

Abstract
One major challenge in the development of cancer therapeutics is the selective delivery of the drugs to their cellular targets. In the case of pancreatic cancer, the σ-2 receptor is a unique target that triggers apoptosis upon activation. We have previously developed a series of chemical compounds with high affinity for the σ-2 receptor and demonstrated rapid internalization of the ligands. One particular specific ligand of the σ-2 receptor, SV119, binds to pancreatic cancer cells and induces target cell death in vitro and in vivo. In this study, we characterized the ability of SV119 to selectively deliver other death-inducing cargos to augment the cytotoxic properties of SV119 itself. When conjugated to SV119, small molecules that are known to interfere with intracellular pro-survival pathways retained their ability to induce cell death, the efficiency of which was enhanced by the combinatorial effect of SV119 delivered with its small molecule cargo.
Our findings define a simple platform technology to increase the tumor-selective delivery of small molecule therapeutics via sigma-2 ligands, permitting chemotherapeutic synergy that can optimize efficacy and patient benefit.
Keywords: apoptosis, drug delivery, pancreatic neoplasms, σ-2 receptor, sigma-2 conjugates
INTRODUCTION
There have been notable improvements since 1975 in the relative 5-year survival rates for many cancers, excluding pancreatic cancer, for which survival has not improved substantially over the past 30 years (1). From 1999 to 2005, the 5-year survival rate was approximately 4–6% (1). This poor outcome reflects primarily the difficulty in achieving early diagnosis and the failure of surgery, radiation and chemotherapy. At diagnosis, only about 15% of patients are eligible for surgical resection (2). After pancreatectomy, and even when combined with chemotherapy, radiotherapy, and immunotherapy the majority of patients develop recurrence and median survival is approximately 2 years (3). Novel therapeutic strategies are desperately needed.
There is considerable interest in stimulating apoptosis and inhibiting the survival machinery as components of cancer therapy (4–6). Many oncogenic transformations result from inactivation or loss of pro-apoptotic genes or the translocation of an anti-apoptotic chromosomal segment to the vicinity of highly active promoters (5;7;8). The σ-2 receptor is a unique target that can facilitate tumor cell apoptosis for pancreatic cancer when engaged with an appropriate ligand(s). The sigma receptor was initially proposed to represent a subtype of opioid receptors (9). Early receptor binding studies using benzomorphan opioids indicated the existence of at least two types of sigma receptors: the σ-1 and σ-2 subtypes (5). These subtypes display different tissue distributions and distinct physiological and pharmacological profiles in both the central and peripheral nervous systems. Although the natural ligand for the σ-2 receptor is still unknown, it has been demonstrated that this receptor is over-expressed in a variety of human and rodent tumors (5;6;10;11), including pancreatic cancer with only weak expression in the normal pancreas (12). As such, synthetic ligands to this receptor could play an important role in cancer diagnosis, imaging, and therapy (13). In fact, we have recently reported on progesterone receptor component 1 (PGRMC1) as being part of the σ-2 receptor complex (14). Equally important, especially with regard to our current study, is the notion that related sigma-2 ligands are rapidly internalized into cancer cells (11).
Here, we assessed the feasibility by chemical conjugation to deliver death-inducing cargo via the σ-2 receptor-specific ligand SV119 into the cancer cells. We focused on various molecules along the apoptosis pathway such as Bim (Bcl-2 interacting mediator of dell death), a BH3-only pro-apoptotic protein in the Bcl-2 family (15–18), carboxyl-terminal modulator protein (CTMP), an Akt inhibitor (19–21), and rapamycin, acting on the PI3K/Akt signaling axis (22;23).
The findings described in our current paper represent a major step in the path to personalized cancer medicine and introduce a platform technology with excellent potential for further expansion and improvement. These novel sigma-2 conjugates are capable of delivering death-inducing payloads selectively into their target cells. In addition, the cancer-selective cell killing properties are further enhanced in combination with standard therapies.
