red), oIPP-FMNred (blue), and FMN (purple), all separated from IDI-2 by treatment with 8 M guanidinium-HCl. (C) UV-visible spectra of an approximately 50 μM solution of T. thermophilus IDI-2 treated with vIPP (orange) and approximately 30 μM solutions of the free adduct at pH 6 (green) and in 6N HCl (magenta), all prepared under anaerobic conditions.
UV-visible studies were also performed with T. thermophilus IDI-2-vIPP and the flavin-vIPP adduct, which were generated, purified, and measured under anaerobic conditions, in order to obtain additional information. The UV-visible absorption spectrum of T. thermophilus IDI-2-vIPP (Fig. 3C, orange line) was similar to those for T. thermophilus and S. shibatae IDI-2-vIPP obtained under aerobic conditions. At pH 6.0, the spectrum of flavin-vIPP (Fig. 3C, green line) had a peak at 370 nm and a shoulder at 303 nm. This spectrum is similar to the corresponding adducts obtained from S. shibatae and T. thermophilus flavin-vIPP isolated under aerobic conditions (15). Thus, the flavin-vIPP adducts are not sensitive to oxygen, unlike the behavior anticipated for reduced flavins alkylated at N5 (21). In addition, N5-alkylated flavins are reported to have absorbance maxima between 320 and 355 nm at pH 5–8, whereas C4a adducts have maxima between 360 and 385 nm with a shoulder at approximately 300 nm (19, 22–24). Thus, spectra obtained at pH 6.0 for the anaerobically produced adduct are consistent with C4a adducts.
The UV-visible spectra of N5 and C4a flavin adducts have been studied by Hemmirich et al. at different pH values (19). According to their observations, changing from pH ∼ 7 to 6N HCl produces a blueshift in the peak maximum for N5 adducts, whereas the maximum for C4a adducts is redshifted. Previously, we had examined the UV-visible spectra of flavin-vIPP and flavin-oIPP in 5% trifluoracetic acid, pH 0.5 (15). However, a lower pH is required to fully protonate the flavin ring. In 6N HCl, the peak maximum shifts from 370 to 396 nm (Fig. 3C, magenta line), and the resulting spectrum is similar to those reported for C4a adducts (23).
Oxidation of N5 and C4a flavin adducts has also been well studied (21, 25). The N5 adducts are unstable in the presence of oxygen, whereas C4a adducts require light to initiate oxidation. During the oxidation reaction, N5 adducts give long-lived semiquinone species that can be detected by an increase in absorbance at 600 nm, whereas C4a flavins do not. The oxidation of the vIPP adduct required light. Continuous irradiation of the adduct at pH 6 with a 300-W slide projector resulted in an increase in the absorbance at approximately 370 and approximately 470 nm, which is consistent with the dealkylation or oxidation of the vIPP adduct to give oxidized FMN. During the course of the irradiation, essentially no change in absorbance was seen at approximately 600 nm, which suggests the absence of a flavin semiquinone (Fig. S4A). Photooxidation of flavin-vIPP in 6N HCl resulted in an increase in absorption at 266 and 396 nm, as expected for formation of protonated FMN (24) (Fig. S4B). The data from our UV-visible and crystallographic studies indicate that vIPP and oIPP alkylate FMNred at C4a.
Kinetic Studies of Gln160 Mutants.
In the X-ray structures of S. shibatae IDI-2·IPP and IDI-2·DMAPP, the side-chain amide moiety of the highly conserved glutamine at position 160 is positioned near the isoprene unit in the substrate on the opposite face from the reduced isoalloxazine unit in FMNred where it might function as a general acid-base catalyst (Fig. 4A). Thibodeaux et al. found support for this proposal by replacing Gln154 in Staphylococcus aureus IDI-2 with asparagine, which resulted in a 150-fold decrease in kcat (10). We created a series of S. shibatae IDI-2 mutants, where Gln160 was replaced with Asn, Glu, His, Leu, and Lys. The Q160N mutant showed a 150-fold decrease in kcat, although kcat/Km only decreased 66-fold (Table 2). Interestingly, decreases in the catalytic efficiencies (kcat/Km) of the mutants—Q160E (10-fold), Q160H (23-fold), and Q160L (28-fold)—were considerably smaller, except for Q160K (130-fold). In addition, the side chain of the Leu mutant cannot participate in a proton transfer step. We suggest that the side chain of Gln160 helps align the substrate in an orientation for optimal activity and that FMNred is the general acid-base catalyst for the proton transfer steps.
