

Abstract
Niemann-Pick type C (NP-C) disease is a neurovisceral lysosomal storage disease characterized by neurological dysfunction, hepatosplenomegaly, and early death. Natural history studies are very difficult to perform due to the low incidence and high heterogeneity of disease in the human population. Sixteen cats with a spontaneously occurring missense mutation in NPC1 were evaluated over time to define the progression of neurological and hepatic disease. Affected cats had remarkably regular onsets of specific signs of cerebellar and vestibular system dysfunction with progressive severity of dysfunction quantified by post-rotatory nystagmus and brain stem auditory evoked response measures. NP-C disease cats also showed increasing serum activity of alanine aminotransferase, asparate aminotransferase, and cholesterol with advancing age. Affected cats lived to a mean age of 20.5 +/- 4.8 weeks. Central nervous system and hepatic lesions were similar to those described in human patients. These data are the first to document progressive hepatic disease in the feline model and demonstrate the importance of liver disease as part of the NP-C disease phenotype. Both neurological and hepatic measures of disease onset and severity can be used as a baseline with which to assess the efficacy of experimental therapies of NP-C disease in the feline model.
Keywords: animal model, lysosomal storage disease, Niemann-Pick type C, central nervous system, hepatic, feline, neurodegeneration, cholesterol
Niemann-Pick type C (NP-C) disease is a neurovisceral lysosomal storage disease characterized biochemically by the endosomal/lysosomal accumulation of unesterified cholesterol, sphingomyelin, and other glycolipids, and clinically by hepatosplenomegaly, progressive neurological dysfunction, and early death (1, 2). Mutations in either the NPC1 or NPC2 gene can result in NP-C disease with NPC1 mutations accounting for 95% of the cases (1, 3, 4). The proteins NPC1 and NPC2 are critical for the movement of unesterified cholesterol and glycosphingolipids from the endosomal/lysosomal compartment to the Golgi apparatus, plasma membrane, and endoplasmic reticulum (5). The mechanisms by which deficient activity of these proteins results in clinical disease are not understood.
NP-C disease has an incidence of 1:150000 and over 260 disease causing mutations have been identified (1). Natural history studies are difficult to perform due to the relatively low incidence and the high heterogeneity of disease. Onset of clinical signs ranges from the neonatal period to 59 years of age (1, 2, 16-18). Neurological deterioration invariably occurs, but the onset and progression varies. Hepatosplenomegaly and neonatal jaundice are common but not always present. Several classification systems based on the age of onset of clinical signs exist with most including an early onset form (early infantile and late infantile), a juvenile-onset form, and an adult-onset form, each having a varied prevalence of specific clinical signs (1, 16, 17). The early-onset form progresses rapidly and is often characterized by severe hepatic dysfunction and psychomotor delay during infancy with subsequent ataxia, marked spasticity, and dementia. The more slowly progressing juvenile-onset form shows intellectual impairment, hepatosplenogemegaly, supranuclear vertical gaze paresis, and ataxia, with a later onset of dementia and, variably, seizures, cataplexy and extrapyramidal deficits. The slowly progressing late-onset form shows cognitive and psychiatric disturbances. No study recounts the progression of clinical signs in a large cohort of patients with the same mutation.
Cats with NP-C disease have a spontaneously-occurring missense mutation in NPC1 (2864G-C) and clinical, neuropathological and biochemical abnormalities similar to those present in juvenile-onset patients making this model homologous to the most common form seen in human patients (6-12). The feline model has been critical for identifying the late endosomal/lysosomal accumulation of gangliosides (GM2 and GM3) and unesterified cholesterol (13), for evaluating the association of GM2 storage with meganeurite formation and abnormal dendritogenesis (10), and for correlating neuroaxonal dystrophy with neurological dysfunction (9). The feline model has also been used to evaluate the efficacy of experimental therapy of NP-C disease (11, 14).
Our goal was to define and quantify the progression of central nervous system and hepatic disease in the feline model of NP-C disease using neurological examinations, electrodiagnostic testing, serum biochemistry, and histopathology. This information will be used to emphasize the liver disease in feline NP-C disease, and, in the future, as a baseline with which to assess the efficacy of experimental therapies of NP-C disease in the feline model.
Methods
Animals
Cats were raised in the animal colony of the School of Veterinary Medicine, University of Pennsylvania, under NIH and USDA guidelines for the care and use of animals in research. Studies were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. All cats were housed at 21°C with ad libitum food and water, 12-hour light cycles, with 12-15 air changes per hour. Peripheral blood leukocytes from all cats were tested at one day of age for the NPC1 missense mutation using a PCR-based DNA test (12). Cats were identified at birth as 1) homozygous for the mutant allele (NP-C), 2) heterozygous for the mutant allele (heterozygote; HET), or, 3) lacking the mutant allele (wild type; WT). For this study, 16 NP-C disease, 20 heterozygotes, and 16 wild-type cats were evaluated.