MATERIALS AND METHODS
σ-2 receptor ligands
Sigma-2-specific ligands SV119, and fluorescently labeled sigma-2 ligand, SW120, were synthesized and prepared as previously described (11;12). The σ-1 receptor ligand, (+)-pentazocine (Sigma Chemical, St. Louis, MO), was used as a control. The peptide-based sigma-2 conjugates were synthesized employing solid phase peptide synthesis (SPPS) using the standard Fmoc/HOBT chemistry using the intermediate SV119-Asp (supplementary Fig. 1). The chemical synthesis of the S2-Rapamycin can be found in the data supplement (supplementary Fig. 2). The conjugates were purified by reverse phase HPLC to >95% purity and, where applicable, the amino acid composition was verified using MALDI-TOF mass spectrometry.
Cell lines
The murine pancreatic adenocarcinoma cell line Panc02, was obtained from Bryan Clary (Duke University) and maintained in RPMI 1640 (24). Human pancreatic adenocarcinoma cell lines (Panc-1, AsPC1, and CFPAC) were obtained from ATCC (Bethesda, MD) and maintained in DMEM (24).
Sigma-2 ligand binding in vitro
Tumor cells were incubated with 10 nM fluorescently labeled σ-2 receptor ligand SW120 for 30 minutes. To demonstrate the specificity of SW120 for σ-2 receptor binding, 10 μM (+)-pentazocine (sigma-1 receptor ligand) was added to the cells 30 minutes prior to SW120 treatment. To study whether Sigma-2-Bim binds to σ-2 receptors in tumor cells, a series of blocking experiments were done with sigma-1 and sigma-2-selective ligands. The tumor cells were preincubated with S2-Bim, SV119, or (+)-pentazocine at the indicated concentrations. The cells were then treated with SW120 for 30 min and the fluorescent intensity of the labeled cells was analyzed by flow cytometry. Quantitative analysis of SW120 displacement was carried out using a nonlinear regression model (25).
Evaluation of cytotoxicity in vitro
Tumor cells were seeded at a density of approximately 2 × 105 cells per 12-well. The various drugs were added to the cultures with a final DMSO concentration of less than 1%. The extent of apoptosis was subsequently assessed by TUNEL and caspase-3 staining employing flow cytometry (FACScan, BD Biosciences) and data analysis with CellQuest software (BD Biosciences) (11).
In vivo assessment of apoptosis
Female C57BL/6 mice (8–12 weeks old) were purchased from the NCI. All mice were injected in the right flank with 200 μL single cell suspension containing 1 × 106 Panc02 cells. Two weeks after tumor implantation, at which point the mean tumor diameter was approximately 5 mm, mice were treated with a single intraperitoneal injection of S2-Bim. Twenty-four hours later, tumors were harvested and minced to 1 mm and digested in a RPMI buffer containing 1 mg/mL collagenase (Sigma-Aldrich, St. Louis, MO) and 0.1 mg/mL DNase (Sigma-Aldrich, St. Louis, MO) for 45 min to obtain a single-cell suspension. After filtering, erythrocyte contaminants were lysed in Ammonium Chloride (ACK) buffer, pelleted, and resuspended in PBS (pH 7.4). Single cell suspensions were fixed with 1% paraformaldehyde. Apoptosis was assessed by staining for activated caspase-3 as described above utilizing flow cytometry.
In vivo assessment of tumor growth and survival
Female C57BL/6 (NCI) and nude mice (nu/nu, Harlan, Indianapolis) (8–12 weeks old) were injected as described above with 1 × 106 Panc02 (C57BL/6) or 2 χ 106 CFPAC cells (nude mice), respectively. Treatment of tumors started ~2 weeks post-implantation, at which point the mean tumor diameter was approximately 6–7 mm. To evaluate the effect of treatment both systemically and on tumors in vivo, several treated mice were sacrificed and blood cytologic (complete blood count) and biochemical analysis (liver enzymes, bilirubin, amylase, lipase, BUN, creatinine, glucose) were performed by the DCM at our institution. For survival studies, tumor bearing mice (n=12 per group) were treated with S2-Bim and S2-BimX. All mice were euthanized when their tumor ulcerated or reached a mean diameter of 15 mm. All studies were performed in accordance with an animal protocol approved by the Washington University Institutional Animal Care Facility.
