

Abstract
Cellular functions of proteins are strongly influenced by their interactions with other proteins. The frequency of protein interactions is a function of the local concentration of two proteins and their affinity for one another. When two proteins are tethered together, the link between them influences their effective concentrations and therefore the frequency of their interaction. Currently no methods exist to systematically vary the effective concentration within this intramolecular interaction. Here we outline a modular, genetically encoded linker, namely, an ER/K [genetically encoded polypeptide motif based on alternating sequence of approximately four glutamic acid (E) followed by approximately four arginine (R) or lysine (K) residues] single α-helix that can be used to regulate the frequency of interaction between two proteins, or between a protein and a peptide, one at each end. We exploit the wide range of interaction affinities between calmodulin and its binding peptides, combined with FRET to determine the effect of the ER/K α-helix in regulating protein interactions. We find that increasing the length of the ER/K α-helix reduces the on rate of the intramolecular interaction without significantly affecting the off rate, regardless of the affinity of the bimolecular interaction. We outline a genetically encoded approach to determine the dissociation constant for both moderate (micromolar Kd) and strong (nanomolar Kd) protein interactions. Our studies demonstrate the use of the ER/K α-helix to systematically engineer FRET biosensors that detect changes in concentration or affinity of interacting proteins, and modulate enzyme autoinhibition. Our findings are consistent with the ER/K α-helix as a worm-like chain with rare, stochastic breaks in the helix backbone that may account for the behavior of myosin VI stepping along actin.
Keywords: systematic protein affinity strength, modulation, cell signaling, biochemistry
Interactions between proteins are essential for cellular function. The association of proteins facilitates their localization to, and therefore function in, specific subcellular compartments (1). Proteins often associate to form a macromolecular complex that can perform functions that the individual components cannot (2). Brief bimolecular interactions among a series of proteins are characteristic of signaling cascades that are essential to propagate and amplify an extracellular signal across the cell and elicit an appropriate response (3). Interactions between proteins are dictated by their binding affinity and the local cellular concentrations of the proteins involved. In addition to bimolecular interactions between proteins, many proteins have intramolecular interactions between distinct domains that regulate their function (4). Such intramolecular interactions within a protein in turn are regulated by the free solution binding affinity of the two domains and the effective concentration experienced by the two domains within the protein.
Regulation of intermolecular and intramolecular protein interactions is important to gain mechanistic insight into cellular processes and is an essential component of pharmacological intervention in disease states. However, proteins have complex structures with predictions of about 1,000 structural families (5) with 10,000 distinct types of interactions between proteins (6). Detailed insight into the intermolecular or intramolecular interaction interface gained from techniques such as X-ray crystallography, NMR, and computational modeling can be used to perturb the interaction by altering the sequence of one or both proteins/domains using site-directed mutagenesis (7, 8). This approach, however, is limited by the effects of the mutation on the structure and folding of the protein domains, which is complicated by the allosteric nature of protein structure (9). Further, site-directed mutagenesis often results in a loss of protein–protein interaction and is therefore usually an all-or-nothing approach to regulation. Given the importance of protein–protein interactions, it is necessary to have an alternate approach that can systematically modulate protein–protein interactions without perturbing the structure of the proteins or protein domains involved.
In this study, we outline the use of a modular protein linker, namely an unusual single ER/K [genetically encoded polypeptide motif based on alternating sequence of approximately four glutamic acid (E) followed by approximately four arginine (R) or lysine (K) residues] α-helix that by virtue of its structural properties can be used to dial-in the frequency of interaction of two proteins or protein domains attached to its ends. We find that varying the ER/K α-helix length systematically alters the frequency of interaction regardless of the free solution binding affinity of the interacting pair attached to the ends of the ER/K α-helix.
Results
Theoretical Estimate of the Effect of the ER/K α-Helix Linker on kclose and kopen.
This study examines the use of the ER/K α-helix as a modular linker that regulates the frequency of interaction of two proteins or protein domains at its ends. In order for the ER/K α-helix to regulate the frequency of interaction, it must limit either the on rate (kclose) or off rate (kopen) of the interaction. Further, this limit should vary systematically with the length and/or structural properties of the ER/K α-helix.