Fig. 4.

Active-site structure and proposed reaction mechanism of IDI-2. (A) The active site of S. shibatae IDI-2·FMNred·IPP complex (IPP-IDI). The substrate and the surrounding residues including FMNred are shown in stick models. Numbers beside yellow dotted lines indicate the distances between atoms. (B) Proposed mechanism for IDI-2, in which N5 of the zwitterionic species of reduced FMN acts as the general acid and the partial negative charge at C4a helps stabilize the 3° carbocationic intermediate. C5H10O4P and OPP in the structures represent the ribitol phosphate and diphosphate groups, respectively.
Table 2.
Kinetic parameters for Q160 mutants of S. shibatae IDI-2
| Enzymes | kcat (× 103 s-1) | Km, μM | kcat/Km, M-1 s-1 |
| Wild type | 29.9 ± 3.5 | 7.39 ± 1.63 | 4050 |
| Q160E | 3.28 ± 0.31 | 8.26 ± 1.41 | 397 |
| Q160H | 1.21 ± 0.11 | 7.13 ± 1.22 | 170 |
| Q160K | ND* | ND* | 31.1† |
| Q160L | 0.221 ± 0.014 | 1.52 ± 0.3 | 145 |
| Q160N | 0.197 ± 0.019 | 3.23 ± 0.75 | 61 |
*ND, not determined.
†Determined from the linear region of a v vs. [S] plot because of an apparently high 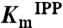 .
.
Discussion
IDI-1 and IDI-2 catalyze the interconversion of IPP and DMAPP by a protonation-deprotonation mechanism. Cysteine and glutamate residues in the active site of IDI-1, which have been implicated as general acid-base catalysts for the proton transfer steps, were identified by site-directed mutagenesis and covalent modification with mechanism-based irreversible inhibitors (2–5). X-ray crystal structures of the enzyme-inhibitor complexes were particularly useful in these studies. The general acid-base catalysts for IDI-2 are not as obvious. Amino acids Ser195, His155, Gln160, and Trp225 form a box that surrounds the isoprenoid moiety in the S. shibatae IDI-2·FMNred·IPP/DMAPP complex as shown in Fig. 4A. The plane defined by carbons C2–C5 in IPP and C1–C5 in DMAPP is stacked over the reduced polycyclic isoalloxazine unit in FMNred, whereas the oxygen and nitrogen atoms in the side-chain amide moiety of Gln160 point downward toward the opposite face of the isoprene unit. The heterocyclic rings of His155 and Trp225, both of which are arranged perpendicular to the plane of the isoprene moiety, form two opposing walls. The methyl group in IPP (the Z-methyl group in DMAPP) points toward the face of the imidazole ring in His155. Whereas the indole nitrogen of Trp225 is located 3.7 Å from C4 of IPP, the perpendicular arrangement of the isoprenyl moiety and the heterocyclic ring places the Trp225 hydrogen at N1 in a position where transfer to C4 of IPP is unlikely. The distance from the Trp225 N1 to IPP C2 is 5.8 Å. Ser195 and the diphosphate residue of IPP form the other two walls. Multiple sequence alignments show that valine is the most common amino acid at position 195 in IDI-2 with serine and threonine found in only 25% of the proteins. The diphosphate moiety in IPP is coordinated with Mg2+ and located in a hydrophilic pocket where it cannot participate in proton transfers to the isoprene unit. Thus, none of the residues forming the walls of the box are likely acid-base catalysts. Site-directed mutagenesis of Gln160 produces only modest reductions in the catalytic efficiency (kcat/Km) for the isomerization reaction, including the Q160L mutant, which shows only a 28-fold decrease. This behavior is inconsistent with Gln160 serving as a catalyst for the protonation-deprotonation steps. Thus, reduced FMN is the likely catalyst for protonation of IPP and DMAPP and for deprotonation of the 3° carbocationic intermediate.