Neurological examination
Physical and neurological examinations were performed weekly from birth until death. The onset and progression of signs of neurological dysfunction (intention tremor, truncal ataxia, inability to walk, inability to stand) were identified and tracked. The effect of rotation on the number of beats of nystagmus (post-rotatory nystagmus (PRN)) was determined at 8, 12, 16, 20, and 24 weeks of age. The number of beats of PRN was counted following moving the cats through five complete revolutions at a frequency of 34 rotations per minute using a metronome to time the rotation speed. This process was repeated three times in each direction (clockwise and counterclockwise) and the number of beats of PRN was then averaged from each time point.
Serum biochemical testing
Phlebotomy was performed at 8, 16, and 24 weeks of age and serum metabolic analyses were performed using a dry chemistry analyzer (Vitros 350, Johnson and Johnson, New Brunswick, NJ).
Brain stem auditory evoked response testing (BAER)
All studies were performed on cats first sedated with intravenous ketamine (2.2 mg/kg; Ketaset, Fort Dodge IA), acepromazine maleate (0.1 mg/kg; acepromazine; Boehringer-Ingelheim, St. Joseph, MO), and atropine sulfate (0.02 mg/kg; Butler Animal Health Supply, Dublin, OH). Then they were anesthetized with intravenous propofol (up to 6 mg/kg; Abbott Laboratories, Chicago, IL), endotracheally intubated, and maintained under anesthesia with isoflurane (IsoVet, Scherin-Plough, Omaha, NE). BAER data were recorded at 8, 16, and 24 weeks of age using 12 mm, 29 gauge subdermal needle recording electrodes and a Nicolet Viking Quest (Nicolet Biomedical, Madison, WI). The active electrode was placed over the osseous bulla of the stimulated ear, the reference electrode over the vertex of the skull, and the ground electrode over the contralateral osseous bulla. Alternating rarefaction and condensation clicks (0.1 ms duration) were delivered to the stimulated ear at 11.1 Hz using a 25 cm plastic tube connected to a plastic earpiece. The earpiece was placed within the external ear canal. The filter settings for the amplifier were 20 Hz and 3 kHz. One thousand evoked responses were averaged for each tracing obtained. An amplifier sensitivity of 1 uV/cm was used to record the responses; the analysis time was 10 ms. Central conduction time was defined as the time between the first and the fifth peak. Wave V/I amplitude was determined by dividing the amplitude of the fifth wave by the amplitude of the first wave and multiplying by 100; amplitude was measured from peak to peak. Both central conduction time and wave V/I amplitude were used as measures of retrocochlear disease. Hearing threshold was defined as the sound intensity at which an evoked waveform was first visible.
Post mortem examination
Cats were killed using an overdose of barbiturates in accordance with the American Veterinary Medical Association guidelines. NP-C disease cats were euthanized when one of the following occurred: 1) need for full body bathing more than two consecutive days in a row (affected cats have difficulty grooming and eating due to tremors), 2) loss of 20% or more of their 4 month-old body weight, 3) development of pressure sores due to recumbancy, or, 4) complete loss of appetite two days in a row. Wild type and heterozygous cats were euthanized between 20 and 29 weeks of age. Immediately before sacrifice, each cat was given 0.5 ml of heparin (1000 units/ml) intravenously. Following sacrifice, the cats were perfused with 500 ml of 0.9% cold saline and samples of brain, liver, spleen, and lung were acquired and frozen. Next, 750 ml of cold 4% paraformaldehyde was perfused into the left ventricle of the heart. Following perfusion, further samples of brain, liver, spleen, and lung were collected and dropped-fixed in paraformaldehyde for 48 hours. Fixed samples were paraffin-embedded, sectioned and stained with hematoxylin/eosin (HE). Immunohistochemistry for glial fibrillary acid protein (anti-cow GFAP; Dako, Denmark) and calbindin (anti-rat CALB; Swant, Switzerland) were also performed on paraffin-embedded brain sections.
Statistical methods
Mean and standard deviation were calculated to describe the data and the unpaired 2-tailed t-test was used to compare data between NP-C disease and wild-type cats, and between wild type and heterozygous cats. Significance values of p<0.05 (†) and p<0.001 (*) are given.
RESULTS
Body weight
Birth weights of wild type, heterozygous, and NP-C disease cats were not significantly different. However, NP-C disease cats weighed significantly (p<0.05) less than wild-type cats beginning at 5 weeks of age (p<0.05) and continued to weigh less until death (Figure 1). Cats heterozygous for the NP-C1 mutation were the same weight as wild-type cats at all ages.
Figure 1.