Statistical analysis
Error bars represent means plus/minus SEM of an experiment with at least three biological replicates. For statistical analysis between two groups, student's t-test was applied. For statistical analysis of differences between groups, ANOVA was performed. For in vivo experiments, Kaplan-Meier survival curves were plotted and differences were compared with a log-rank test. A p-value of less than 0.05 was considered significant for all analyses.
RESULTS
Chemical linkage of sigma-2-selective compounds with pro-apoptotic molecules does not alter target cell specificity
We have previously reported that apoptosis-inducing peptides such as the BH3 domain of the Bcl-2 antagonist Bim (18) and a peptide derived from the Akt-inhibitor CTMP [CTMP-4, Ref. (21)] can be delivered to cancer cells via the HIV-1-derived transduction domain TAT. Since this delivery mode is non-selective, we assessed the possibility to deliver pro-apoptotic effector molecules via SV119 (Fig. 1A) preferentially to pancreatic cancer cells that are characterized by an abundance of receptors for this ligand.
Figure 1. Chemical structures of the relevant drugs used in this study.

The parental compound SV119 (A) served as the basic structure for the generation of other conjugates. The basic structural organization of peptide-linked SV119 is shown in (B). The peptide sequences were as follows: S2-Bim (EIWIAQELRRIGDEFNAYYAR-OH), the inactive form S2-BimX (EIWIAQEARRIGAEFNAYYAR-OH), S2-CTMP-4 (LDPKLMKEEQMSQAQLFTRSFDDGL-OH), and the inactive derivative S2-CTMP-4X (LDPKLMKEEQMSQAQLATRSADDGL-OH). The conversion to functionally inactive peptide variants was done by Alanine substitution and is underlined. (C) The SV119-based conjugate S2-rapamycin. The sigma-1-specific ligand (+)-pentazocine (D) served as a negative control for binding studies in which the fluorescently-labeled sigma-2 ligand SW120 (E) was employed.
The following compounds were generated as described using standard chemistry (26;27): S2-Bim and S2-CTMP-4 (Fig. 1B, only the precursor structure without the corresponding peptide is shown) as well as S2-Rapamycin (Fig. 1C) along with their non-functional, inactive variants (see also supplementary Fig. 1 and 2). The binding capacity of the conjugates to the σ-2 receptor was compared to non-conjugated SV119. As a negative control, the sigma-1-specific ligand (+)-pentazocine (Fig. 1D) was employed (12).
Competitive binding assays were performed using the fluorescently-labeled sigma-2 ligand SW120 [(11;25) and (28)] (Fig. 1E). Compared to the non-conjugated SV119 compound, S2-Bim and its inactive variant S2-BimX displaced SW120 in a dose-dependent fashion (Fig. 2). As expected, the sigma-1-specific ligand (+)-pentazocine was unable to compete off SW120 and served as a negative control. Similar results were obtained when the same assays were repeated with SV119 conjugated to CTMP-4 and rapamycin (data not shown). These results suggest that covalent attachment of additional small molecule cargoes to the parental molecule SV119 does not alter the specificity of the compounds to the σ-2 receptor.
Figure 2. Chemical linkage of a Bim peptide to SV119 does not interfere with binding of the conjugate to the σ-2 receptor.