Previous work with mechanism-based inhibitors indicates that the polycyclic isoalloxazine unit in FMNred is in intimate contact with the inhibitors (12–15). This result was confirmed by our X-ray studies with S. shibatae IDI-2 reacted with vIPP and oIPP. Analysis of fits to the electron density map suggested that the inhibitors were attached to C4a of reduced FMN. However, the resolution of the X-ray data was not sufficiently high to allow us to rigorously exclude the possibility that the inhibitors were attached at N5. We conducted a series of UV-visible spectroscopic studies on the flavin-inhibitor adducts produced by T. thermophilus and S. shibatae IDI-2. Spectra for the FMNred-vIPP adducts from both proteins recorded at pH 6 are similar when isolated under aerobic conditions (see Fig. 3B and ref. 15) and similar to the adduct from T. thermophilus when formed and isolated under strictly anaerobic conditions (Fig. 3C). In all three experiments, spectra with peaks at approximately 370 nm and shoulders at approximately 303 nm characteristic of C4a adducts were seen. In spectra of the anaerobic T. thermophilus adduct taken in 6N HCl, the peak at 370 nm shifted to 396 nm. The bathochromic shift in strongly acidic solutions is characteristic of C4a-flavin adducts, in contrast to the hypsochromic shift observed for N5 adducts. In addition, the adducts were stable in the presence of oxygen at pH 6 or, in 6N HCl, under conditions where N5 adducts typically oxidize to give long-lived semiquinones (21). Although the possibility of an N5 to C4a rearrangement cannot be ignored, it is typically slow (21) and is unlikely to account for our observations.
The pro-R proton at C2 of IPP is removed during isomerization to DMAPP and a proton is added to the re face of the double bond in DMAPP during isomerization to IPP (18). The conformation of the isoprenoid residue in the IDI-2·FMNred·IPP/DMAPP complex places the pro-R proton at C2 in IPP and the re face of the double bond in DMAPP in contact with the polycyclic isoalloxazine unit in FMNred. This orientation predicts a suprafacial transposition of the hydrogen atoms at C2 and C4 with the reduced flavin serving as the acid or base catalyst for the isomerization. In contrast, IDI-1 catalyzes an antarafacial transposition of the hydrogens (1). The mechanics of the proton transfers are unclear. The protons at N1 and N5 in FMNred are the most likely acids for protonation of the double bonds in IPP and DMAPP. The C2-C3-C4 carbons in the isoprene unit, which undergo changes in bonding during isomerization, are located on the convex surface of the reduced isoalloxazine moiety in a plane almost parallel with and approximately 3.1 Å above C4a in the X-ray structure of the IDI-2·FMNred·IPP/DMAPP complex. C3 is almost directly above C4a, whereas C2 and C4 are located 3.3 and 3.8 Å, respectively, from N5. In contrast, C2 and C4 are 4.8 and 3.8 Å, respectively, from N1.
N1, which is considerably more acidic than N5 in the dominant neutral diketo tautomer of FMNred, is not as well-positioned as N5 to facilitate protonation and deprotonation during the interconversion of IPP and DMAPP. If one considers the locations of hydrogen atom attached to N1, the distances to C2 and C4 in IPP increase to 5.6 and 4.3 Å, respectively. We and other researchers have proposed that a likely candidate for protonation of the isoprenoid double bond is N5 in the zwitterionic tautomer of reduced FMN (8, 10, 15, 16) (Fig. 4B). In this form, positively charged deprotonated N5 has an estimated pKa of approximately 4 in solution (26). The negative charge in the zwitterion is distributed over the π-network connecting O2 and O4 in the pyrimidine ring and is stabilized by the side-chain ammonium group in the conserved Lys193 located near N1 (16). This interaction should enhance the acidity of the N5 protons in the enzyme-bound cofactor. In addition, the hydrogen at N5 on the convex surface of the isoalloxazine moiety points toward IPP and is located < 3 Å from C2 and C4.