Previous studies are consistent with the ER/K α-helix behaving as a worm-like chain (WLC) with a persistence length (Lp) of approximately 15 nm (10). A Monte Carlo simulation of the ER/K α-helix, modeled as an ideal WLC with Lp = 15 nm, provided a distribution of conformations adopted by different length ER/K α-helices (Fig. 1 A and B, Left). Extensive conformational sampling (> 20 million) was used to estimate the probability of the two ends of the ER/K α-helix being close enough to facilitate interaction between two proteins. This probability is defined as the fraction of total number of conformations sampled with end-to-end distance less than 2 nm. This length was used as the distance between the ends of the ER/K α-helix when two small proteins [ca. 20 kDa with hydrodynamic radius (Rh) ca. 2 nm] bind to each other. However, the conclusions of different models do not vary for lengths ranging from 1 to 3 nm. This probability was computed over a range of ER/K α-helix lengths (L) and was found to be vanishingly small (< 10-5) for L comparable to the Lp. Mehraeen et al. (12) have previously reported theoretical distributions for end-to-end distance distributions for WLCs. Their theoretical distributions show that the cumulative distribution of end-to-end distances less than 2 nm are also on the order of 10-5 for L ∼ Lp. These results suggest that, for a previously estimated Lp of 15 nm (11) and L ranging from 10–30 nm, the ER/K α-helix could limit the kclose of interaction of proteins at its ends. Given the rarity of Monte Carlo derived conformations with end-to-end distances less than 2 nm, this approach cannot accurately estimate the effect of ER/K α-helix length on the frequency of interaction. The theoretical distributions of Mehraeen et al. (12) suggest that, for an ideal WLC with Lp = 15 nm, the number of conformations with end-to-end distance less than 2 nm increases with ER/K α-helix length ranging from 10 to 30 nm. However, given the low frequency of occurrence of these conformations, it is possible that any deviations of the ER/K α-helix from an ideal WLC will likely influence this trend. Nonetheless, the ER/K α-helix is a good candidate linker to limit kclose.
Fig. 1.

ER/K α-helix as an ideal WLC. (A) Ten nanometer helix, (B) 30 nm helix. Sample conformations derived from a metropolis Monte Carlo simulation of the backbone of the ER/K α-helix. (Left) Helix free in solution. (Right) Ends of the helix are connected by a stiff spring (100 pN/nm) with resting length 2 nm. Monte Carlo simulations are used to estimate the probability of interaction between the ends, which in turn influences the on rate of the intramolecular interaction (kclose). Mean bending energy stored in open and closed conformations is used to predict the effect of the helix on the off rate (kopen).
The ER/K α-helix could also alter kopen. For the ER/K α-helix as an ideal WLC, the additional energy stored in the closed state relative to the open state decreased from 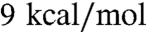 (1 kcal = 4.18 kJ) for L = 10 nm to 5 kcal/mol for L = 20 nm and 3 kcal/mol for L = 30 nm (see SI Data). For comparison, the strength of a single protein salt bridge between proteins has been estimated to range from 0.5 to 4 kcal/mol (13). The probability of dissociation of the protein–protein interaction will therefore increase as the energy needed to hold the ER/K α-helix in the closed state increases. Therefore, the ER/K α-helix modeled as an ideal WLC is a good candidate to increase kopen relative to that of the free bimolecular interaction.
(1 kcal = 4.18 kJ) for L = 10 nm to 5 kcal/mol for L = 20 nm and 3 kcal/mol for L = 30 nm (see SI Data). For comparison, the strength of a single protein salt bridge between proteins has been estimated to range from 0.5 to 4 kcal/mol (13). The probability of dissociation of the protein–protein interaction will therefore increase as the energy needed to hold the ER/K α-helix in the closed state increases. Therefore, the ER/K α-helix modeled as an ideal WLC is a good candidate to increase kopen relative to that of the free bimolecular interaction.
Design of a Systematic Protein Affinity Strength Modulation (SPASM) FRET sensor.
We have termed our approach SPASM. Fig. 2A shows a schematic of a single polypeptide chain used to detect ER/K α-helix modulation of the interaction between calmodulin (CAM) and its binding peptide (peptide). This single polypeptide chain is termed the SPASM FRET sensor to emphasize the presence of the ER/K α-helix and the FRET pair for sensing the frequency of interaction of the protein and protein/peptide at the ends of the ER/K α-helix. From the N to the C terminus, the sensor contained enhanced CFP (eCFP, the FRET donor), CAM, ER/K α-helix, mCitrine (mCit, the FRET acceptor), and a peptide with known affinity for binding Ca2+-activated CAM. All components were separated by a (Gly-Ser-Gly) × 4 [(GSG) × 4] linker to ensure rotational freedom.
Fig. 2.

SPASM sensor design. (A) Schematic of protein domains (His6 is at the N terminus) in the SPASM sensor. A 12 amino acid Gly-Ser-Gly (GSG)4 linker is placed between different domains to ensure rotational freedom. (B) Structural model of the SPASM sensor in the open (Left) and closed (Right) conformation. (C) Sequences of two ER/K α-helices. Note that the 20-nm helix used in this study is the first 130 amino acids of the 30-nm helix sequence. (D, Left) Coomassie gel staining of purified SPASM sensors with 30 nm (lanes 1 and 2) and 20 nm (lanes 3 and 4) ER/K α-helices. Lanes 2 and 4 were treated with TEV-protease, which cleaves at its recognition site engineered between the CAM and ER/K α-helix. (Right) Laser fluorescence gel scan (457-nm eCFP excitation with 520BP40 emission). This condition captures both eCFP and mCitrine fluorescent bands. (E) Absorption spectrum of purified SPASM sensor showing peaks at 433 nm (eCFP) and 514 nm (mCit). Note that mCit has approximately twofold higher extinction coefficient compared to eCFP.