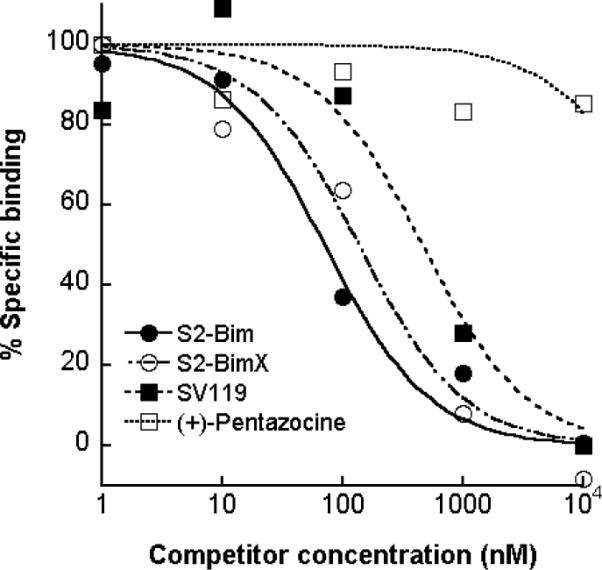
Human CFPAC cells were either left untreated or preincubated with increasing concentrations of (+)-pentazocine (a sigma-1-specific ligand used as a negative control), SV119, or the conjugates S2-Bim and S-2-BimX, followed by treatment with 50 nM SW120 (30 min). The cells were then washed, detached and the mean fluorescent intensity of the cells was determined by flow cytometry and normalized to the cells treated with SW120 alone. The corresponding IC50 values are: (+)-pentazocine, 49170 nM; SV119, 460 nM; S2-BimX, 136 nM and S2-Bim, 71 nM.
Sigma-2 conjugates augment target cell killing compared to the delivery vehicle alone
The drug conjugates were next tested for their ability to induce cancer cell death in vitro. As a surrogate of target cell apoptosis, activation of caspase-3 was used as previously described (12). The mouse pancreatic cancer cell line Panc02 was challenged in vitro with equimolar doses (10 μM) of S2-conjugates and caspase-3 induction was assessed 24 hours later. Relative to the non-modified parental sigma-2 delivery agent, all compounds linked to an “active” cargo exerted an increase in target cell apoptosis (Fig. 3). Apoptosis induction increased between 1.3-fold (S2-CTMP-4, Fig. 3A) and 3.5-fold (S2-Rap, Fig. 3B) with S2-Bim resulting in an intermediate increase of 1.6-fold compared to SV119 alone (Fig. 3C). The most dramatic effect in overall potency was achieved with S2-Bim with an 11% increase in caspase-3 activation compared to SV119 alone (Fig. 3C). Of note, this activity was not matched by TAT-Bim, a recently described very potent, non-selective inducer of target cell death (18), with S2-Bim being 13% more powerful.
Figure 3. Sigma-2 conjugates are more potent inducers of target cell apoptosis than the delivery vehicle alone.

Mouse Panc02 cells were treated with the indicated reagents and analyzed for apoptosis induction by intracellular staining for active caspase-3 using flow cytometry. (A) Sigma-2-CTMP-4, (B) Sigma-2-Rapamycin, and (C) Sigma-2-Bim were compared to the non-conjugated parental S2-ligand SV119. DMSO served as a negative control for all assays. Also, TAT-Bim (A) and TAT-CTMP-4 (B) were included as positive controls and have been described in earlier studies (18;21). *, p < 0.03; **, p < 0.0004.
Sigma-2 conjugates mediate cell death via interference with intracellular signaling pathways
To gain insight into the respective killing mechanisms, we studied the signaling pathways that are targeted by the various effector molecules delivered to the cells via the SV119 moiety. Caspase-3 activation is closely linked to the antagonizing interaction of Bim with Bcl-2 and has been already described above (Fig. 3C). CTMP interferes with activation of Akt while rapamycin blocks activation of the p70 S6 kinase as part of the mTOR complex.
CFPAC cells were treated with S2-CTMP-4 and the activation status of Akt was monitored 24 hours post-treatment. Assessed on the basis of its phosphorylation status, only S2-CTMP-4 and TAT-CTMP-4 (included as a control), were capable of interfering with Akt activation (Fig. 4). Likewise, when S2-Rapamycin was employed, a significant reduction in phosphorylation of its downstream target was observed (data not shown). In both cases, SV119 alone was incapable of altering the activation status of these two central kinases.
Figure 4. The S2-CTMP-4 conjugate directly interferes with the activation status of its intracellular target.

CFPAC cells were treated with the S2-CTMP-4 and the activation (phosphorylation) status of its downstream target (Akt) was analyzed using Bioplex™ according to the manufacturer's instructions (Bio-Rad, Hercules, CA). DMSO served as a negative control. *, p = 0.01.