Upon protonation of the double bond, the resulting 3° carbocation would be stabilized by the partial negative charge at C4a, which augments the π-cation stabilization provided by His155 and Trp225. This stabilization may be enhanced by a charge transfer interaction between the highest occupied molecular orbital of anionic reduced flavin and the lowest unoccupied molecular orbital of the intermediate, as proposed for the purple intermediate of d-amino acid oxidase (27). In the zwitterionic mechanism, suprafacial proton transfer would be catalyzed with N5 functioning as an acid and a base in the protonation-deprotonation sequence, perhaps with a minor change in the position of the carbocation relative to the flavin between the protonation and deprotonation steps. It is likely that the C4a adduct we observed for vIPP is formed by collapse of the allylic cation-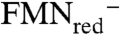 ion pair. Although it does not constitute proof, the activity seen for IDI-2·1-deazaFMNred and the absence of activity for IDI-2·5-deazaFMNred are consistent with the zwitterionic mechanism (6, 28).
ion pair. Although it does not constitute proof, the activity seen for IDI-2·1-deazaFMNred and the absence of activity for IDI-2·5-deazaFMNred are consistent with the zwitterionic mechanism (6, 28).
The role of FMNred in acid-base catalysis is an unrecognized function for the cofactor. Recently, Beyer and coworkers reported two FADred-dependent proteins—i.e., CrtY, lycopene cyclase, from Pantoea ananatis (29) and CRTISO, lycopene cis-trans isomerase, from tomato (30). An acid-base role of the flavin cofactor was proposed for CRTISO, whereas the suggested function of FADred in CrtY was to stabilize the positively charged transition state or intermediate. Thus, the roles proposed for reduced flavins involved in isomerization and cyclization reactions initiated by protonation of the trisubstituted carbon-carbon double bonds found in isoprenoid metabolites may be more widespread that initially anticipated.
Methods
UV-Visible Assays.
S. shibatae IDI-2 were incubated with the irreversible inhibitors in 50 mM MOPS-NaOH buffer, pH 6.0, containing 50 μM enzyme, 100 μM oIPP or vIPP, 1 mM NADH, 1 mM MgCl2, 0.2% CHAPS at 60 °C for 30 min. The reaction was stopped by chilling, and the enzyme was washed with 10 mM NH4HCO3 in three cycles of concentration and dilution using Amicon Ultra 10K centrifugal filters. The solvent was then exchanged with 1.25 mM NH4HCO3 followed by two cycles of concentration and dilution. UV-visible absorption spectra were measured by using a SHIMADZU UV-2350 spectrophotometer. A portion of the enzyme was denatured by adding 3× the original volume of 10 mM Tris·HCl, pH 7.4, containing 8 M guanidinium-HCl, followed by incubation at 30 °C for 5 min. The denatured enzyme was removed by ultrafiltration with spin columns before UV-visible spectra of flavin-inhibitor adducts were measured. All manipulations with S. shibatae IDI-2 were performed aerobically.