CAM is a signaling molecule that binds to Ca2+ and switches from an inactive state [apocalmodulin (Apo CAM)] to an active state (CAM) (14). Ca2+ binding causes a dramatic increase in its binding affinity for a variety of effectors including protein kinases, phosphatases (calcineurin), ion channels, and active transporters (Ca2+ pump). CAM binding to these proteins triggers their activation in response to Ca2+ influx. A wide variety of small peptides that bind specifically to Ca2+ bound CAM and Apo CAM have been identified, and their affinities for binding CAM have been systematically quantified (15). CAM-peptide interactions are a model system to study ER/K α-helix modulation of protein–protein interactions. CAM-peptide binding affinity can be easily controlled in vitro by the presence or absence (chelation with EGTA) of Ca2+. The wide range of interaction affinities derived from CAM effector binding sites, both in the Ca2+ bound and Apo states, can be used to quantify the contribution of the ER/K α-helix to the on and off rates of protein–protein interactions.
FRET was used as a measure of the interaction between CAM and Ca2+. FRET occurs between eCFP and mCitrine, variants of CFP and YFP, which are used because of their increased brightness and fluorophore stability (16). When CFP and YFP are in close proximity (≤ 8 nm) excitation of CFP results in significant FRET from CFP to YFP. FRET between CFP and YFP has been used extensively to assess interaction between two proteins that are either free in solution or tethered together with a linker (17, 18). In the absence of Ca2+, the ER/K α-helix was designed to separate eCFP and mCit with the sensor adopting an “open” state (Fig. 2B, Left). In the presence of Ca2+, the high affinity of the peptide for CAM was designed to hold the ER/K α-helix in a “closed” state (Fig. 2B, Right) and bring eCFP and mCit within FRET range. The emission spectrum of the sensor sample following excitation of the donor, referred to as the FRET spectrum, is a function of the relative number of sensor molecules in the open and closed states. There are several ER/K α-helical protein motifs with L = 10, 20, and 30 nm in naturally occurring proteins (10). In this study, a 10-nm helix from myosin VI, and a 20- and 30-nm helix derived from the Kelch-motif family protein were used (Fig. 2C) for our SPASM sensor constructs. The sensor was purified using a combination of affinity (Ni-nitrilotriacetate) and gel filtration, and the purity was assessed using laser fluorescence gel scanning and Coomassie staining of the PAGE run samples (Fig. 2D), in addition to the sample absorption spectrum showing peaks at 433 nm (eCFP) and 514 nm (mCit) (Fig. 2E) (see SI Materials and Methods for details).
FRET Ratio (R) Varies Linearly with Fraction of Sensor in the Closed State.
For an ideal WLC with Lp = 15 nm, the mean end-to-end distances for ER/K α-helices with L = 10, 20, and 30 nm are 9, 16, and 23 nm, respectively (19). Therefore, in the absence of CAM-peptide interactions, no significant FRET between eCFP and mCit is expected (R0 ∼ 5 nm; ref. 20). Accordingly, sensor constructs with 10-, 20-, and 30-nm helices that have the CAM-binding peptide substituted by an extended linker (GSG) × 4 with no known affinity for CAM showed no measurable FRET (Fig. 3A). For these constructs, selective excitation of the donor (eCFP at 433 nm) resulted in donor emission (eCFP emission maximum at 475 nm) without significant acceptor emission (mCit emission maximum at 525 nm) (Fig. 3A). A CAM-binding peptide that selectively binds to the activated Ca2+ bound CAM underwent a dramatic increase in FRET when CAM was transitioned from the Apo (Fig. 3B, EGTA) to Ca2+ bound state (Fig. 3B, calcium). The change in signal was found to be similar for 10-, 20-, and 30-nm ER/K α-helices. It must be noted that the concentrations of sensors used in these experiments ranged from 5 to 50 nM, well below the previously reported free solution Kd (equilibrium dissociation constant) of this CAM-peptide interaction (ca. 1 µM) (21). Therefore, the change in FRET is derived primarily from intramolecular interactions with minimal contribution from intermolecular interactions. This observation was also confirmed by showing that serial dilution of the sample does not significantly alter the ratio of mCit (acceptor) to eCFP (donor) emission (see below).
Fig. 3.