In an attempt to demonstrate broader applicability, we assessed the killing capacity of S2-Bim using additional pancreatic cancer cell lines. All the cell lines under investigation responded to this novel drug candidate in a dose-dependent fashion being consistently more potent than SV119 alone and the non-selective death inducer TAT-Bim (supplementary Fig. 3). Surprisingly, the covalently linked inactive form of Bim (S2-BimX) had virtually no activity in all cell lines tested. The reason for this loss of activity in vitro is currently unknown and might be partially rescued under in vivo settings (see below).
Sigma-2-Bim induces apoptosis in vivo and reduces tumor growth
According to the activity profiles of our various sigma-2 conjugates, S2-Bim appeared to have the greatest potential as a cancer therapeutic. Consequently, this drug was tested next in vivo, i.e. using a syngeneic and a xenogenic subcutaneous mouse model of pancreatic cancer. In an initial study, mouse pancreatic tumor cells (Panc02) were engrafted into the flanks of wild-type C57BL/6 mice. After the tumors were established, the effect of S2-Bim was assessed following a single i.p. injection by immunohistochemistry (TUNEL staining) and by staining for activated caspase-3. Twenty-four hours post-treatment, large areas of TUNEL-positive cells were identified (Fig. 5A). Furthermore, the activity of S2-Bim was dose-dependent as doubling the concentration to 400 μg per animal increased the number of caspase-3-positive tumor cells by nearly 100% (Fig. 5B).
Figure 5. The sigma-2-Bim conjugate induces target cell death and reduces tumor growth in preclinical mouse models of pancreatic adenocarcinoma.

C57BL/6 mice bearing established tumor grafts (Panc02) were treated with a single dose of S2-Bim via i.p. injection (400 μg). Tumors were removed 24 h later and prepared for TUNEL staining (A). In a similar setting, Panc02 tumors were removed 24 h following i.p. injection of S2-Bim. They were then processed for caspase-3 activation using flow cytometry (B). The in vivo activity of S2-Bim was studied in a syngeneic model (Panc02) where tumor bearing C57/BL6 mice were treated every other day with the indicated therapeutics (C, arrows), and a xenogeneic model (CFPAC), with a daily treatment schedule (D, arrows). Tumor growth was determined by caliper measurement and is represented as mean diameter (n = 12 per group). Comparisons between two experimental groups were done using a one-tailed Student's t test. (C) *, ns; ** p = 0.03 and (D)#, ns (vs. S2-BimX); * p < 0.05; ** p < 0.0001 (all others vs. DMSO control).
In vivo efficacy was studied next in which C57BL/6 mice with established Panc02 tumors were treated i.p. with S2-Bim for two weeks every other day. The controls, vehicle alone and the conjugate incorporating the inactive variant of the Bim peptide S2-BimX, had no effect on the tumor growth (Fig. 5C), in agreement with the lack of in vitro cell killing activity (supplementary Fig. 3). In contrast, S2-Bim prevented tumor growth completely within the first week of treatment (Fig. 5C). However, at his dosing regimen the initial response to the drug ceased and the tumor growth resumed at a similar rate as the controls. Of note, while all the control mice (vehicle and S2-BimX) died at 43 days post tumor inoculation, almost half (>40%) were still alive at the end of the experiment (supplementary Fig. 4).
Using a xenograft tumor model, CFPAC-bearing nude mice were treated with S2-Bim for one week on a daily schedule. Tumor growth of mice receiving vehicle only progressed rapidly whereas mice treated with S2-BimX displayed a somewhat reduced tumor growth rate, similar to SV119 alone (Fig. 5D). In stark contrast, the tumor growth curve of mice receiving S2-Bim displayed a negative slope (tumor regression) during the treatment period (Fig. 5D). However, after the treatment was discontinued, the tumor growth progressed, comparable to that of the control group.
