

Abstract
Background
Breast cancer stem cells (BCSCs) are the source of breast tumors. Compared with other cancer cells, cancer stem cells show high resistance to both chemotherapy and radiotherapy. Targeting of BCSCs is thus a potentially promising and effective strategy for breast cancer treatment. Differentiation therapy represents one type of cancer stem-cell-targeting therapy, aimed at attacking the stemness of cancer stem cells, thus reducing their chemo- and radioresistance. In a previous study, we showed that down-regulation of CD44 sensitized BCSCs to the anti-tumor agent doxorubicin. This study aimed to determine if CD44 knockdown caused BCSCs to differentiate into breast cancer non-stem cells (non-BCSCs).
Methods
We isolated a breast cancer cell population (CD44+CD24- cells) from primary cultures of malignant breast tumors. These cells were sorted into four sub-populations based on their expression of CD44 and CD24 surface markers. CD44 knockdown in the BCSC population was achieved using small hairpin RNA lentivirus particles. The differentiated status of CD44 knock-down BCSCs was evaluated on the basis of changes in CD44+CD24- phenotype, tumorigenesis in NOD/SCID mice, and gene expression in relation to renewal status, metastasis, and cell cycle in comparison with BCSCs and non-BCSCs.
Results
Knockdown of CD44 caused BCSCs to differentiate into non-BCSCs with lower tumorigenic potential, and altered the cell cycle and expression profiles of some stem cell-related genes, making them more similar to those seen in non-BCSCs.
Conclusions
Knockdown of CD44 is an effective strategy for attacking the stemness of BCSCs, resulting in a loss of stemness and an increase in susceptibility to chemotherapy or radiation. The results of this study highlight a potential new strategy for breast cancer treatment through the targeting of BCSCs.
Keywords: Breast cancer stem cells, Breast cancer cells, CD44, Differentiation, Differentiation therapy, Knockdown
Background
The existence of breast cancer stem cells (BCSCs) in malignant breast tumors has been demonstrated in many previous studies [1-4]. These stem cells exhibit a range of phenotypes, including CD44+CD24-, CD44+CD24-/dim, CD44+CD24-/dimESA+ and CD44+CD24-Lin- [1-4]. These cells possess specific characteristics, such as anti-tumor-drug and radiation resistance [5]. Because they can escape the effects of chemotherapy or radiation therapy, relapse remains a possibility. The resistance of these cells may be mediated by signaling through the Wnt pathway [6]. They also express high levels of anti-apoptotic proteins, such as survivin and Bcl [7], and evidence suggests that alterations in DNA repair and cell cycle kinetics may be involved in their resistance to radiation and chemotherapy [8]. In addition, BCSCs have been shown to be resistant to hormone therapy [9-11]. The discovery of this cancer stem cell population in breast tumors has thus opened up several potential approaches for breast cancer treatment, especially in terms of BCSC-targeting therapy.
The resistance of BCSCs to radiation and chemotherapy means that there is a need to develop agents able to attack this cell population. Because of their stemness, targeting therapies have usually been designed to regulate the self-renewal characteristics, as well as the differentiation of stem cells. Several strategies designed to target BCSCs are currently available, and can be divided into two main groups: those directly targeting BCSCs and those that indirectly targeting BCSCs through the cell microenvironment.
A number of developmental pathways responsible for regulating stemness have been elucidated during the past decade, including Wnt, Notch, and Hedgehog, and several studies have demonstrated that disrupted regulation of these pathways can lead to the development of breast cancer in mice [12-15] and humans [16-18]. HER2 signaling represents one of most significant advances in breast cancer research. Trials of agents targeting HER2, such as trastuzumab and lapatinib, have shown improved overall survival of patients with advanced disease [19] as well as reduced tumor recurrence [20]. Another study found that HER2-targeting agents reduced the BCSC population [19].
However, despite the remarkable clinical efficacy of HER2-targeting agents, a third of HER2-positive tumors do not respond to these agents as well as expected on the basis of their reduced resistance, and almost 50% of patients who respond to HER2-targeted agents relapse within a year [21]. The reason for this phenomenon is unclear. Moreover, nearly 50% of patients are negative for HER2 [22]. Thus the search for new therapeutic strategies continues worldwide.
The adhesion molecule CD44 is a cell surface transmembrane glycoprotein involved in lymphocyte activation, recirculation and homing, adhesion of extracellular matrix, angiogenesis, and cell proliferation, differentiation, and migration [23]. These properties are associated with the pathologic activities of cancer cells. Previous research demonstrated that knockdown of CD44 in BCSCs sensitized them to the anti-tumor drug doxorubicin [24], suggesting that CD44 knockdown affected the stemness or differentiation of these cells.
The current study therefore aimed to investigate the effects of CD44 knockdown on the stemness and differentiation of BCSCs in severe combined immunodeficient (SCID) mice in terms of gene expression, cell cycle, and tumorigenesis, in comparison with breast cancer non-stem cells (non-BCSCs). The results will facilitate the development of BCSC-targeting differentiating gene therapy for breast cancer treatment.
Materials and methods
Primary culture of breast cancer cells from malignant breast tumors
Primary culture of breast cancer cells from malignant breast tumors was carried out as previously described [4,24]. Briefly, tumor biopsies were obtained from consenting hospital patients then transferred to our laboratory. Biopsy samples were washed three to four times with phosphate-buffered saline (PBS) supplemented with 1 × antibiotics and an antimycotic (Sigma-Aldrich, St Louis, MO), and homogenized into small fragments (approximately 1-2 mm3) using scissors. Homogenized samples were seeded in 35-mm culture dishes (Nunc, Roskilde, Denmark) in M171 medium (Invitrogen, Carlsbad, CA) containing mammary epithelial growth supplement (Invitrogen, Carlsbad, CA), and incubated at 37°C in 5% CO2. The medium was refreshed every third day, and the cells were sub-cultured continuously until most cells resembled epithelial-like cells. Ten patients were included in this study, and cancer cells were isolated from all 10 tumors by primary culture.
Isolation of four cancer cell populations based on CD44 and CD24 expression
All primary cultures were analyzed for the presence of BCSCs by flow cytometry. The samples with the highest percentage of BCSCs were used to isolate four cancer cell populations, based on their expression of CD44 and CD24. CD44+CD24- cell populations were classed as BCSCs, while CD44+CD24+, CD44-CD24+ and CD44-CD24- cell populations were classed as non-BCSCs or differentiated cells. In subsequent experiments, the term "non-BCSCs" or "differentiated cells" refers to a mixture of the CD44+CD24+, CD44-CD24+ and CD44-CD24- cell populations. These four cancer cell populations were isolated based on their cell surface expression of CD44 and CD24 using a magnetic-activated cell sorting (MACS) system with anti-CD44 and anti-CD24-biotin combined anti-biotin microbeads (Miltenyi Biotec Inc., CA). Positive selection was performed using MS columns, and negative selection using LD columns (Miltenyi Biotec Inc., CA). Cultured cells were detached by trypsin/EDTA 0.25% (Sigma-Aldrich, St Louis, MO). CD44+CD24+ cells were isolated in two steps: cells were initially stained with CD44 microbeads and CD44+ cells were collected; the CD44+ cells were then stained with anti-CD24-biotin, followed by anti-biotin microbeads to isolate CD44+CD24+ cells. CD44+CD24-, CD44-CD24+ and CD44-CD24- cells were similarly isolated by combining positive collections or depletions based on CD44 and CD24 expression, using the same techniques. The phenotypes of all isolated cells were confirmed by flow cytometry using a BD FACSCalibur machine (BD Biosciences, Franklin Lakes, New Jersey) with anti-CD44-phycoerythrin (PE) and anti-CD24-fluorescein isothiocyanate (FITC) monoclonal antibodies (BD Biosciences, Franklin Lakes, New Jersey). Their purities were confirmed by flow cytometry, and samples with > 90% purity were used for further experiments.