UV-visible assays for T. thermophilus IDI-2 were performed under anaerobic conditions as previously described (15), with the following modifications. At least 7 d before running an assay, all reagents were made anaerobic by degassing under vacuum followed by purging with argon and stored in an anaerobic (< 5 ppm O2) chamber (Coy Laboratories) until needed. Solutions containing enzyme and vIPP were purged the day of experimentation. Inhibition reactions were conducted in 100 mM Hepes buffer, pH 7.0, containing 100 μM T. thermophilus IDI-2·FMN, 2 mM MgCl2, and 8 mM Na2S2O4, and were initiated by addition of 400–800 μM vIPP. After overnight incubation at 37 °C in the anaerobic chamber, the samples were concentrated to approximately 10–20 μL and washed 3× with oxygen-free 10 mM NH4HCO3, pH 7.8, by using a Microcon 30 kDa molecular mass cutoff filter. The enzyme solution was concentrated and then diluted to 50 μM with 5% glycerol in 10 mM NH4HCO3, pH 7.8. A portion of the concentrated solution was diluted to approximately 30 μM in 20 mM NaH2PO4, pH 6.0, containing 8 M guanidinium-HCl. The denatured protein was removed by ultrafiltration. UV-visible measurements were conducted with the flavin-inhibitor adduct in the filtrate. Another portion of the concentrated solution of inactivated enzyme was diluted to approximately 30 μM with 6N HCl and centrifuged at 16,000 × g to remove denatured protein. The UV-visible spectrum of the filtrate was recorded. All manipulations for sample preparation were performed in an anaerobic chamber at 20 °C. The samples were removed from the chamber for spectroscopic measurements by using screw-top cuvettes sealed with a septum. Oxidation reactions were performed by removal of the screw top, stirring of the sample (with a parylene-coated micro stir bar, a generous gift from V&P Scientific, Inc., San Diego, CA) and exposure to light from a 300 W slide projector. Spectra were collected, and the total time of light exposure (not oxygen exposure) was recorded. UV-visible spectra were collected by using an Agilent 8453 diode array spectrophotometer or Beckman Coulter DU 730 spectrophotometer.
Coordinates.
Each of the adduct-complex structures—i.e., vIPP-IDI and oIPP-IDI—was constructed with the C4a adduct of the most likely chemical structures (structures 3 and 10 in Fig. S2, respectively), although the possibility of other isomeric C4a-adduct structures cannot be excluded (see SI Methods). It is possible that adducts with multiple isomeric structures are bound in the crystals.
Supplementary Material
Acknowledgments.
We thank Dr. Retsu Miura, Kumamoto University, for helpful discussions. This work was partly supported by grants-in-aid for scientific research from Japan Society for the Promotion of Science (23580111, to H.H.) and from Asahi Glass Foundation (H.H.) and by National Institutes of Health Grant GM 21552 from the Institute for General Medical Sciences (to C.D.P.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3B03 (vIPP-IDI), 3B04 (oIPP-IDI), 3B05 (IPP-IDI), and 3B06 (DMAPP-IDI)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115749108/-/DCSupplemental.
References
- 1.Kuzuyama T, Hemmi H, Takahashi S. Mevalonate pathway in bacteria and archaea. In: Mander L, Liu H-W, editors. Comprehensive Natural Products II Chemistry and Biology. Vol 1. Oxford: Elsevier; 2010. pp. 493–516. [Google Scholar]
- 2.Street IP, Coffman HR, Baker JA, Poulter CD. Identification of Cys139 and Glu207 as catalytically important groups in the active site of isopentenyl diphosphate:dimethylallyl diphosphate isomerase. Biochemistry. 1994;33:4212–4217. doi: 10.1021/bi00180a014. [DOI] [PubMed] [Google Scholar]