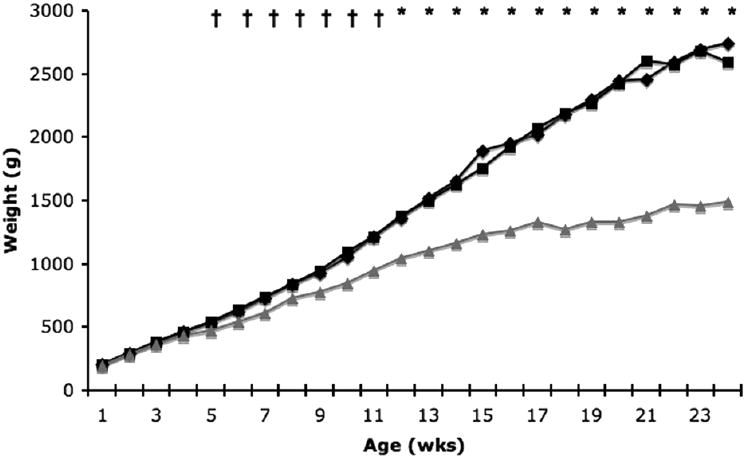
Weekly body weights of wild type (WT; n=16), heterozygote (HET; n=20), and Niemann-Pick type C (NP-C; n=16) disease cats. NP-C disease cats weighed significantly less than WT cats beginning at 5 weeks of age and continued to weigh less throughout life (†p<0.05; *p<0.001). (diamond, WT; square, HET; triangle, NP-C).
Onset of neurological dysfunction
NP-C disease cats had a progressive age of onset of specific nervous system signs all of which could be explained by cerebellar and vestibular dysfunction. Intention tremors and mild truncal ataxia were first seen at 6.4 +/- 0.8 weeks of age. Ataxia increased in severity and resulted in falling when running/playing at 11.8 +/- 1.9 weeks of age and increased in severity so that no cats could walk without falling at 15.3 +/- 2.6 weeks of age. Cats were unable to take even one step without falling at 17 +/- 2.1 weeks and were no longer able to stand without assistance at 19 +/- 3 weeks of age. Wild type and heterozygous cats developed no neurological deficits.
Post-rotatory nystagmus
Post-rotatory nystagmus (PRN) in wild type, heterozygous, and NP-C disease cats were not significantly different at eight weeks of age. However, NP-C disease cats had significantly more PRN at 12 and 16 weeks of age (p<0.05), and at 20 and 24 weeks of age (p<0.001) weeks of age when compared to wild-type cats (Figure 2). In wild type and heterozygous cats, PRN significantly decreased between 8 and 12 weeks of age (p<0.001). In contrast, no significant differences in PRN were found between NP-C disease cats of different ages.
Figure 2.
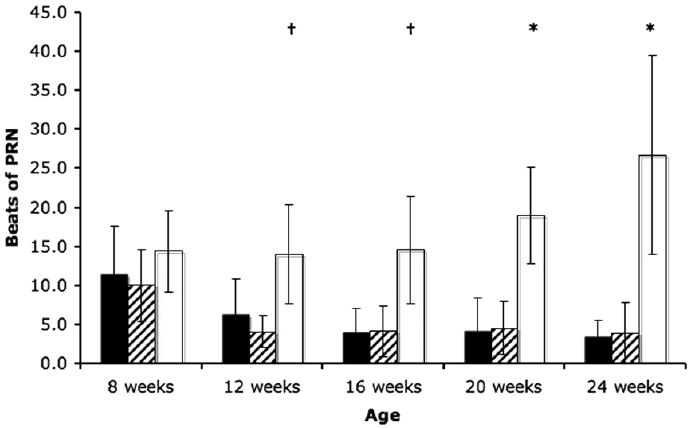
Post-rotatory nystagmus (PRN) in WT (n=16), HET (n=20), and NP-C disease (n=16) cats. NP-C disease cats had significantly more post-rotatory nystagmus beginning at 12 weeks of age and continuing throughout life (†p<0.05; *p<0.001) compared to WT cats. A significant decrease in PRN (p<0.001) was seen between 8 and 12 weeks of age in WT and HET cats. (black, WT; hatched, HET; white, NP-C).
Brain stem auditory evoked response testing (BAER)
NP-C disease cats had significantly greater (p<0.05) central conduction time at 16 and 24 weeks of age compared to wild-type cats (Figure 3a). Although wild type and heterozygous cats had a significant decrease in central conduction time between 8 and 16 weeks of age, NP-C disease cats showed no similar decrease with advancing age.
Figure 3.