FRET as a readout of protein interaction. (A) Emission spectra of control SPASM sensors with CAM linked to a 12 amino acid Gly-Ser-Gly peptide (GSG)4 by 10-, 20-, and 30-nm ER/K α-helices. Spectra are taken at 4 mM Ca2+. Excitation of eCFP (430 nm) results in eCFP emission (475 nm) without significant mCit emission (525 nm). (B) Sample emission spectrum of SPASM sensor with CAM linked by a 20-nm ER/K α-helix to a C15W peptide (21) that interacts with 1 μM affinity to Ca2+-activated CAM. Excitation of eCFP (430 nm) results in eCFP emission (475 nm) with no detectable FRET for mCit in the absence of Ca2+ (chelation by 1 mM EGTA) or strong FRET (large mCit peak at 525 nm) in the presence of 4 mM Ca2+.
The relative number of sensors in the closed state, also termed fc for fraction closed [fraction open (fo) is 1-fc], is related to the ratio of the intramolecular on and off rates by the following equation
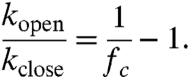 |
[1] |
Here kopen and kclose are the intramolecular off and on rates, respectively. In order to derive fc from the FRET spectrum, the equivalent bimolecular system was used where fc can be varied systematically by changing the sensor concentration (see SI Discussion). A tobacco etch virus (TEV) protease site was built into the sensor between the CAM and the ER/K α-helix. Following purification of the sensor as a single polypeptide chain (see Materials and Methods), TEV protease was used to obtain stoichiometrically equivalent amounts of eCFP-tagged CAM and mCit-tagged binding peptide that participate in a bimolecular interaction. Complete digestion with TEV was verified by laser fluorescence gel scanning (Fig. 2D). TEV-digested sample was concentrated to over 10-fold (16 μM) above the Kd (ca. 1 µM) to facilitate the bimolecular interaction. Bimolecular interactions between Ca2+-activated CAM and peptide resulted in high FRET (Fig. 4A). Serial dilutions of the sample were used to reduce the equilibrium fraction bound in accordance with the Kd, and therefore the fraction of molecules in the high FRET state, resulting in a reduction in mCit emission with decreasing concentration of the sample (Fig. 4B). Given the Kd of the interaction, the fc for a given concentration of the sample is directly calculated from Eq. 2 (note, Fig. 4B top vs. bottom abscissa).
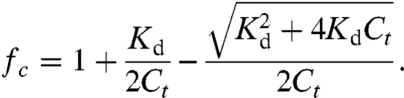 |
[2] |
Fig. 4.

FRET ratio varies linearly with fraction of interacting protein in the bound state. (A) Emission spectra of SPASM sensors digested with TEV-protease to cleave a site between CAM and the ER/K α-helix, resulting in a bimolecular interaction between eCFP-tagged CAM and mCit-tagged C15W peptide (ca. 1-µm affinity). Sample spectra corresponding to 160- and 16-μM interaction affinities are shown. (B) Measured FRET ratio (mCit/eCFP) as a function of sensor concentration (top abscissa) and equilibrium fraction bound (bottom abscissa). Dotted line represents fit to Eq. S2. Linear regression fit of FRET ratio to equilibrium fraction bound has R2 of 0.996.
The FRET ratio (mCitmax/eCFPmax) varied approximately linearly with fc (Fig. 4B, R2 = 0.993 for linear regression fit). The approximate linear relationship between FRET ratio (R) and fc is consistent with a two-state model of the FRET sensor (see Materials and Methods). R for a given fc was therefore expressed as a linear combination of the Robs of the unbound (open) state (Ropen) and the bound (closed) state (Rclosed).
| [3] |
Robs, unlike the FRET spectrum, is normalized for the number of sensor molecules present in the sample. Although Rclosed and Ropen cannot be directly measured, they can be estimated from the equivalent bimolecular interaction as discussed in the next section.
FRET Ratio (R) Can Be Used to Measure Kd of Medium Affinity Interactions (ca. Micromolar).