- 3.Street IP, Poulter CD. Isopentenyldiphosphate:dimethylallyldiphosphate isomerase: Construction of a high-level heterologous expression system for the gene from Saccharomyces cerevisiae and identification of an active-site nucleophile. Biochemistry. 1990;29:7531–7538. doi: 10.1021/bi00484a023. [DOI] [PubMed] [Google Scholar]
- 4.Wouters J, et al. Catalytic mechanism of Escherichia coli isopentenyl diphosphate isomerase involves Cys-67, Glu-116, and Tyr-104 as suggested by crystal structures of complexes with transition state analogues and irreversible inhibitors. J Biol Chem. 2003;278:11903–11908. doi: 10.1074/jbc.M212823200. [DOI] [PubMed] [Google Scholar]
- 5.Wouters J, Oudjama Y, Stalon V, Droogmans L, Poulter CD. Crystal structure of the C67A mutant of isopentenyl diphosphate isomerase complexed with a mechanism-based irreversible inhibitor. Proteins. 2004;54:216–221. doi: 10.1002/prot.10573. [DOI] [PubMed] [Google Scholar]
- 6.Hemmi H, Ikeda Y, Yamashita S, Nakayama T, Nishino T. Catalytic mechanism of type 2 isopentenyl diphosphate:dimethylallyl diphosphate isomerase: Verification of a redox role of the flavin cofactor in a reaction with no net redox change. Biochem Biophys Res Commun. 2004;322:905–910. doi: 10.1016/j.bbrc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. An unusual isopentenyl diphosphate isomerase found in the mevalonate pathway gene cluster from Streptomyces sp strain CL190. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothman SC, Helm TR, Poulter CD. Kinetic and spectroscopic characterization of type II isopentenyl diphosphate isomerase from Thermus thermophilus: Evidence for formation of substrate-induced flavin species. Biochemistry. 2007;46:5437–5445. doi: 10.1021/bi0616347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thibodeaux CJ, Mansoorabadi SO, Kittleman W, Chang WC, Liu HW. Evidence for the involvement of acid/base chemistry in the reaction catalyzed by the type II isopentenyl diphosphate/dimethylallyl diphosphate isomerase from Staphylococcus aureus. Biochemistry. 2008;47:2547–2558. doi: 10.1021/bi701467g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thibodeaux CJ, Chang WC, Liu HW. Linear free energy relationships demonstrate a catalytic role for the flavin mononucleotide coenzyme of the type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase. J Am Chem Soc. 2010;132:9994–9996. doi: 10.1021/ja104090m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma NK, Pan JJ, Poulter CD. Type II isopentenyl diphosphate isomerase: Probing the mechanism with alkyne/allene diphosphate substrate analogues. Biochemistry. 2010;49:6228–6233. doi: 10.1021/bi100844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston JB, Walker JR, Rothman SC, Poulter CD. Type-2 isopentenyl diphosphate isomerase mechanistic studies with cyclopropyl and epoxy analogues. J Am Chem Soc. 2007;129:7740–7741. doi: 10.1021/ja072501r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker JR, Rothman SC, Poulter CD. Synthesis and evaluation of substrate analogues as mechanism-based inhibitors of type II isopentenyl diphosphate isomerase. J Org Chem. 2008;73:726–729. doi: 10.1021/jo702061d. [DOI] [PubMed] [Google Scholar]
- 14.Hoshino T, Tamegai H, Kakinuma K, Eguchi T. Inhibition of type 2 isopentenyl diphosphate isomerase from Methanocaldococcus jannaschii by a mechanism-based inhibitor of type 1 isopentenyl diphosphate isomerase. Bioorg Med Chem. 2006;14:6555–6559. doi: 10.1016/j.bmc.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Rothman SC, Johnston JB, Lee S, Walker JR, Poulter CD. Type II isopentenyl diphosphate isomerase: Irreversible inactivation by covalent modification of flavin. J Am Chem Soc. 2008;130:4906–4913. doi: 10.1021/ja7108954. [DOI] [PubMed] [Google Scholar]