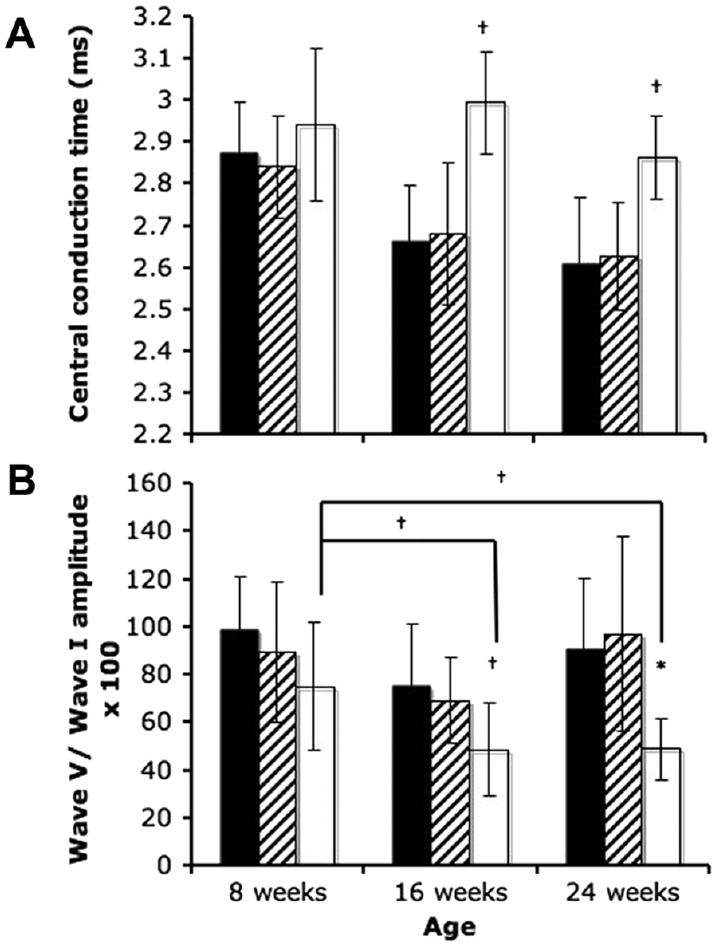
Central conduction time (A) and wave V/I amplitude ratio (B) in WT (n=15), HET (n=16), and NP-C disease (n=11) cats. NP-C disease cats had significantly greater latency between the first and fifth wave of the BAER at 16 and 24 weeks of age (a; †p<0.05). WT cats showed a significant decrease in central conduction time between 8 and 16 weeks and 8 and 24 weeks of age. NP-C disease cats had significantly smaller wave V/I amplitude at 16 and 24 weeks of age compared to WT animals (b; †p<0.05; *p<0.001). Affected cats showed a significant decrease in wave V/I amplitude between 8 and 16 weeks of age and between 8 and 24 weeks of age. (black, WT; hatched, HET; white, NP-C).
NP-C disease cats also had significantly lower (p<0.05) wave V/I amplitude ratio at 16 and 24 weeks of age compared to wild-type cats (Figure 3b). In NP-C disease cats, a significant decrease in V/I amplitude was found between 8 weeks and 16 weeks, and between 8 and 24 weeks of age. No differences between ages were found in wild type and heterozygous cats.
Finally, there was no significant difference in hearing threshold among wild type, heterozygous, and NP-C disease cats at any age (data not shown).
Clinical pathology
NP-C disease cats had significantly greater (p<0.05) serum concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) at 8, 16, and 24 weeks of age compared with wild-type cats (Figure 4). As NP-C disease cats aged, significant increases in ALT and AST were found between 8 and 16 weeks of age and between 8 and 24 weeks of age. NP-C disease cats had significantly greater alkaline phosphatase (ALKP) activity (p<0.05) at 16 and 24 weeks of age compared to wild type and heterozygous cats. No significant differences in these three enzymes were found between wild type and heterozygous cats at any age.
Figure 4.
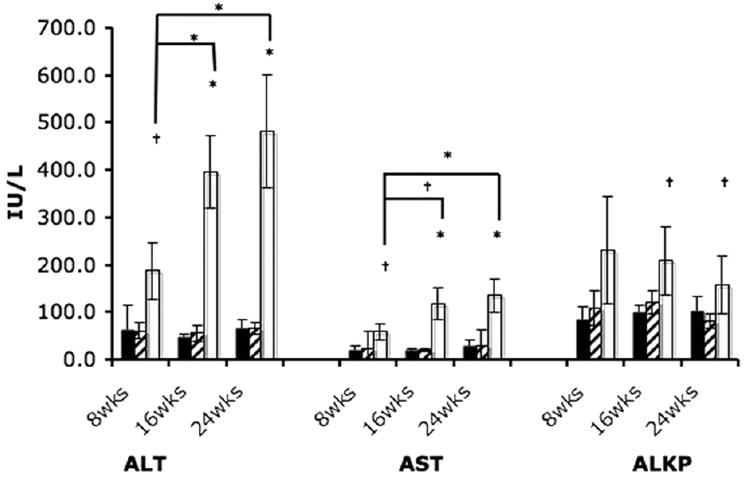
Hepatic enzyme activity in WT (n=13), HET (n=18), and NP-C disease (n=16) cats. NP-C disease cats had significantly greater alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity at all ages examined compared to WT cats (†p<0.05; *p<0.001). Significant increases in ALT and AST were found in NP-C disease cats over time between 8 and 16 weeks of age and between 8 and 24 weeks of age. Significantly greater alkaline phosphatase (ALKP) activity was found in NP-C disease cats at 16 and 24 weeks of age compared to WT cats. (black, WT; hatched, HET; white, NP-C).