Knockdown of CD44+CD24- cells with small hairpin RNA using lentivirus particles
CD44 small hairpin RNA (shRNA) lentivirus particles (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were stably transfected, according to the manufacturer's instructions. Briefly, CD44+CD24- cells were plated on day 1 into 12-well plates with complete medium (DMEM/F12 supplemented with 10% fetal bovine serum and 1 × antibiotic-mycotic) and incubated overnight. The medium was replaced on day 2 with fresh complete medium supplemented with 5 μg/ml polybrene (Sigma-Aldrich, St Louis, MO) for 6 h, after which 20 μl MEM with 25 mM HEPES containing 105 infectious units of virus were directly added into the culture. The plate was shaken to mix the virus particles and incubated overnight at 37°C in 5% CO2. The medium was changed on day 3 to fresh complete medium without polybrene. Successfully transduced cells were selected by culturing in complete medium supplemented with 10 μg/ml puromycin dihydrochloride (Sigma-Aldrich, St Louis, MO) for 12 h, followed by 5 μg/ml puromycin dihydrochloride for 1 week. CD44 knockdown BCSCs were confirmed by determination of CD44 expression by flow cytometry and immunocytochemistry. Samples with > 90% purity were used for further experiments for evaluating tumorigenesis in SCID mice and investigating gene expression and cell cycle.
Flow cytometry
Cells were washed twice in PBS supplemented with 1% bovine serum albumin (Sigma-Aldrich, St Louis, MO). The cell surface Fc receptor was blocked using IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) on ice for 15 min. Cells were stained for 30 min at 4°C with anti-CD44-PE and anti-CD24 FITC monoclonal antibodies (BD Biosciences, Franklin Lakes, New Jersey). After washing, cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, New Jersey) using CellQuest Pro software at 10,000 events.
Gene expression analysis
Ten random colonies formed after plating CD44 knockdown BCSCs at low density were used for the analysis of gene expression. To evaluate the differentiated status of BCSCs, expression of 15 genes related to the properties of cancer stem cells and cancer/normal cells, as well as some genes related to signaling pathways over-expressed in cancer stem cells, were analyzed in comparison with BCSCs and non-BCSCs. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; [Genebank:NM_002046]) was used as an internal control for all experiments. All primers used in this study were designed using Primer Blast software (NCBI). Primer pairs were chosen to give polymerase chain reaction (PCR) products of 100-350 bp. The universal primer for each forward and reverse primer was then added. The universal primer sequence was suggested by the manufacturer, using the GenomeLab GeXP genetic analysis system (Beckman-Coulter, Brea, California). All primer sequences are listed in Table 1. All primers were checked for specificity and working status by in silico PCR and in vitro reverse-transcription PCR using a universal RNA template (Clontech, CA, USA). Only primer pairs that gave the intended PCR products were used in subsequent experiments. Two multiplex PCR reactions were used to evaluate the change in stemness: one multiplex with 17 genes included Bcl-2 [Genebank:NM_000633], Fos [Genebank: NM_005252], ICAM1 [Genebank:NM_000201], CCND1 [Genebank: NM_053056], MMP7 [Genebank:NM_002423], Myc [Genebank:NM_002467], PRKCE [Genebank:NM_005400], TP53 [Genebank:NM_000546], VCAM1 [Genebank:NM_001078], IL4R [Genebank:NM_000418], PTCH1 [Genebank:NM_000264], HSPB1 [Genebank: NM_001540], PTGS2 [Genebank: NM_000963], HSF1 [Genebank:NM_005526], LEF1 [Genebank:NM_016269], TCF7 [Genebank:NM_003202], and FASN [Genebank:NM_004104] and the other with five genes included Muc-1 [Genebank:NM_002456], cyclin E2 [Genebank:NM_004702], EGFR [Genebank:NM_005228], Myc [Genebank:NM_002467], and cyclin D1 [Genebank:BC001501].
Table 1.
Primer sequences used in this research.
| Gene | Sequence | GeXP PCR product (bp) |
|---|---|---|
| Multiplex 1: 17 genes (+ GAPDH and KanR) | ||
| Bcl-2 (B-cell lymphoma 2) | F:AGGTGACACTATAGAATATGGAACTGTACGGCCCCAGCA | 140 |
| [Genebank:NM_000633] | R:GTACGACTCACTATAGGGAAGGGTGATGCAAGCTCCCACCA | |
| Fos (FBJ murine osteosarcoma viral oncogene homolog) | F:AGGTGACACTATAGAATAGTTATAAAAGCAGTGGCTGCGGC | 153 |
| [Genebank: NM_005252] | R:GTACGACTCACTATAGGGAAGCACGGTCACTGCTCGTTCG | |
| ICAM1 (intercellular adhesion molecule 1) | F:AGGTGACACTATAGAATAGTGCTATTCAAACTGCCCTGA | 169 |
| [Genebank:NM_000201] | R:GTACGACTCACTATAGGGAGCGTAGGGTAAGGTTCTTGC | |
| CCND1 (cyclin D1) | F:AGGTGACACTATAGAATAGCATGTTCGTGGCCTCTAAG | 177 |
| [Genebank: NM_053056] | R:GTACGACTCACTATAGGGACAGGTTCCACTTGAGCTTGTT | |
| MMP7 (matrix metalloproteinase-7) | F:AGGTGACACTATAGAATATTGGCTTTGCGCGAGGAGCT | 181 |
| [Genebank:NM_002423] | R:GTACGACTCACTATAGGGACTGCTACCATCCGTCCAGCGT | |