The use of R to measure fc was independently validated using another approach. Given the linear relationship between fc and FRET ratio (Robs), the data for the bimolecular interaction (Fig. 5A) can be fit to Eqs. 2 and 3 with Kd, Ropen, and Rclosed as fit parameters. The fit yielded a Kd of 1.3 μM, which agrees well with a previous measurement of 1.7 μM for this CAM-peptide interaction pair (21). Although Ropen and Rclosed were allowed to float for the fit, they are constrained by Robs at high and low Ct. However, the Kd measurement and the quality of the fit (R square) were not affected significantly (< 10% difference) when Ropen and Rclosed were constrained to Robs at low and high concentrations, respectively. Therefore, a single parameter fit of the experimental data to Eqs. 2 and 3 was sufficient to measure Kd. This approach to measure Kd using R vs. C can be universally applied to any protein–peptide or protein–protein interaction. A single concentrated protein sample (ca. 20-fold higher than the expected Kd with 50 μL volume) is sufficient to obtain the entire curve using serial dilution (Fig. 5 represents the mean ± SD for four samples; see Materials and Methods). Coexpression of the protein and protein/peptide with both a TEV-protease site and an ER/K α-helix between them provides an easy method to obtain stoichiometrically equivalent amounts of the two binding partners from a single purified sample. The ER/K α-helix acts as a spacer that allows proper folding of the two proteins. In order to accurately estimate the Kd of two interacting proteins using this approach, it is essential to measure Robs over a range of bimolecular concentrations that bracket the Kd. For very high-affinity interactions (< 10 nM) the FRET spectrum from the fluorometer has lower signal to noise and the accuracy of Robs is reduced. For such high-affinity interactions, competitive binding of either of the fluorescently tagged proteins to a nonfluorescent partner with known affinity can be used, as described in the next section.
Fig. 5.

Quantifying the equilibrium dissociation constant using FRET; Determination of the effective concentration of interacting partners in a SPASM sensor. (A) FRET ratio (mCit/eCFP) as a function of protein concentration. Blue dots, intact SPASM sensor with C15W peptide in the presence of 4 mM Ca2+. Red dots, SPASM sensor with C15W peptide digested with TEV-protease to cleave SPASM sensor into eCFP-tagged CAM and mCit-tagged C15W peptide. Black line, bimolecular interaction fit to Eqs. 2 and 3 to calculate Kd of bimolecular interaction. (B) Competitive binding of VIP (50 nM affinity) to TEV-cleaved SPASM sensor with CAM and Trp3 peptide (0.3 nM Kd). Inverse FRET ratio (eCFP/mCit) increases as eCFP-CAM/mCit-peptide interaction is disrupted by VIP. (C) Competitive binding of free unlabeled CAM to intact SPASM sensor linked to C15W peptide by 10 nm (red), 20 nm (green), and 30 nm (blue) ER/K α-helices. Inverse FRET ratio (eCFP/mCit) increases as the open conformation of the sensor is favored by increased unlabeled CAM concentration. (A–C, Insets) Low concentration data shown on log axis for clarity. (D) Effective concentration of CAM-peptide interaction within the SPASM sensor measured by competitive binding to free unlabeled CAM. Black squares, peptides that bind to CAM with indicated affinities in the presence of Ca2+. Blue diamonds, Apo CAM binding to peptide with 0.7-μM affinity. All data shown are mean ± SD of at least four different experiments.
FRET Ratio (R) Can Be Used to Measure the Kd of High-Affinity interactions (ca. Nanomolar).
Competitive binding assays have been used to determine the Kd of high-affinity interactions. However, the techniques for detecting competitive binding require multiple steps, for instance, centrifugation followed by either quantitative analysis of PAGE gels by Coomassie stain, immunoblotting, or a radioimmunoassay (22). In this study, FRET was used as a quantitative readout of competitive binding. Inverse FRET ratio (IR), defined as the ratio of eCFP (475 nm) to mCit (525 nm) emission maxima, decreases as the vasoactive intestinal peptide (VIP) competes for binding in the bimolecular FRET interaction between eCFP-tagged CAM and mCit-tagged Trp3 (Fig. 5B). We measured a Kd of 53 nM for the VIP-based on a 0.3-nM affinity of the Trp3 peptide (see Materials and Methods), which is nearly identical to the previously reported 54-nM affinity for this interaction (23).
Effective Concentration of the Intramolecular Interaction Decreases with Increasing ER/K α-Helix Length.
Studies on the equivalent bimolecular interaction can be used to measure the FRET ratio corresponding to the open (Ropen) and closed (Rclosed) states, which in turn can be used to calculate fc (Eq. 3) and hence kopen/kclose for different SPASM sensors. One drawback of this approach is that, as fc approaches one, the kopen/kclose is very sensitive to the accuracy of fc (Eq. 1); fc approaches one for protein/peptide pairs that interact with high affinity (ca. nanomolar). In addition, for such high-affinity interactions we cannot use the serial dilution approach, outlined earlier, to measure Ropen and Rclosed. Hence, an alternative approach to measure fc-based competitive binding of the sensor to free unlabeled CAM was used (Fig. 5C, Materials and Methods). This technique of measuring fc can be applied to both weak- and strong-affinity interactions between the protein and protein/peptide within the sensor.