At 8, 16, and 24 weeks of age, NP-C disease cats had significantly lower (p<0.05) serum albumin concentrations (2.2 +/- 0.2 g/dl; 2.4 +/- 0.2 g/dl; 2.6 +/- 0.3 g/dl) compared to wild-type cats (2.6 +/- 0.4 g/dl; 2.9 +/- 0.3 g/dl; 3.1 +/- 0.1 g/dl). NP-C disease cats also had significantly greater (p<0.05) serum cholesterol concentrations at 8, 16, and 24 weeks of age, and significantly greater serum bilirubin concentrations at 16 and 24 weeks of age compared to wild-type cats (Table 1). Serum cholesterol concentrations in NP-C disease cats were significantly greater at 16 and 24 weeks of age compared to 8 week-old NP-C disease cats (p<0.05). No significant differences were found in serum albumin, cholesterol, or total bilirubin concentrations between wild type and heterozygous cats at any age.
Table 1.
Serum cholesterol and total bilirubin concentrations.
| Wild-type | Heterozygote | NP-C disease | |
|---|---|---|---|
| Cholesterol (mg/dl) | |||
| 8 weeks | 146 +/-50 | 132 +/-38 | 170 +/-35† |
| 16 weeks | 105 +/-10 | 112 +/-13 | 244 +/-34* † |
| 24 weeks | 115 +/-19 | 120 +/-49 | 237 +/-29* † |
| Total bilirubin (mg/dl) | |||
| 8 weeks | 0.5 +/-0.2 | 0.5 +/-0.2 | 0.6 +/-0.2 |
| 16 weeks | 0.4 +/-0.1 | 0.3 +/-0.1 | 0.9 +/-0.4* |
| 24 weeks | 0.4 +/-0.1 | 0.3 +/-0.1 | 0.9 +/-0.1* |
p<0.05;
p<0.001.
Finally, to assess the cause of the decreased albumin and the function of the liver in NP-C disease cats, urine protein/creatinine ratios and serum bile acids were measured. These tests were only run for two 25 week-old NP-C disease cats. Urine protein/creatinine ratio was normal (0.15 and 0.18; reference range <0.2) (15). Mild elevations in both pre-prandial (8.6 μmol/L and 22.0 μmol/L; reference <2.0 μmol/L) and post-prandial bile acids (18.4 μmol/L and 37.8 μmol/L; reference <10 μmol/L) were found.
Survival times
Mean survival time for NP-C disease cats was 20.5 weeks +/- 4.8 weeks (range = 11 – 29 weeks). All cats were euthanized due to progressive tremors that made it difficult for them to eat and groom. At the time of euthanasia, no cats were able to remain sternal without being supported.
Histology
Hepatomegaly was noted at gross post mortem examination in all NP-C disease cats. The liver was weighed in two affected (121 g and 132 g; both 12% body weight) and two wild-type cats (63.9 g and 71 g; 3% and 2% body weight). Similarly, the brain was weighed in two affected (20.5 g and 21 g; both 2% body weight) and two wild-type cats (29.2 g and 30 g; 0.9% and 1% body weight). Histological evaluation showed that the cytoplasm of hepatocytes and Kuppfer cells had severe and extensive vacuolization and dilated hepatic sinusoids containing clusters of macrophages with granular to foamy vacuolated cytoplasm. The brain had multifocal neuronal cytoplasmic vacuolization with gliosis. Neuronal necrosis was seen with severe loss of Purkinje cells visible on both hematoxylin/eosin and calbindin-stained slides (Figure 5). Thinning of the granular cell layer of the cerebellum was also seen. The white matter of the cerebellum showed swollen axons with dilated myelin sheaths, numerous spheroids, and myelomacrophages. Astrogliosis of both the gray and white matter was present and numerous swollen macrophages were seen surrounding vessels. The spleen and lung showed multifocal histiocytosis with macrophages containing small clear vacuoles.
Figure 5.