| Myc (V-myc myelocytomatosis viral oncogene homolog (avian)) | F:AGGTGACACTATAGAATACTCTCCTTGCAGCTGCTTAGA | 187 |
| [Genebank:NM_002467] | R:GTACGACTCACTATAGGGACCTCGTCGCAGTAGAAATACG | |
| PRKCE (Protein kinase C epsilon type) | F:AGGTGACACTATAGAATAAAGGTCCCTACCTTCTGCGA | 197 |
| [Genebank:NM_005400] | R:GTACGACTCACTATAGGGACCAGTACTTTGGCGATTCCT | |
| TP53 (Cellular Tumor Antigen p53) | F:AGGTGACACTATAGAATATTCCCTGGATTGGCAGCCAGACTG | 201.5 |
| [Genebank:NM_000546] | R:GTACGACTCACTATAGGGATCCATTGCTTGGGACGGCAAGG | |
| VCAM1 (vascular cell adhesion molecule 1) | F:AGGTGACACTATAGAATAAGGTGACGAATGAGGGGACCACATC | 215.4 |
| [Genebank:NM_001078] | R:GTACGACTCACTATAGGGAAGCCTCCAGAGGGCCACTCAAA | |
| IL4R (IL-4 receptor) | F:AGGTGACACTATAGAATAAAGTGGCACAACTCCTACAGG | 231 |
| [Genebank:NM_000418] | R:GTACGACTCACTATAGGGACCCTGAGCATCCTGGATTATT | |
| PTCH1 (Protein patched homolog 1) | F:AGGTGACACTATAGAATAGCCTTCGCTCTGGAGCAGATTT | 240 |
| [Genebank:NM_000264] | R:GTACGACTCACTATAGGGAGTTGGTCTCGAGGTTCGCTGCTT | |
| HSPB1(heat shock 27 kDa protein 1) | F:AGGTGACACTATAGAATAGCACACTGACCGTGGAGGCC | 241 |
| [Genebank: NM_001540] | R:GTACGACTCACTATAGGGAGAACACACAGGTGGCGGGGG | |
| PTGS2 (prostaglandin-endoperoxide synthase 2) | F:AGGTGACACTATAGAATACAGCTCCACAGCCAGACGCC | 249 |
| [Genebank: NM_000963] | R:GTACGACTCACTATAGGGATCCTGTCCGGGTACAATCGCACT | |
| HSF1 (Heat shock factor protein 1) | F:AGGTGACACTATAGAATAGCCTTCCTGACCAAGCTGTGGACC | 271 |
| [Genebank:NM_005526] | R:GTACGACTCACTATAGGGATCTCTCTGGCTTGACCAGGCCG | |
| GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) | F:AGGTGACACTATAGAATAAAGGTGAAGGTCGGAGTCAA | 277.2 |
| [Genebank:NM_002046] | R:GTACGACTCACTATAGGGAGATCTCGCTCCTGGAAGATG | |
| LEF1 (Lymphoid enhancer-binding factor-1) | F:AGGTGACACTATAGAATAGGTGCAGCCATCCCATGCGGT | 290.2 |
| [Genebank:NM_016269] | R:GTACGACTCACTATAGGGAAGGGTTGCCTGAATCCACCCGTG | |
| TCF7 (Transcription factor 7) | F:AGGTGACACTATAGAATAGGCGAGGAGCAGGACGACAAGAG | 305 |
| [Genebank:NM_003202] | R:GTACGACTCACTATAGGGATTTGTACATGCCGCTGGTGCAC | |
| FASN (Fatty acid synthase) | F:AGGTGACACTATAGAATAGAGTCGGAGAACTTGCAGGAGT | 307 |
| [Genebank:NM_004104] | R:GTACGACTCACTATAGGGAGTGTGTTCCTCGGAGTGAATCT | |
| KanR (Kanamycin resistance) | F:AGGTGACACTATAGAATAATCATCAGCATTGCATTCGATTCCTGTTTG | 325.4 |
| R:GTACGACTCACTATAGGGAATTCCGACTCGTCCAACATC | ||
| Multiplex 1: 5 genes (+ GAPDH and KanR) | ||
| Muc-1 (Mucin 1, transmembrane, transcript variant 1) | F:AGGTGACACTATAGAATAGACGTCAGCGTGAGTGATGT | 177.7 |
| [Genebank:NM_002456] | R:GTACGACTCACTATAGGGAGACAGCCAAGGCAATGAGAT | |
| Cyclin E2 (Cyclin E2 (CCNE2), transcript variant 3) | F:AGGTGACACTATAGAATAGAGCCCGAAGAGCACTGAAAAACC | 194.6 |
| [Genebank:NM_004702] | R:GTACGACTCACTATAGGGAGAGGAATTGGCTAGGGCAATCAA | |
| EGFR (EGF receptor, transcript variant 1) | F:AGGTGACACTATAGAATAGAAAGGCAGCCACCAAATTA | 208.4 |
| [Genebank:NM_005228] | R:GTACGACTCACTATAGGGAACTATCCTCCGTGGTCATGC | |
| Myc (V-myc myelocytomatosis viral oncogene homolog (avian)) | F:AGGTGACACTATAGAATAGAGCAACGTCTCCACACATCAGCAC | 217 |
| [Genebank:NM_002467] | R:GTACGACTCACTATAGGGAGAGTTTTGTGTGTTCGCCTCTTGAC | |
| Cyclin D1 (G1/S-specific cyclin-D1) | F:AGGTGACACTATAGAATACGTGGCCTCTAAGATGAAGG | 252.5 |
| [Genebank:BC001501] | R:GTACGACTCACTATAGGGATGCGGATGATCTGTTTGTTC | |
| GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) | F:AGGTGACACTATAGAATAAAGGTGAAGGTCGGAGTCAA | 277.2 |
| [Genebank:NM_002046] | R:GTACGACTCACTATAGGGAGATCTCGCTCCTGGAAGATG | |
| KanR (Kanamycin resistance) | F:AGGTGACACTATAGAATAATCATCAGCATTGCATTCGATTCCTGTTTG | 325.4 |
| R:GTACGACTCACTATAGGGAATTCCGACTCGTCCAACATC | ||
F: Forward; R: Reverse.
RNA was isolated from all cell samples using an RNA isolation kit (Fermentas, Glen Burnie, Maryland, USA). Gene expression levels of 15 genes involved in drug resistance, cell cycle and signaling pathways were assayed using the capillary GenomeLab GeXP genetic analysis system (Beckman-Coulter, Brea, California). A multiplex panel was designed to assess the genes. In addition to the genes of interest, each panel contained an internal control gene (kanamycin resistance, KanR) and a normalization gene (GAPDH). cDNA was synthesized from 500 ng total RNA using the GenomeLab GeXP Start Kit (Beckman-Coulter, Brea, California). PCR and multiplex detection were performed according to the manufacturer's instructions. GeXP data were analyzed after normalization of all genes of interest against the geometric mean of the normalization gene.
Cell cycle assay
Cell cycle analysis was carried out according to the following protocols. Cells from each group were washed twice in PBS and fixed in cold 70% ethanol for at least 3 h at 4°C. Cells were then washed twice in PBS and stained with 1 ml of PI (20 μg/ml). A 50-μl volume of RNase A (10 μg/ml) was added to the samples and incubated for 3 h at 4°C. Stained cells were analyzed by flow cytometry using CellQuest Pro software (BD Biosciences, Franklin Lakes, New Jersey).