An alternate measure of the strength of interaction between the two proteins is the effective concentration (Ceff). Effective concentration is the concentration of the protein/peptide pair in the equivalent bimolecular interaction that has an fc equal to that of the intramolecular interaction within the SPASM sensor. The effective concentration can be estimated from the competitive binding assay (see Materials and Methods). The higher the Ceff or fc, the stronger the interaction between the protein–peptide pair within the sensor and vice versa. The advantage of using Ceff over fc is that, as fc approaches one, small changes in fc can cause large changes in Ceff. Therefore, comparing interaction strengths based on Ceff is easier than with fc. Fig. 5D is a summary of Ceff for the intramolecular interaction between CAM and corresponding peptide, measured using five different interaction affinities (four different peptides with one in the Apo CAM state), for three ER/K α-helices with length 10, 20, and 30 nm. Regardless of the affinity of interaction between CAM and peptide, or the state of activation of CAM, the Ceff for a given interaction was found to decrease with increasing ER/K α-helix length (Fig. 5D). Specifically, Ceff decreased about 10-fold when helix length increased from 10 to 20 nm and about threefold when the helix length increased from 20 to 30 nm. Hence the ER/K α-helix can be used to systematically effect large changes in Ceff between interacting proteins. Further, we estimated the off rate (kopen) of the CAM-Trp3 peptide sensor by monitoring the quench of FRET signal following incubation of vast excess of unlabeled CAM (Fig. S1). These measurements (see SI Data) show the ER/K α-helix does not affect the off rate (kopen). Therefore the effect of the ER/K α-helix on the effective concentration is mediated through the on rate (kclose).
Discussion
Structural Role of the ER/K Motif in Naturally Occurring Proteins.
Glu4(Arg/Lys)4 (ER/K) is a naturally occurring motif that we had identified to be present in numerous proteins from divergent species with diverse functions (10). We have previously used a variety of biophysical approaches to summarily characterize its behavior as a semirigid spacer between protein domains (11). In this study, we find that, although the ER/K α-helix predominantly adopts an extended conformation (Fig. 3A), rare stochastic breaks in it enable two protein domains placed one at each end to interact (see SI Data). Although the frequency of breaks increases linearly with increasing ER/K α-helix length, the probability of end-to-end interaction decreases due to the lack of spatial coordination between the breaks (see Fig. S2 for simplified model). Taken together, the rare stochastic breaks result in an approximately 30-fold decrease in effective concentration of the ends when the ER/K α-helix length is increased threefold (Fig. 5D). In the context of the ER/K motif in naturally occurring proteins, the findings of this study have profound implications for the contribution of this domain to protein function. Specifically, if the protein domains separated by the ER/K α-helix have moderate (ca. micromolar) to high (ca. nanomolar) affinity for each other, the length of the ER/K motif will dictate the frequency of the interaction. For proteins such as the molecular motor myosin, kinases, or translation initiation factors, the frequency of interaction is likely to influence enzymatic activity (10). In this study, we quantify the effective concentrations for a range of bimolecular interaction affinities (0.2 nM–1 μM) that will serve as a measure of effective concentration of different interactions separated by an ER/K α-helix of defined length (Fig. 5D). Further, we isolate ER/K α-helix dependent on-rates that when combined with the kinetics of individual interactions will enable predictions of the preferred conformation (open vs. closed) of the protein.
We have previously reported that the ER/K α-helix within myosin VI forms an extended structure that extends the lever arm stroke of this molecular motor (10). The lever arm stroke of several myosins, including myosin VI has been recorded using optical tweezers (24). The upper bound on the stroke time of the lever arm is 100 μs, which is determined by the time resolution of the optical trapping measurements (10 kHz). The fact that myosin VI has a large stroke is therefore consistent with the time between helix breaks being larger than 100 μs, such that helix breaks do not influence the stroke size. In a processive myosin VI dimer, the lever arm stroke of the leading head is followed by a dwell, when the trailing head searches for the next binding site. Stochastic breaks in the ER/K α-helix during this dwell would allow the trailing head to bind actin at different sites, corresponding to the location of the break in the helix. Interestingly, the model with stochastic breaks is consistent with the wide distribution of step sizes in myosin VI, relative to myosin V, which does not have an ER/K α-helix (25). Therefore, the influence of the ER/K α-helix linker on protein function appears to be different at different timescales.
Engineering FRET Biosensors.