- 16.Unno H, et al. New role of flavin as a general acid-base catalyst with no redox function in type 2 isopentenyl-diphosphate isomerase. J Biol Chem. 2009;284:9160–9167. doi: 10.1074/jbc.M808438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Ruyck J, Pouyez J, Rothman SC, Poulter D, Wouters J. Crystal structure of type 2 isopentenyl diphosphate isomerase from Thermus thermophilus in complex with inorganic pyrophosphate. Biochemistry. 2008;47:9051–9053. doi: 10.1021/bi801159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao CL, Kittleman W, Zhang H, Seto H, Liu HW. Stereochemical analysis of isopentenyl diphosphate isomerase type II from Staphylococcus aureus using chemically synthesized (S)- and (R)-[2-2H]isopentenyl diphosphates. Org Lett. 2005;7:5677–5680. doi: 10.1021/ol0524050. [DOI] [PubMed] [Google Scholar]
- 19.Hemmirich P, Ghisla S, Hartmann U, Müller F. Chemistry and molecular biology of flavin in the “fully reduced” state. In: Kamin H, editor. Flavins and Flavoproteins: Proceedings of the Third International Symposium of Flavins and Flavoproteins. Baltimore: University Park; 1971. pp. 83–105. [Google Scholar]
- 20.Foresman JB, Wong MW, Wiberg KB, Frisch MJ. Theoretical investigation of the rotational barrier in allyl and 1,1,3,3-tetramethylallyl ions. J Am Chem Soc. 1993;115:2220–2226. [Google Scholar]
- 21.Walker WH, Hemmeric P, Massey V. Light-induced alkylation and dealkylation of flavin nucleus Stable dihydroflavins: Spectral course and mechanism of formation. Eur J Biochem. 1970;13:258–266. doi: 10.1111/j.1432-1033.1970.tb00926.x. [DOI] [PubMed] [Google Scholar]
- 22.Dudley KH, Ehrenberg A, Hemmerich P, Muller F. Spectra and structures of components of the flavin redox system. Helv Chim Acta. 1964;47:1354–1383. [Google Scholar]
- 23.Scolanagelschneider G, Brustlein M, Hemmerich P. 4a-Alkyldihydroflavin—Coenzyme synthesis and modification of flavodoxin. Eur J Biochem. 1976;69:305–314. doi: 10.1111/j.1432-1033.1976.tb10887.x. [DOI] [PubMed] [Google Scholar]
- 24.Hemmerich P, Haas W. Recent developments in the study of “fully reduced flavin”. In: Yagi K, editor. Reactivity of Flavins: The Proceedings of the Symposium Dedicated to the Late Professor Leonor Michaelis under the Auspices of the Japanese Biochemical Society. Tokyo: University of Tokyo; 1975. pp. 1–13. [Google Scholar]
- 25.Blankenhorn G, Hemmerich P. Oxidative dealkylation of 4a-alkylated 4a,5-dihydroflavins: Properties of 4a-alkylated flavin radicals. Tetrahedron. 1979;35:1129–1134. [Google Scholar]
- 26.Macheroux P, Ghisla S, Sanner C, Ruterjans H, Muller F. Reduced flavin: NMR investigation of N5-H exchange mechanism, estimation of ionisation constants and assessment of properties as biological catalyst. BMC Biochem. 2005;6:26. doi: 10.1186/1471-2091-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizutani H, et al. Three-dimensional structure of the purple intermediate of porcine kidney D-amino acid oxidase. Optimization of the oxidative half-reaction through alignment of the product with reduced flavin. J Biochem. 2000;128:73–81. doi: 10.1093/oxfordjournals.jbchem.a022732. [DOI] [PubMed] [Google Scholar]
- 28.Kittleman W, Thibodeaux CJ, Liu YN, Zhang H, Liu HW. Characterization and mechanistic studies of type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase from Staphylococcus aureus. Biochemistry. 2007;46:8401–8413. doi: 10.1021/bi700286a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu QJ, et al. The lycopene cyclase CrtY from Pantoea ananatis (formerly Erwinia uredovora) catalyzes an FADred-dependent non-redox reaction. J Biol Chem. 2010;285:12109–12120. doi: 10.1074/jbc.M109.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu QJ, Ghisla S, Hirschberg J, Mann V, Beyer P. Plant carotene cis-trans isomerase CRTISO: A new member of the FADred-dependent flavoproteins catalyzing non-redox reactions. J Biol Chem. 2011;286:8666–8676. doi: 10.1074/jbc.M110.208017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.