Tumorigenesis assay
Non-obese diabetic (NOD)/SCID mice (5-6 weeks old) (NOD.CB17-Prkdcscid/J) (Charles River Laboratories) were used in this research. Four experiments were performed to evaluate the effects of CD44 knockdown on the tumorigenic potential of BCSCs. In the first experiment, BCSCs were injected into 12 mice with three mice/dose at 106, 105, 104 and 103 cells/mouse. Similarly, CD44 knockdown BCSCs and non-BCSCs were injected at the same doses in the second and third experiments. The fourth experiment comprised negative control mice injected with PBS. The first and second, and third and fourth experiments, respectively, were performed using the right and left sides of the same mice. All mice were housed in clean cages and maintained according to institutional guidelines on animal welfare. Mice were followed up for 2 months to detect tumors.
Statistical analysis
All experiments were performed in triplicate. Differences between mean values were assessed by t-tests and analysis of variance. A P value of ≤ 0.05 was considered to be significant. Data were analyzed using Statgraphics software (v 7.0; Statgraphics Graphics System, Warrenton, VA).
Results
Isolation and culture of BCSCs and non-BCSCs
Primary cultures were derived from 10 breast tumor samples, including eight well-developed tumors with many cells expanding from the tissues and two samples that were infected with micro-organisms. The primary cultures were sub-cultured to produce a large number of cells, which were then evaluated for the presence of BCSCs. BCSCs comprised 4.32 ± 1.78% of all the samples (range 2.51-7.09%) (Figure 1A, B). The sample with the highest BCSC population was used to isolate four populations based on the expression of CD44 and CD24 markers (CD44+CD24-, CD44+CD24+, CD44-CD24+, and CD44-CD24- cells) by magnetic cell sorting. The purities of the isolated cell populations assessed by flow cytometry ranged from 95.14-99.99% (Figure 1C-F). The four cultured cell populations had homogeneous shapes (Figure 2) and were cultured and subcultured to produce sufficient cells for further experiments.
Figure 1.
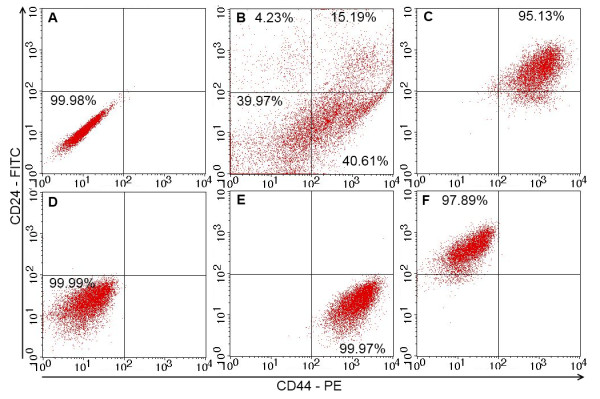
Expression of CD44 and CD24 in four different cell populations analyzed by flow cytometry. (A) Unstained cells. The breast cancer cell population (B) was sorted into four populations: CD44+CD24+ (C), CD44-CD24+ (D) and CD44-CD24- (E), CD44+CD24- (BCSCs) (F). All sorted cell populations exhibited high degrees of purity.
Figure 2.

Primary cells, breast cancer cells and four sub-populations of CD44+CD24+, CD44+CD24-, CD44-CD24- and CD44-CD24+ cells. Cells expanding from the tissue (A) showed at least two different shapes (epithelial and stromal) and became homogeneous after sub-culturing (B). These cells were sorted into four sub-populations of CD44-CD24+ (C), CD44-CD24- (D), CD44+CD24- (E) and CD44+CD24+ cells (F).
Expression of CD44 in CD44 knockdown BCSCs
Following shRNA lentivirus transfection and selection with puromycin for 1 week, CD44 knockdown BCSCs showed decreased CD44 expression compared with BCSCs before knockdown, as demonstrated by immunocytochemistry and flow cytometry (Figure 3). Thus CD44 shRNA lentivirus combined with puromycin selection efficiently silenced CD44 mRNA expression in treated cells. Protein quantification by flow cytometry demonstrated that the percentage of CD44-positive cells in BCSCs before and after CD44 knockdown was reduced from 96.32% ± 3.33% to 0.12% ± 0.03% (n = 3) (Figure 3D, H). This level of suppression was greater than that achieved in a previous study using transfection of small interfering RNA (siRNA) [24].
Figure 3.

CD44 expression before (A, B, C and D) and after CD44 knockdown by shRNA (E, F, G and H) combined with selection using puromycin dihydrochloride for 1 week. Cells were evaluated by immunocytochemistry after staining with anti-CD44 and fluorescein-isothiocyanate (FITC)-conjugated secondary antibody and were observed under a monochromatic fluorescence microscope (Carl Zeiss, Oberkochen, Germany) with white light, Hoechst 33342 and a FITC filter (A and E, B and F, C and G, respectively) and by flow cytometry after staining with anti-CD44-FITC (D and H).
Gene expression in CD44 knockdown BCSCs compared with BCSCs and non-BCSCs
The expression of important genes related to stemness, anti-tumor drug resistance, and metastasis in BCSCs was altered in CD44 knockdown BCSCs, as shown in Figures 4 and 5. Muc-1, MMP9, and Myc expression levels were strongly reduced by CD44 knockdown, bringing them in line with levels in non-BCSCs. Levels of several other genes such as EGFR and cyclin D1 also fell. High Bcl-2 expression in BCSCs is particularly associated with chemoresistance, and its expression also decreased to the level in non-BCSCs after knockdown of CD44. The expression of genes related to stemness, such as LEF1, also decreased. LEF1, TCF7, and Myc are members of the Wnt signaling pathway; Bcl-2, MMP7, and Myc are members of the PI3K/AKT signaling pathway; HSF1, TP53, and MYC are members of the Stress pathway; and PTCH1, PKRCE, PTGS2, and IL4R are members of the Hedgehog signaling pathway. Expression of all these genes was reduced to levels similar to those seen in non-BCSCs.
Figure 4.
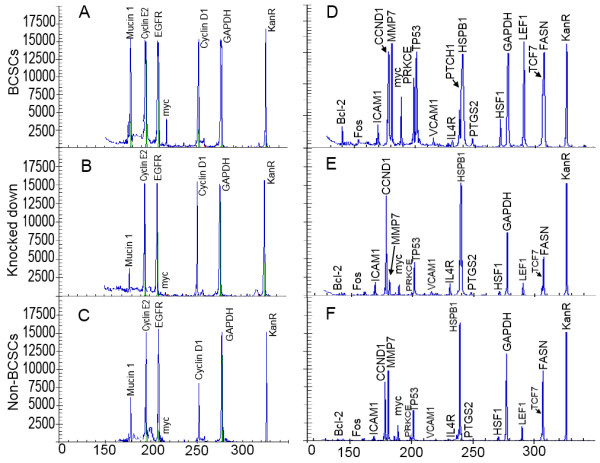
Similar gene expression patterns in CD44 knockdown BCSCs (B, E) and non-BCSCs (C, F) compared with BCSCs (A, D), in genes related to cell viability, proliferation, metastasis and anti-tumor drug resistance, analyzed using a genetic analysis system (GeXP, Beckman-Coulter, Brea, California). Knocked down: CD44 knockdown BCSCs.