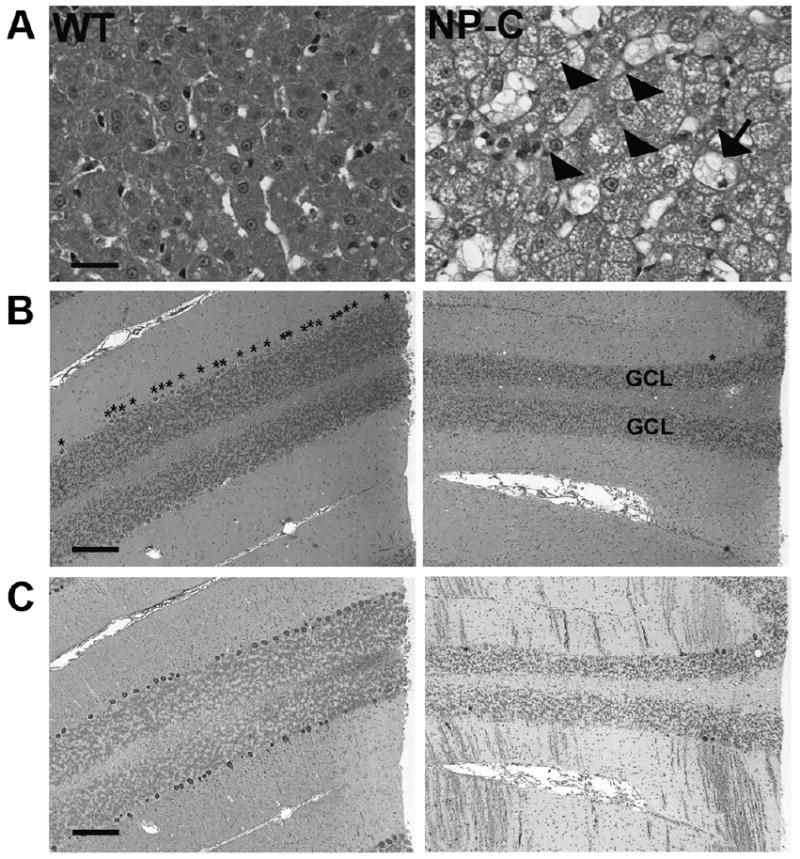
Hepatic (400×) and cerebellar (50×) histology of age-matched WT and NP-C disease cats. A)Liver from a 22 week old NP-C disease cat showed vacuolization of both Kuppfer cells (arrow) and hepatocytes (arrowheads). A decrease in cerebellar Purkinje cell number was seen on both (B) hematoxylin/eosin (* above Purkinje cells) and (C) calbindin stained sections (Purkinje cell soma and process stained brown). Thinning of the granular cell layer (GCL) was also present. Bar = 25 um (liver). Bar = 200 um (cerebellum).
DISCUSSION
Feline NP-C1 disease is due to a missense mutation, a guanine to cytosine substitution (2864G-C; C955S), which is similar to most of the juvenile onset patients (12). Previous studies performed in seven NP-C disease cats with the same mutation described signs of cerebellar dysfunction developing at approximately eight weeks of age with most animals either dying or being euthanized due to the severity of disease progression before eight months of age (6-8). No descriptive statistical data for age of onset or progression of signs of neurological dysfunction were previously published. Our study showed a regular onset of progressive signs of cerebellar and vestibular system dysfunction beginning at about six weeks of age that progressed until cats were unable to remain in sternal recumbancy. A quantitative measure of vestibular dysfunction was developed based on the number of beats of PRN. NP-C disease cats had significantly greater PRN beginning at 12 weeks of age and this difference between affected and wild-type cats continued until at least 24 weeks of age. Although there was an apparent trend for increasing beats of PRN in NP-C disease cats with increasing age, the large variance found in affected cats did not make this increase statistically significant. Interestingly, wild type and heterozygous cats had more PRN at 8 weeks of age than at later ages, which likely represents ongoing development of the vestibular system. In contrast, NP-C disease cats showed no decrease in PRN between 8 and 12 weeks of age. Central conduction time is a measure of transit time from the earliest stimulation of the acoustic nerve to stimulation of the inferior colliculus (19). NP-C disease cats showed a delay in central conduction time at 16 and 24 weeks of age compared to wild-type cats. Like the PRN data, wild-type cats showed a significant decrease in central conduction time developing after 8 weeks of age that likely represents continued maturation of the acoustic system. A decrease in the wave V/I amplitude ratio has been used an indicator of retrocochlear (cranial nerve VII or brain stem) pathology (20). NP-C disease cats developed a significant decrease in this ratio between 8 and 16 weeks of age. Taken together, the decrease in central conduction time and in amplitude of waves V/I suggest eighth nerve and/or brain stem disease (20), and the lack of a difference in hearing threshold in NP-C disease cats compared to wild-type cats suggests the absence of functionally significant cochlear pathology.
In addition to cerebellar and vestibular dysfunction, human patients may show vertical supranuclear gaze palsy, dysarthria, dysphagia, cataplexy, seizures, and cognitive and psychiatric disturbances (2). All cats maintained oculocephalic eye movements in all directions; however, NP-C disease cats did not readily follow targets in either the horizontal or vertical direction. It could not be determined whether the failure of “tracking” represented an abnormality of voluntary control of ocular muscles or a disinterest associated with the severity of disease. Affected cats showed no obvious abnormalities of behavior that could be interpreted as cognitive or psychiatric dysfunction, however, impairment of motor ability due to cerebellar dysfunction was severe and, therefore, limited the behavioral observations that could be made. No generalized seizures or cataleptic episodes were observed, however, as affected cats aged, they developed abnormal episodic rhythmic chewing motions when held which may indicate focal seizures. Electroencephalographic studies are in progress to evaluate further the presence or absence of epileptiform activity in affected cats.