Since the first genetically encoded protein FRET biosensor to detect calcium signaling over a decade ago (26), a large number of similar designs have been developed to directly detect the activation of proteins involved in cell signaling pathways (18, 27, 28). These FRET biosensor designs are based on a single polypeptide chain with CFP and YFP variants separated by two protein domains that interact in response to cell signaling events such as calcium influx or phosphorylation of one of the protein domains. The two protein domains in these biosensors are separated by a short (approximately three) amino acid linker composed of Gly and Ser. As a consequence, the CFP/YFP pair is in close proximity with substantial FRET under basal conditions. Activation of the sensor causes the protein domains to interact, which results in a decrease in distance and rotational freedom between the FRET pair and therefore increased FRET. The change in FRET signal, assessed from the emission spectrum following donor excitation, is limited by the close proximity of the CFP/YFP pair enforced by the short linker between the protein domains. Enhancing the change in FRET signal involves either changing the specific linker sequence by trial and error (29) or altering the arrangement of protein elements in the biosensor using circular permutation (30). In this study, we find that the ER/K α-helix minimizes baseline FRET by virtue of its extended structure (Fig. 3A), resulting in a substantial increase in the dynamic range of the biosensor following activation (Fig. 3B). The availability of different length ER/K α-helices allows systematic alteration of the effective concentration of the interacting proteins (Fig. 5B), such that the ER/K α-helix provides an affinity barrier that separates the protein domains in the inactive conformation. The biosensor constructs with the ER/K α-helix are designed with (Gly-Ser-Gly)4 linkers between the proteins to ensure rotational freedom between the protein domains. This feature, combined with the wide range of conformations explored by the ER/K α-helix (Fig. 1), should allow protein domains with diverse binding interfaces to interact without steric hindrance. Further, the plasmid constructs for the biosensors containing the ER/K α-helix have been designed with unique restriction enzyme sites flanking different protein domains to allow easy cut-and-paste cloning using PCR.
Determining the Dissociation Constant of Protein–Protein Interactions.
Several approaches exist to determine the dissociation constant (Kd) of a pair of interacting proteins. In this study, we use the ER/K α-helix to make stoichiometric amounts of the interacting partners. TEV-protease is used to separate the interacting pair. We show that the FRET ratio correlates linearly with equilibrium fraction bound (Fig. 4B). A straightforward dilution series with FRET ratio measured on a fluorometer is therefore sufficient to measure the Kd of moderate affinity (ca. micromolar) interactions (Fig. 5A). This technique can also be extended to measure the Kd of high-affinity interactions (ca. nanomolar) using a competitive binding assay (Fig. 5B). Key advantages of this approach over other spectroscopic techniques (measurement of tryptophan fluorescence or dansyl labeling) is that it is both genetically encoded and generally applicable to any protein interacting pair. The use of the ER/K α-helix involves expressing a single polypeptide chain with both proteins in stoichiometric amounts.
Protein Engineering with the ER/K α-Helical Linker.
Three key features of the ER/K α-helix for protein engineering are (i) it is genetically encoded, (ii) varying length ER/K α-helices are readily available from naturally occurring ER/K motifs of different lengths, and (iii) the ER/K α-helix by virtue of its alternating charge is unlikely to interact with the protein domains at its ends and is stable under extremes of pH, salt concentration, and temperature. Regardless of the type of linker used to regulate interactions between proteins, detailed characterization of the effect of the linker on the on and off rates of the interaction is necessary before it can be generally applied. In this study, we use the wide range of interaction affinities between calmodulin and its binding peptides to characterize the effect of different length ER/K α-helices. Specifically, we find that the ER/K α-helix influences the on rate without significantly impacting the off rate relative to the bimolecular interaction. Our detailed characterization, combined with the unique properties of the ER/K α-helix, should enable its use as a modular linker in regulating protein–protein interactions.
Materials and Methods
Reagents.
Vasoactive intestinal peptide (synthetic, human sequence) was purchased from Sigma. All other reagents, unless otherwise stated, were also purchased from Sigma.
Molecular Biology.
All elements were cloned into the multiple cloning sites of the pET47b vector (Novagen). From N to C terminus, the constructs consist of eCFP, CAM, ER/K α-helix, mCitrine, and CAM-binding peptides (see SI Materials and Methods for details). All elements were separated by a (Gly-Ser-Gly) × 4 linker.
Protein Expression and Purification.
Protein expression and purification are based on previously published protocols (31). Details can be found in SI Materials and Methods.
Data Acquisition and Analysis.
FRET spectra of samples diluted in buffer A with indicated Ca2+ (CaCl2) or EGTA (stock pH 8) concentration were acquired on a Fluorolog fluorometer (Horiba Scientific). Quartz microcuvettes (Starna Cell, 16.40F-Q-10/Z8.5) were used with sample volume of 50 μL. For FRET spectra, samples were excited at 430 nm with 2- to 8-nm bandpass on excitation and emission (4- to 8-nm bandpass) was acquired from 450–650 nm. Further details can be found in SI Materials and Methods. Data from fluorometer was analyzed using custom Matlab software (Mathworks).
Supplementary Material
Acknowledgments.
The authors thank Mark Tsuchida and Pehr Harbury for useful discussions. S.S. is supported by an American Cancer Society Postdoctoral Fellowship and J.A.S. by Grant GM33289 from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 20279.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1116066108/-/DCSupplemental.