Sigma-2-Bim exhibits limited and only transient off-site toxicities
In an attempt to assess the potential systemic toxicity of the S2-Bim conjugate, tumor-bearing C57BL/6 mice were injected i.p. with a single dose of the drug and several key organs were analyzed for apoptosis induction via staining for active caspase-3. The following day, a definitive increase in tumor cell apoptosis was detectable for S2-Bim with only background signals for the vehicle and inactive sigma-2 compound S2-BimX (Fig. 6A). Also, significant caspase-3 activity was noted in the pancreas, which occurred only transiently (see below) and further suggests that our delivery vehicle has specificity for this particular organ. All the other organs analyzed were essentially unaffected by this treatment regimen (Fig. 6A).
Figure 6. Systemic sigma-2-Bim exhibits only minor and transient organ toxicities.

(A) C57BL/6 mice bearing established tumor grafts (Panc02) were treated with a single dose of S2-Bim (400 μg) via i.p. injection. DMSO and the inactive conjugate S2-BimX were used as controls. The next day, organ damage was assessed by staining for activated caspase-3 using flow cytometry (n = 3). Data are expressed as means +/− 1.0 SE (n = 3). Amylase and lipase levels were chosen to monitor toxicity of S2-Bim toward the pancreas. Peripheral blood was drawn from normal mice one week (B) and two weeks (C) following a single treatment with 400 μg S2-Bim. DMSO and the inactive conjugate S2-BimX were used as controls. Serum chemistry evaluations were performed by the animal care facility at Washington University. *, p < 0.05; ns, not significant.
Since we noticed a slight toxicity of S2-Bim to the pancreas, we assessed next serum amylase and lipase levels. These would rise in case of organ damage to the pancreas following drug treatment. And indeed, at one week following a single injection of S2-Bim into normal mice, we detected a rise in the levels of these biomarkers, ranging from 7- to 10-fold relative to their baseline levels (Fig. 6B). However, the mice demonstrated no clinically measurable signs of distress and recovered from this insult quickly as by one week after drug cessation both serum concentrations reached baseline levels (Fig. 6C). The same is true for our long-term treatment studies where the mice remained healthy by weight and appetite throughout the course of our experiments (Fig. 5 C and D) and we did not encounter unanticipated deaths. In support of these findings, necropsy revealed no gross or microscopic organ damages.
Sigma-2-Bim augments standard of care therapy
Monotherapies are rarely successful when it comes to the treatment of malignant diseases. For this reason, we asked if and to what extent a dual therapy would potentially increase the effectiveness of S2-Bim. Two of the most widely applied treatment regiments are radiation and conventional chemotherapy. When CFPAC cells were treated in vitro with a combination of gemcitabine and S2-Bim, an increase of ~10 to 15% cell death was induced compared to the same treatment but replacing the active with the inactive form of the sigma-2 compound (Fig. 7A). Cotreatment of S2-Bim and radiation in vitro resulted in an almost 100% increase in TUNEL-positive target cells from 30% to nearly 60% (Fig. 7B).
Figure 7. Sigma-2-Bim supports combination therapy in vitro.

The effect of S2-Bim in combination with radiation and standard chemotherapy were assessed using the human pancreatic cancer cell line Panc-1. (A) As a chemotherapeutic reagent, the cells were either treated with gemcitabine alone (30 nM) or in combination with different concentrations of S2-Bim and S2-BimX. (B) Combination with radiation therapy (2000 rad) was studied as above alone or in combination with different concentrations of S2-Bim and S2-BimX. DMSO-treated cells served as a control. Apoptosis induction was monitored by TUNEL staining using flow cytometry. Data are expressed as means +/− SEM (n = 3). *, p < 0.02; **, p < 0.0002; ***, p < 0.00002.
DISCUSSION
A fundamental limitation of most conventional cancer therapeutics is their non-directed delivery to the target cells. Non-selective delivery often represents the primary reason for toxic side effects and limited treatment efficiencies. If the delivery component would be structurally separated from the therapy component of a drug conjugate, one could build on these modular reagents to tailor therapies that would target particular defects associated with a specific type of cancer subtype. Such a cancer-selective delivery agent could open up the door for retesting many of the drugs that already proved efficacious in vitro but proved to be too toxic for in vivo applications.