Figure 5.

Expression levels of genes related to metastasis (A), cell viability, proliferation and anti-tumor drug resistance (B) in BCSCs, non-BCSCs and CD44 knockdown BCSCs normalized to GAPDH. Knockdown: CD44 knockdown BCSCs.
Cell cycle in CD44 knockdown BCSCs compared with BCSCs and non-BCSCs
The cell cycle was affected by knock-down of CD44, as shown in Figure 6. The percentage of cells in G2/M phase was significantly higher in BCSCs compared with non-BCSCs (28.60 ± 0.60% vs. 23.41% ± 0.50%, P < 0.05) while the number of cells in S phase was lower (13.93 ± 0.69% vs 20.08 ± 0.31%, P < 0.05). In contrast, the numbers of cells in G1/G0 phase were similar in BCSCs and non-BCSCs (57.47 ± 0.23% vs 56.51 ± 0.55%, P > 0.05). G2/M phase and S phase in CD44 knockdown BCSCs approached those in non-BCSCs. G2/M phase in CD44 knockdown BCSCs decreased and was similar to non-BCSCs (24.23 ± 0.34% vs 23.41 ± 0.50%, respectively) while S phase increased from 13.93 ± 0.69% in BCSCs to 16.98 ± 0.95% in CD44 knockdown BCSCs, compared with 20.08 ± 0.31% in non-BCSCs (Figure 6). The percentages of cells in G1/G0 phase in BCSCs, non-BCSCs and CD44 knockdown BCSCs were similar. These results suggest that CD44 knockdown decreased proliferating ability and extended S phase to increase the similarities with non-BCSCs.
Figure 6.

Similar cell cycles in non-BCSCs (B, E, H), and CD44 knockdown BCSCs (C, F, I) cells compared with BCSCs (A, D, G). G1/G0 phase was relatively unchanged, but G2/M phase decreased and S phase increased in CD44 knockdown BCSCs.
Tumorigenesis of CD44 knockdown BCSCs compared with BCSCs and non-BCSCs in NOD/SCID mice
The tumor-causing potential of the cells was evaluated to assess the differentiated phenotype after CD44 knockdown. BCSCs caused tumors in 66.67% (2/3) of mice with 103 cells, while 106 non-BCSCs were needed to cause tumors in 25% of mice (1/3). The tumor-causing potential was reduced in the CD44 knockdown BCSCs, with doses of 104 cells causing tumors in 0% of mice, compared with 100% of mice before CD44 down-regulation (Figure 7). At doses of 105 grafted cells, BCSCs were capable of generating tumors in up to 100% of mice (3/3), compared with only 33.33% (1/3) of mice in the case of CD44 knockdown BCSCs. Figure 7A shows that injection with 106 BCSCs caused large tumors (on the right), while 106 CD44 knockdown BCSCs failed to produce any tumors (on the left). This suggests that knockdown of CD44 caused differentiation and loss of the stemness characteristics of BCSCs.
Figure 7.
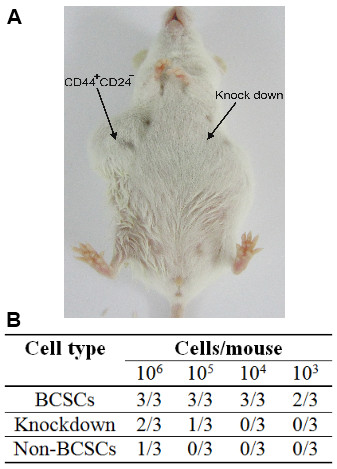
Tumorigenic capacities of CD44 knockdown BCSCs, BCSCs and non-BCSCs in NOD/SCID mice. CD44 knockdown BCSCs and non-BCSCs were similar, but there was a significant difference in tumorigenic capacity between CD44 knockdown BCSCs and BCSCs (B). Injection of 106 BCSCs caused large tumors (right), while 106 CD44 knockdown BCSCs failed to produce any tumors (left) (A).
Discussion
The effectiveness of breast cancer treatment currently remains low. This may be attributable to the existence of a small population of cancer stem cells with high resistance to chemotherapy and radiation therapy, which can thus be responsible for high rates of relapse after treatment, as well as for metastasis. Cancer stem-cell-targeting therapy thus represents a promising potential therapy for the treatment of breast cancer. In this study, we evaluated the role of CD44 in maintaining stemness and inhibiting the differentiation of BCSCs. Previous studies suggested that downregulation of CD44 allowed BCSCs to differentiate into cancer non-BCSCs or normal cells in breast tissue. To confirm this, we initially isolated BCSCs from malignant breast tumors based on their CD44 and CD24 expression pattern.
To determine the contribution of CD44 to the characteristics of BCSCs, we performed CD44 knockdown using a shRNA lentiviral vector and puromycin selection. This method was more effective than siRNA for generating a stable and pure cell population lacking CD44 expression, which could then be compared with non-knockdown cells.
The stemness of the CD44 knockdown cells was evaluated based on three criteria: the expression of genes related to stem cells, metastasis, and drug resistance; changes in the cell cycle; and the ability to form tumors in vivo in a NOD/SCID mouse model.
CD44 knockdown cells showed dramatically changed gene expression patterns compared with the original cells. Genes associated with the metastatic ability of cancer stem cells, especially Muc-1, MMP9 and Myc, were strongly reduced by CD44 knockdown. Mucin 1 is encoded by the Muc-1 gene. Mucin-1 protects the body from infection by binding pathogens to oligosaccharides via the extracellular domain, thus preventing the pathogen from reaching the cell surface [25,26]. Mucin-1 also functions in a cell-signaling capacity [27]. Over-expression of Muc-1 is often associated with colon, breast, ovarian, lung and pancreatic cancers [28]. Mucin-1 plays important roles in cancer development and metastasis by inhibiting the anti-tumor immune response [29], promoting the growth of cancer cells by binding to EGFR in an epidermal growth factor-dependent manner [30], preventing cell death by inhibition of p53-mediated apoptosis and promotion of p53-mediated cell cycle arrest [31], and promoting cancer metastasis [32-34]. Because Mucin-1 promotes the expression of Myc [35], levels of Myc expression were also decreased in association with Mucin-1 down-regulation, with consequent effects on the metastatic ability of BCSCs. A recent study by Fessler et al. showed that Mucin-1 was a determinant of trastuzumab (Herceptin) resistance in breast cancer cells, as well as being associated with resistance to taxol, doxorubicin, and cyclophosphamide [36]. Low expression of Mucin-1 would thus be expected to decrease metastasis and drug resistance in BCSCs.