Elevations in serum liver enzymes, bile acids, total bilirubin, and cholesterol, and decreased serum albumin have been previously reported in feline NP-C disease cats but there is are no data evaluating changes in these serum activity and concentrations over time (11). The activity of the enzymes ALT and AST were significantly greater in NP-C disease cats at all ages examined compared to wild-type cat. ALT is a cytosolic enzyme with a half life of hours in the cat (21). It is considered to be specific for increases in hepatocellular permeability and its rise over time in NP-C disease cats suggests ongoing and progressively worsening hepatocellular damage. AST is located in both the cytosol and mitochondria and serum levels increase with hepatocellular or muscle disease. Both increases in ALT and AST suggest progressive hepatocellular disease. Finding decreased albumin, without albuminuria, and increased bile acids in affected cats confirms the presence of hepatocellular dysfunction. In contrast, serum ALKP activity does not increase with hepatocellular permeability but increases with cholestasis and with osteoblastic activity associated with growth. The presence of concurrent hyperbilirubinemia with increased ALKP suggests cholestasis although no icterus was identified in these cats; no bone pathology in NP-C disease has been described. Although histological evaluation of the liver showed no ductular or canalicular bile stasis, hepatocellular bile accumulation is difficult to see histologically in cats and we, therefore, suggest that affected cats have some degree of abnormal bilirubin metabolism or hepatobiliary bile stasis.
Progressive neonatal liver failure and cholestasis have been described in neonatal NP-C disease in human patients (22-25). In one study of 52 children, 34 had cholestatic liver disease and approximately half of these children had persistent elevations of liver enzymes although neurological dysfunction remained the predominant disease manifestation (24). In mice with NP-C disease, progressive increases in ALT and AST have been reported to occur throughout life (26). The presence of increasing serum liver enzyme activity and of liver dysfunction in the feline model supports that liver disease is an important part of NP-C disease in the feline model, as it is in the neonatal disease in human patients and in the murine model.
NP-C disease cats were euthanized when the severity of the ataxia impaired urination/defecation, eating, and grooming to the extent that cats required repetitive bathing. No cats lost substantial weight, developed pressure sores, or were anorexic. The majority of affected cats were continuing to gain weight at the time of death albeit much more slowly than unaffected littermates. With aggressive nursing care, it is expected that affected cats could be maintained for longer periods of time.
Finally, cats heterozygous for the NPC1 mutation demonstrate abnormal cholesterol and sphingomyelin concentration within the brain as well as evidence of mild neuropathology (27). Our study shows that these biochemical and histological abnormalities did not result in measurable abnormalities using neurological examination, electrodiagnostic testing, or serum metabolite analyses.
The availability of a well-characterized feline NP-C disease model has several benefits. The feline model has a missense mutation with a juvenile onset of neurological signs making it homologous to the most common disease form seen in human patients. The size of the cat allows for safe and repeated sampling of blood and other body fluids for biochemical and pharmacological measurements, and the large size of the cat brain permits the study of regional neuropathology using imaging as has been reported in other feline disease models (28). The lesions of the central nervous system including the abnormality of ectopic dendritogenesis, more closely resemble those found in human patients than do those in the murine model (13). Finally, the ability to genotype animals at birth, and the availability of ante mortem quantitative measures of disease progression will prove useful for evaluating the efficacy of experimental therapies.
Acknowledgments
The authors thank Dr. Mary Beth Callan for her critical advice on the manuscript.
Financial Support: Ara Parseghian Medical Research Foundation and Dana’s Angels Research Trust (Vite), RR02512 (Haskins).
Abbreviations
- ALKP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BAER
brain stem auditory evoked response
- CALB
calbindin
- GM
ganglioside monosialo
- HE
hematoxylin and eosin
- HET
heterozygote
- NP-C
Niemann-Pick type C
- NPC1
Niemann-Pick disease type C1 gene or protein
- PRN
post-rotatory nystagmus
References
- 1.Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 2.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette Mackie EJ, Pentchev PG. Niemann-Pick Disease Type C: A Lipid Trafficking Disorder. In: Valle D, Beaudet AL, Vogelstein B, K KW, Antonarakis SE, Ballabio A, editors. Metabolic and Molecular Bases of Inherited Disease. 8. Ch. 145. McGraw-Hill Companies, Inc; New York: 2001. www.ommbid.com. [Google Scholar]