References
- 1.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 2.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 3.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009;21:462–469. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pufall MA, Graves BJ. Autoinhibitory domains: Modular effectors of cellular regulation. Annu Rev Cell Dev Biol. 2002;18:421–462. doi: 10.1146/annurev.cellbio.18.031502.133614. [DOI] [PubMed] [Google Scholar]
- 5.Chothia C. Proteins. One thousand families for the molecular biologist. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- 6.Aloy P, Russell RB. Ten thousand interactions for the molecular biologist. Nat Biotechnol. 2004;22:1317–1321. doi: 10.1038/nbt1018. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker BA, Panchenko AR. Deciphering protein–protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comput Biol. 2007;3:e43. doi: 10.1371/journal.pcbi.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoemaker BA, Panchenko AR. Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol. 2007;3:e42. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 10.Sivaramakrishnan S, Spink BJ, Sim AY, Doniach S, Spudich JA. Dynamic charge interactions create surprising rigidity in the ER/K alpha-helical protein motif. Proc Natl Acad Sci USA. 2008;105:13356–13361. doi: 10.1073/pnas.0806256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sivaramakrishnan S, et al. Combining single-molecule optical trapping and small-angle X-ray scattering measurements to compute the persistence length of a protein ER/K alpha-helix. Biophys J. 2009;97:2993–2999. doi: 10.1016/j.bpj.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehraeen S, Sudhanshu B, Koslover E, Spakowitz A. End-to-end distribution for a wormlike chain in arbitrary dimensions. Phys Rev E Nonlin Soft Matter Phys. 2008;77:061803. doi: 10.1103/PhysRevE.77.061803. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin RL. Energetics of protein folding. J Mol Biol. 2007;371:283–301. doi: 10.1016/j.jmb.2007.05.078. [DOI] [PubMed] [Google Scholar]
- 14.Chin D, Means AR. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 15.O’Neil KT, DeGrado WF. How calmodulin binds its targets: Sequence independent recognition of amphiphilic alpha-helices. Trends Biochem Sci. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- 16.Sample V, Newman RH, Zhang J. The structure and function of fluorescent proteins. Chem Soc Rev. 2009;38:2852–2864. doi: 10.1039/b913033k. [DOI] [PubMed] [Google Scholar]
- 17.Kenworthy AK. Imaging protein–protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Allen MD. FRET-based biosensors for protein kinases: Illuminating the kinome. Mol Biosyst. 2007;3:759–765. doi: 10.1039/b706628g. [DOI] [PubMed] [Google Scholar]
- 19.Howard J. Mechanics of Motor Proteins and the Cytoskeleton. Sunderland, MA: Sinauer; 2001. Chap 6. [Google Scholar]
- 20.Siegel RM, et al. Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci STKE. 2000;2000:lp1. doi: 10.1126/stke.2000.38.pl1. [DOI] [PubMed] [Google Scholar]
- 21.Vorherr T, et al. Interaction of calmodulin with the calmodulin binding domain of the plasma membrane Ca2+ pump. Biochemistry. 1990;29:355–365. doi: 10.1021/bi00454a008. [DOI] [PubMed] [Google Scholar]
- 22.Zettner A. Principles of competitive binding assays (saturation analysis). 1. Equilibrium techniques. Clin Chem. 1973;19:699–705. [PubMed] [Google Scholar]
- 23.Malencik DA, Anderson SR. Binding of hormones and neuropeptides by calmodulin. Biochemistry. 1983;22:1995–2001. doi: 10.1021/bi00277a040. [DOI] [PubMed] [Google Scholar]
- 24.Sung J, Sivaramakrishnan S, Dunn AR, Spudich JA. Single-molecule dual-beam optical trap analysis of protein structure and function. Methods Enzymol. 2010;475:321–375. doi: 10.1016/S0076-6879(10)75014-X. [DOI] [PubMed] [Google Scholar]
- 25.Rock RS, et al. Myosin VI is a processive motor with a large step size. Proc Natl Acad Sci USA. 2001;98:13655–13659. doi: 10.1073/pnas.191512398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyawaki A, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 27.Kiyokawa E, Aoki K, Nakamura T, Matsuda M. Spatiotemporal regulation of small GTPases as revealed by probes based on the principle of Forster Resonance Energy Transfer (FRET): Implications for signaling and pharmacology. Annu Rev Pharmacol Toxicol. 2011;51:337–358. doi: 10.1146/annurev-pharmtox-010510-100234. [DOI] [PubMed] [Google Scholar]
- 28.Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 30.Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spink BJ, Sivaramakrishnan S, Lipfert J, Doniach S, Spudich JA. Long single alpha-helical tail domains bridge the gap between structure and function of myosin VI. Nat Struct Mol Biol. 2008;15:591–597. doi: 10.1038/nsmb.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.