EGFR and cyclin D1 expression were also reduced in CD44 knockdown cells. EGFR is often strongly expressed in many cancers [37,38], including breast cancer. However, BCSCs that weakly express this gene are unaffected by drugs that attack the EGFR, such as gefitinib, erlotinib, and cetuximab. The reduction of CD44 expression increased EGFR expression to a level similar to that in non-BCSCs, which are sensitive to chemotherapy. Cyclin D1 is encoded by the G1/S-specific CCND1 gene, and increased expression of cyclin D1 thus caused cells to move rapidly into S phase. However, cyclin E2 expression was not increased by CD44 knockdown, and cells were therefore mainly stopped in phase G1/S. The results of cell cycle analysis were in accord with these explanations.
Gene expression analysis also showed down-regulation of Bcl-2 by CD44 knockdown. Bcl-2 is capable of inhibiting anticancer drug-induced apoptosis mediated by the voltage-dependent anion channel in the outer mitochondrial membrane, and over-expression of Bcl-2 and Bcl-XL might confer resistance to chemotherapy [39]. Cells with low Bcl-2 gene expression are more sensitive to chemotherapy. Previous studies showed that CD44 knockdown cells were more sensitive to doxorubicin than BCSCs, similar to breast cancer cells [24]. FASN was also down-regulated in CD44 knockdown BCSCs. FASN expression is up-regulated in the early steps of breast cancer and represents a therapeutic target for breast cancer metastasis [40,41] and liposarcoma [42]. Inhibition of FASN suppressed the growth of cancer stem-like cells in breast cancer [43] and colon cancer [44], and induced apoptosis in diffuse large B-cell lymphoma [45] and in gastric-tumor-bearing mice [46]. CD44 knockdown was also associated with down-regulation of heat shock transcription factor 1 (HSF1) to a level similar to that seen in non-BCSCs. HSF1 is a major transactivator of genes coding for heat shock proteins. HSF1 is involved in tumor initiation, maintenance, and progression by regulating the expression of heat shock proteins [47]. Down-regulation of HSF1 decreased cell proliferation and enhanced sensitivity to hyperthermia in human melanoma cell lines [48]. It has thus been considered as a promising target for anti-cancer treatment [49], especially in breast cancer [50].
LEF1 up-regulates Oct4 promoter activity and physically interacts with Nanog; these comprise two key components of embryonic stem cell pluripotency. Down-regulation of LEF1 by siRNA induced differentiation of mouse embryonic stem cells [33]. The low expression of LEF1 in non-BCSCs and the down-regulation of LEF1 by CD44 knockdown indicated the differentiation of BCSCs into non-BCSCs. Moreover, LEF1, TCF7 and Myc are all members of the Wnt signaling pathway, and their down-regulation thus represents suppression of the Wnt signaling pathway. Similarly Bcl-2, MMP7, and Myc are members of the PI3K/AKT pathway; HSF1, TP53, and Myc belong to the Stress signaling pathway; and PTCH1, PKRCE, PTGS2 (Cox-2), and IL4R are members of the Hedgehog, calcium and protein kinase C, and Jak-Stat pathways, respectively. Their reduced expression following CD44 knockdown demonstrated its effect on several signaling pathways. The Wnt, Hedgehog and Jak-Stat pathways are important pathways in stem cells and cancer stem cells and have been considered as promising therapeutic targets [51-54]. Down-regulation of some key signaling pathway genes proved that CD44 knockdown BCSCs underwent phenotypic changes from cancer stem cells to cancer cells or normal cells. Down-regulation of the Stress and calcium protein kinase C pathways might increase the sensitivity of CD44 knockdown BCSCs to some anti-tumor drugs, such as doxorubicin, because the Stress and protein kinase C pathways help cancer cells to cope with stress and changes of environment.
Changes in the gene expression profiles of CD44 knockdown BCSCs drove the cell cycle towards that seen in non-BCSCs. S-phase cells were increased and G2/M cells decreased in CD44 knockdown BCSCs and non-BCSCs compared with BCSCs. These cell cycle results resembled those found in cancer stem cells from solid tumors isolated on the basis of CD133 expression, showing that cancer stem cells were mainly in G2/M phase [55]. Karimi-Busheri et al. compared the cell cycles of mammosphere-forming and adhesive cells in cancer stem cells isolated from MCF-7 cell lines; most adhesive cells were in S phase, while mammosphere-forming cells were concentrated in the G2/M phase [56]. Thus, CD44 knockdown appeared to drive BCSCs toward a non-BCSC phenotype and differentiation.
The main physiological difference between cancer stem cells and the remaining cells in the tumor is their different tumorigenic potentials when transplanted into mice. Tumorigenic potential thus represents the gold standard for demonstrating a change in stemness of BCSCs. The tumorigenic potential of CD44 knockdown BCSCs in this study was reduced to that of non-BCSCs. Many previous studies found that as few as 50-100 BCSCs were adequate to generate tumors in NOD/SCID mice [3], while others found that 103 cancer stem cells could cause tumors [4,57]. In the current study, grafts of 103 BCSCs were capable of producing tumors in SCID mice, whereas CD44 knockdown BCSCs required at least 106 cells, similar to the situation for non-BCSCs. The altered biological characteristics of these cells indicated that CD44 knockdown changed the stem cell phenotype with high tumor-causing potential into cells with lower tumor-causing potential, representing differentiation of the cancer stem cells.
Differentiation therapy targeting cancer stem cells is currently under investigation by many groups, particularly focusing on the use of chemicals to cause stem cell differentiation. Takehara et al. showed that BCSCs differentiated when treated with acetaminophen, which also inhibited tumor formation in a nude mouse model [58]. Estrogen also causes BCSCs to differentiate, as demonstrated by a reduction in the number of cancer stem cells in tumors positive for estrogen receptors [59]. All-trans retinoic acid (ATRA) can prevent breast cancer recurrence by inducing BCSC differentiation and cell cycle arrest [2,60]. BCSCs may also be affected in terms of stem cell self-renewal, differentiation, motility and mesenchymal phenotype after treatment with the polyamine analog [1 N, 12 N] bis (ethyl)-cis -6.7-dehydrospermine (PG11047) [61]. Similarly, Roy et al. found that ATRA, trichostatin A and vorinostat caused dose-dependent decreases in the BCSC population, and showed that these differentiating agents reduced the number of BCSCs within the MCF7 cell line. Mammosphere formation in primary breast cancers (n = 3) was decreased by ≥ 25% by ATRA treatment combined with 6 Gy irradiation, compared with irradiation alone [62].
The results of the current study showed that CD44 plays an important role in the maintenance of BCSC stemness. Because inhibition of CD44 expression caused differentiation of BCSCs as well as reduced anti-tumor drug resistance [24], it is possible that gene therapy designed to interfere with CD44, as well as other factors that could reduce CD44 expression, represent promising therapeutic strategies for treating breast cancer, especially in combination with radiation or other anti-tumor agents.