- 3.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, 3rd, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 4.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, Dietschy JM. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J Neuropathol Exp Neurol. 2005;64:323–333. doi: 10.1093/jnen/64.4.323. [DOI] [PubMed] [Google Scholar]
- 6.Lowenthal AC, Cummings JF, Wenger DA, Thrall MA, Wood PA, de Lahunta A. Feline sphingolipidosis resembling Niemann-Pick disease type C. Acta Neuropathol. 1990;81:189–197. doi: 10.1007/BF00334507. [DOI] [PubMed] [Google Scholar]
- 7.Brown DE, Thrall MA, Walkley SU, Wenger DA, Mitchell TW, Smith MO, Royals KL, March PA, Allison RW. Feline Niemann-Pick disease type C. Am J Pathol. 1994;144:1412–1415. [PMC free article] [PubMed] [Google Scholar]
- 8.Munana KR, Luttgen PJ, Thrall MA, Mitchell TW, Wenger DA. Neurological manifestations of Niemann-Pick disease type C in cats. J Vet Intern Med. 1994;8:117–121. doi: 10.1111/j.1939-1676.1994.tb03208.x. [DOI] [PubMed] [Google Scholar]
- 9.March PA, Thrall MA, Brown DE, Mitchell TW, Lowenthal AC, Walkley SU. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997;94:164–172. doi: 10.1007/s004010050689. [DOI] [PubMed] [Google Scholar]
- 10.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 11.Somers KL, Brown DE, Fulton R, Schultheiss PC, Hamar D, Smith MO, Allison R, Connally HE, Just C, Mitchell TW, Wenger DA, Thrall MA. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis. 2001;24:427–436. doi: 10.1023/a:1010588112003. [DOI] [PubMed] [Google Scholar]
- 12.Somers KL, Royals MA, Carstea ED, Rafi MA, Wenger DA, Thrall MA. Mutation analysis of feline Niemann-Pick C1 disease. Mol Genet Metab. 2003;79:99–103. doi: 10.1016/s1096-7192(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 13.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283–1287. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- 15.King JN, Tasker S, Gunn-Moore DA, Strehlau G BENRIC (benazepril in renal insufficiency in cats) Study Group. Prognostic factors in cats with chronic kidney disease. J Vet Intern Med. 2007;21:906–916. [PubMed] [Google Scholar]
- 16.Fink JK, Filling-Katz MR, Sokol J, Cogan DG, Pikus A, Sonies B, Soong B, Pentchev PG, Comly ME, Brady RO. Clinical spectrum of Niemann-Pick disease type C. Neurology. 1989;39:1040–1049. doi: 10.1212/wnl.39.8.1040. [DOI] [PubMed] [Google Scholar]
- 17.Iturriaga C, Pineda M, Fernandez-Valero EM, Vanier MT, Coll MJ. Niemann-Pick C disease in Spain: clinical spectrum and development of a disability scale. J Neurol Sci. 2006;249:1–6. doi: 10.1016/j.jns.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 18.Higgins JJ, Patterson MC, Dambrosia JM, Pikus AT, Pentchev PG, Sato S, Brady RO, Barton NW. A clinical staging classification for type C Niemann-Pick disease. Neurology. 1992;42:2286–2290. doi: 10.1212/wnl.42.12.2286. [DOI] [PubMed] [Google Scholar]
- 19.Buchwald JS, Huang CL. Far-field acoustic response: Origins in the cat. Science. 1975;189:382–384. doi: 10.1126/science.1145206. [DOI] [PubMed] [Google Scholar]
- 20.Silman S, Silverman CA. Auditory Diagnosis: Principles and Applications. Academic Press; San Diego: 1991. Braistem auditory evoked potentials; pp. 151–297. [Google Scholar]
- 21.Lassen ED. Laboratory evaluation of the liver. In: Thrall MA, editor. Veterinary Hematology and Clinical Chemistry. Lippincott Williams, Wilkins; Philadelphia: 2004. pp. 355–375. [Google Scholar]
- 22.Rutledge JC. Progressive neonatal liver failure due to type C Niemann-Pick disease. Pediatr Pathol. 1989;9:779–784. doi: 10.3109/15513818909022387. [DOI] [PubMed] [Google Scholar]
- 23.Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet. 1988;33:331–348. doi: 10.1111/j.1399-0004.1988.tb03460.x. [DOI] [PubMed] [Google Scholar]
- 24.Kelly DA, Portmann B, Mowat AP, Sherlock S, Lake BD. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J Pediatr. 1993;123:242–247. doi: 10.1016/s0022-3476(05)81695-6. [DOI] [PubMed] [Google Scholar]
- 25.Ashkenazi A, Yarom R, Gutman A, Abrahamov A, Russell A. Niemann-Pick disease and giant cell transformation of the liver. Acta Paediatr Scand. 1971;60:285–294. doi: 10.1111/j.1651-2227.1971.tb06658.x. [DOI] [PubMed] [Google Scholar]
- 26.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 2005;42:886–893. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- 27.Brown DE, Thrall MA, Walkley SU, Wurzelmann S, Wenger DA, Allison RW, Just CA. Metabolic abnormalities in feline Niemann-Pick type C heterozygotes. J Inherit Metab Dis. 1996;19:319–330. doi: 10.1007/BF01799262. [DOI] [PubMed] [Google Scholar]
- 28.Vite CH, Magnitsky S, Aleman D, O’Donnell P, Cullen K, Ding W, Pickup S, Wolfe JH, Poptani H. Apparent diffusion coefficient reveals gray and white matter disease, and T2 mapping detects white matter disease in the brain in feline alpha-mannosidosis. AJNR. Am J Neuroradiol. 2008;29:308–313. doi: 10.3174/ajnr.A0791. [DOI] [PMC free article] [PubMed] [Google Scholar]