This study used CD44 shRNA lentiviral particles to generate a stable CD44 knockdown BCSC population, with clear modifications in gene expression and cell characteristics. However, reverse-transcribed DNA can randomly insert into the cell genome and potentially disturb the function of cellular genes, leading to the activation of oncogenes and thus promoting the development of cancer. However, previous studies found that lentiviral vectors had a low tendency to integrate in places that caused cancer [63], and one study found no increase in tumor incidence and no earlier onset of tumors in a mouse strain following the use of lentiviral vectors [64]. In addition, we randomly evaluated 10 colonies and found similar results in terms of gene expression. Given that lentivirus integration is random, the 10 colonies would be exceptionally unlikely to contain similar insertion sites to disrupt the function of the genes.
Conclusions
Down-regulation of CD44 caused changes in the phenotype of BCSCs. CD44 knockdown BCSCs lost the BCSC phenotype and showed reduced expression of genes related to stemness, metastasis and tumorigenesis, especially Muc-1 and Bcl-2. In addition, the cell cycle changed to resemble that seen in differentiated cells (non-BCSCs), and anti-tumor drug resistance and tumorigenic potential in NOD/SCID mice were both reduced. These data indicate that BCSCs were differentiated into non-BCSCs by CD44 knockdown. These results suggest that a combination of differentiation therapy aimed at down-regulation of CD44 in BCSCs, together with chemical or radiation therapies, represents a promising therapeutic strategy for the treatment of breast cancer, and also suggest that RNAi gene therapy could provide a novel differentiation strategy.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors read and approved the final manuscript. PVP carried out studies including primary culture, isolation of breast cancer stem cells, gene expression analysis and designed the study and drafted the manuscript in cooperation with all other authors. NLCP and NTN performed the knockdown of CD44 by shRNA. NHT and TTD participated in the in vivo animal experiments. DVL and KDT participated in the cell cycle and flow cytometry analysis. NKP participated in designing the study and drafted the manuscript in cooperation with all other authors, and also checked PCR primers.
Contributor Information
Phuc V Pham, Email: pvphuc@hcmuns.edu.vn.
Nhan LC Phan, Email: plcnhan@hcmus.edu.vn.
Nhung T Nguyen, Email: ntnhung@hcmus.edu.vn.
Nhung H Truong, Email: thnhung@hcmuns.edu.vn.
Thuy T Duong, Email: dtthuy@hcmuns.edu.vn.
Dong V Le, Email: levandong@yahoo.com.
Kiet D Truong, Email: dtkiet87@yahoo.com.
Ngoc K Phan, Email: pkngoc@hcmuns.edu.vn.
Acknowledgements
This work was funded by grants from the Vietnam National Project about Breast Cancer, Ministry of Science and Technology, Vietnam (Code: DTDL.2011-T/30). We thank the Oncology Hospital at Ho Chi Minh for supplying the malignant breast cancer tumors.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Wicinski J, Cervera N, Monville F, Finetti P, Bertucci F, Wicha MS, Birnbaum D, Charafe-Jauffret E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle. 2009;8(20):3297–302. doi: 10.4161/cc.8.20.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10(1):R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuc PV, Khuong TTT, Dong LV, Kiet TD, Giang TT, Ngoc PK. Isolation and characterization of breast cancer stem cells from malignant tumours in Vietnamese women. JCAB. 2010;4(12):163–16. [Google Scholar]
- Dave B, Chang J. Treatment resistance in stem cells and breast cancer. J Mammary Gland Biol Neoplasia. 2009;14:79–82. doi: 10.1007/s10911-009-9117-9. [DOI] [PubMed] [Google Scholar]
- Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. 2007;104(2):618–23. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Yang D, Lippman ME. Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Semin Oncol. 2003;30(5 Suppl 16):133–42. doi: 10.1053/j.seminoncol.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7(7):989–99. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat ML, Shackleton M, Stingl J, Vaillant F, Forrest NC, Eaves CJ, Visvader JE, Lindeman GJ. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98(14):1011–4. doi: 10.1093/jnci/djj267. [DOI] [PubMed] [Google Scholar]
- Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xeno-grafts. Proc Natl Acad SciUSA. 2008;147(9):4056–66. doi: 10.1073/pnas.0706216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad SciUSA. 2009;106(33):13820–5. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorechovsky' I, Benediktsson KP, Toftgård R. The patched/hedgehog/smoothened signalling pathway in human breast cancer: No evidence for H133Y SHH, PTCH and SMO mutations. Eur J Cancer. 1999;35(5):711–3. doi: 10.1016/S0959-8049(99)00017-9. [DOI] [PubMed] [Google Scholar]
- Soriano JV, Uyttendaele H, Kitajewski J, Montesano R. Expression of an activated Notch4(int-3) oncoprotein disrupts morphogenesis and induces an invasive phenotype in mammary epithelial cells in vitro. Int J Cancer. 2000;86:652–659. doi: 10.1002/(SICI)1097-0215(20000601)86:5<652::AID-IJC8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148(3):567–78. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly OG, Pinson KI, Skarnes WC. The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development. 2004;131(12):2803–15. doi: 10.1242/dev.01137. [DOI] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–11. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7(3):86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani G, Fasolo A, De Benedictis E, Gianni L. Trastuzumab as adjuvant systemic therapy for HER2-positive breast cancer. Nat Clin Pract Oncol. 2009;6(2):93–104. doi: 10.1038/ncponc1298. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Carlsson J, Nordgren H, Sjöström J, Wester K, Villman K, Bengtsson NO, Ostenstad B, Lundqvist H, Blomqvist C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer. 2004;90(12):2344–8. doi: 10.1038/sj.bjc.6601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthases: potential therapeutic targets in cancer. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5(1):3–14. doi: 10.2174/1568006053005056. [DOI] [PubMed] [Google Scholar]
- Phuc PV, Nhan PLC, Nhung TH, Tam NT, Hoang NM, Tue VG, Thuy DT, Ngoc PK. Downregulation of CD44 reduces doxorubicin resistance of CD44+CD24- breast cancer cells. Onco Targets Ther. 2011;4:71–8. doi: 10.2147/OTT.S21431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Hollingsworth MA. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006;16(9):467–476. doi: 10.1016/j.tcb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Moncada DM, Kammanadiminiti SJ, Chadee K. Mucin and Toll-like receptors in host defense against intestinal parasites. Trends Parasitol. 2003;19(7):305–311. doi: 10.1016/S1471-4922(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, Sutton P, McGuckin MA, Van Nhieu, Guy Tran. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 2009;5(10):e1000617. doi: 10.1371/journal.ppat.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendler SJ. MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia. 2001;6(3):339–353. doi: 10.1023/A:1011379725811. [DOI] [PubMed] [Google Scholar]
- Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- Pochampalli MR, el Bejjani RM, Schroeder JA. MUC1 is a novel regulator of ErbB1 receptor trafficking. Oncogene. 2007;26(12):1693–1701. doi: 10.1038/sj.onc.1209976. [DOI] [PubMed] [Google Scholar]
- Wei X, Xu H, Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancel Cell. 2005;7(2):167–178. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Bharti A, Li Y, Kufe D. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion. J Biol Chem. 1997;272(19):12492–4. doi: 10.1074/jbc.272.19.12492. [DOI] [PubMed] [Google Scholar]
- Huang C, Qin D. Role of Lef1 in sustaining self-renewal in mouse embryonic stem cells. J Genet Genomics. 2010;37(7):441–9. doi: 10.1016/S1673-8527(09)60063-1. [DOI] [PubMed] [Google Scholar]
- Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SIL, Gendler SJ, Mukherjee P. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30(12):1449–59. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Raina D, Chen W, Li G, Huang L, Kufe D. MUC1 oncoprotein functions in activation of fibroblast growth factor receptor signaling. Mol Cancer Res. 2006;4(11):873–883. doi: 10.1158/1541-7786.MCR-06-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler SP, Wotkowicz MT, Mahanta SK, Bamdad C. MUC1 is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res Treat. 2009;118(1):113–24. doi: 10.1007/s10549-009-0412-3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–8. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F, Abramowitz L, Benabderrahmane D, Duval X, Descatoire VR, Hénin D, Lehy TRS, Aparicio T. Growth factor receptor expression in anal squamous lesions: modifications associated with oncogenic human papillomavirus and human immunodeficiency virus. Human Pathology. 2009;40(11):1517–27. doi: 10.1016/j.humpath.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Verrier F, Deniaud A, Lebras M, Metivier D, Kroemer G, Mignotte B, Jan G, Brenner C. Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene. 2004;23:8049–8064. doi: 10.1038/sj.onc.1208001. [DOI] [PubMed] [Google Scholar]
- Lupu R, Menendez JA. Targeting fatty acid synthase in breast and endometrial cancer: An alternative to selective estrogen receptor modulators? Endocrinology. 2006;147(9):4056–66. doi: 10.1210/en.2006-0486. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Guerrico A, Lupu R. Fatty Acid Synthase (FASN) as a Therapeutic Target for Breast Cancer Metastasis. Cancer Res. 2009;69(24 Suppl) Abstract nr 6160. [Google Scholar]
- Olsen AM, Eisenberg BL, Kuemmerle NB, Flanagan AJ, Morganelli PM, Lombardo PS, Swinnen JV, Kinlaw WB. Fatty acid synthesis is a therapeutic target in human liposarcoma. Int J Oncol. 2010;36(5):1309–14. doi: 10.3892/ijo_00000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, Wakabayashi G, Koeda K, Kashiwaba M, Suzuki K, Chiba T, Endo M, Fujioka T, Tanji S, Mo YY, Cao D, Wilber AC, Watabe K. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2010. in press . [DOI] [PMC free article] [PubMed]
- Murata S, Yanagisawa K, Fukunaga K, Oda T, Kobayashi A, Sasaki R, Ohkohchi N. Fatty acid synthase inhibitor cerulenin suppresses liver metastasis of colon cancer in mice. Cancer Sci. 2010;101(8):1861–5. doi: 10.1111/j.1349-7006.2010.01596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin S, Hussain AR, Ahmed M, Bu R, Ahmed SO, Ajarim D, Al-Dayel F, Bavi P, Al-Kuraya KS. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol Cancer Ther. 2010;9(5):1244–55. doi: 10.1158/1535-7163.MCT-09-1061. [DOI] [PubMed] [Google Scholar]
- Dowling S, Cox J, Cenedella RJ. Inhibition of fatty acid synthase by Orlistat accelerates gastric tumor cell apoptosis in culture and increases survival rates in gastric tumor bearing mice in vivo. Lipids. 2009;44(6):489–98. doi: 10.1007/s11745-009-3298-2. [DOI] [PubMed] [Google Scholar]
- Meng L, Gabai VL, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene. 2010;29(37):5204–13. doi: 10.1038/onc.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Fujimoto M, Hayashida N, Takii R, Nakai A, Muto M. Silencing HSF1 by short hairpin RNA decreases cell proliferation and enhances sensitivity to hyperthermia in human melanoma cell lines. J Dermatol Sci. 2010;60(3):187–92. doi: 10.1016/j.jdermsci.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets. 2009;13(4):469–78. doi: 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]
- Calderwood SK. Heat shock proteins in breast cancer progression--a suitable case for treatment? Int J Hyperthermia. 2010;26(7):681–5. doi: 10.3109/02656736.2010.490254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe Naoko, Harris Pamela J, Warren Ronald Q, Percy Ivy S. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature Reviews Clinical Oncology. 2011;8(2):97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- Suling Liu, Wicha Max S. Targeting Breast Cancer Stem Cells. JCO. 2010;28(25):4006–4012. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget. 2010;1(7):563–77. doi: 10.18632/oncotarget.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vargas H, Ouzounova M, Le Calvez-Kelm F, Lambert MP, McKay-Chopin S, Tavtigian SV, Puisieux A, Matar C, Herceg Z. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics. 2010;6(4):428–39. doi: 10.4161/epi.6.4.14515. [DOI] [PubMed] [Google Scholar]
- Tirino V, Desiderio V, d'Aquino R, De Francesco F, Pirozzi G, Graziano A, Galderisi U, Cavaliere C, De Rosa A, Papaccio G, Giordano A. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One. 2008;3(10):e3469. doi: 10.1371/journal.pone.0003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Busheri F, Rasouli-Nia A, Mackey JR, Weinfeld M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010;12(3):R31. doi: 10.1186/bcr2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- Takehara M, Hoshino T, Namba T, Yamakawa N, Mizushima T. Acetaminophen-induced differentiation of human breast cancer stem cells and inhibition of tumor xenograft growth in mice. Biochem Pharmacol. 2011;81(9):1124–35. doi: 10.1016/j.bcp.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Simões BM, Vivanco MD. Cancer stem cells in the human mammary gland and regulation of their differentiation by estrogen. Future Oncol. 2011;7(8):995–1006. doi: 10.2217/fon.11.80. [DOI] [PubMed] [Google Scholar]
- Li RJ, Ying X, Zhang Y, Ju RJ, Wang XX, Yao HJ, Men Y, Tian W, Yu Y, Zhang L, Huang RJ, Lu WL. All-trans retinoic acid stealth liposomes prevent the relapse of breast cancer arising from the cancer stem cells. J Control Release. 2011;149(3):281–91. doi: 10.1016/j.jconrel.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Cirenajwis H, Smiljanic S, Honeth G, Hegardt C, Marton LJ, Oredsson SM. Reduction of the putative CD44+CD24- breast cancer stem cell population by targeting the polyamine metabolic pathway with PG11047. Anticancer Drugs. 2010;21(10):897–906. doi: 10.1097/CAD.0b013e32833f2f77. [DOI] [PubMed] [Google Scholar]
- Roy R Willan P Clarke R Farnie G Differentiation therapy: targeting breast cancer stem cells to reduce resistance to radiotherapy and chemotherapy Breast Cancer Res 201012Suppl 1O5. 10.1186/bcr249620497593 [DOI] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, Schmidt M, von Kalle C, Howe S, Thrasher AJ, Aiuti A, Ferrari G, Recchia A, Mavilio F. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110(6):1770–8. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, Doglioni C, von Kalle C, Naldini L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24(6):687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]