In our current paper, we investigated this concept by chemically linking a targeting vehicle with high specificity to pancreatic cancer cells with a variety of small molecules that would, upon delivery to the target cell, exert their respective intracellular effector functions (apoptosis induction). It should be noted here that our chosen delivery agent has a limited, intrinsic cytotoxicity toward pancreatic cancer cells. This was, however, considered during the design phase of our dual domain therapeutics, as only the combined activities of the cancer-selective delivery vehicle SV119 and a secondary, complementary death stimulus were expected to result in the most effective drug conjugate. Indeed, we demonstrated here that the overall potency of our various drug conjugates enhanced the intrinsic killing capability of the delivery agent SV119 via specific engagement of the respective therapy component intended to induce a particular apoptotic signaling pathway.
A key requirement for a universally applicable drug delivery platform requires that the target specificity is maintained following covalent linkage of the parental structure to a particular effector moiety. The additional effector molecules did not abrogate binding of the conjugates to pancreatic cancer cells. However, when SV119 was combined with the inactive peptide mutant(s) of, e.g. Bim (S2-BimX), a complete loss of killing activity was noticed in vitro. This was unexpected, since we assumed that this variant should have at least SV119 baseline activity. The same reagent elicited SV119 baseline activity in a pancreatic xenograft model using CFPAC cells and highlights the notion that the bioactivity of these drugs might be variable and depend, at least in part, on the specific cell type in question and/or on the type of application employed (in vitro vs in vivo). A potential explanation for this finding might relate to the non-conservative substitution of the charged amino acid Asparagine with a hydrophobic Alanine located within the Bim peptide. The exact nature of the differential activity profile is currently being investigated, since it might provide clues for the future design of improved variants of this class of cancer drugs.
Therapeutically, the most promising results were obtained in a xenograft model of pancreatic cancer using S2-Bim. As long as the sigma-2 conjugate was administered, established human tumors responded with shrinkage. This was certainly encouraging as it has rarely been seen that this type of cancer is treatable. A somewhat different picture was obtained in a syngeneic tumor model. Tumor growth was halted for nearly a week but resumed while treatment was still ongoing. At the present time, we can only speculate as to why the same drugs displayed differential activity profiles in different animal models. For example, mouse Panc02 cells grow much more aggressively than human CFPAC cells (29). This feature could lead to a faster development of resistance to our cancer drugs and would represent a characteristic that is cell line specific. On the other hand, it could well be possible that an immune component might be involved that counteracts tumor eradication in a normal host (C57BL/6-Panc02). Since these two alternative mechanisms are critical for the evaluation of our investigative drugs, we are currently in the process of dissecting which of the mechanistic paths is the dominating factor.
While the cytotoxic benefit of S2-Bim was greatest on the target cells, we did notice mild elevations of biochemical markers of tissue injury, i.e. amylase and lipase. In our model, rising levels of these biomarkers were only transient and restricted to the pancreas. As with many cytotoxic drugs, we expected to find evidence of mild organ damage, especially of the liver but did not detect elevations of the respective biochemical markers. Even though our sigma-2 ligands are highly tumor-selective, we cannot rule out binding to non-transformed, normal cells or tissues. In fact, we did indeed identify binding at lower levels to many other cell/organ types including the liver and the intestine. Along this line, we are currently in the process of evaluating relevant pharmacological parameters of selected drug conjugates such as half-life and maximum tolerated dose (MTD). Results from these ongoing studies will help us in the transition process from the preclinical to the clinical arena.
Peptides are not ideal partners for the formulation of effective anti-cancer drugs. One of the main reasons is their susceptibility to proteolytic degradation. Along this line, Abbott has recently developed the Bim peptidomimetics ABT-737 and ABT-263 (30–32), with the latter even being orally bioavailable. Based on the data demonstrated here it seems feasible to combine sigma-2 ligands with other small molecular therapeutics and assess their potency in future studies.
Monotherapy is rarely sufficient to combat aggressive human malignancies. With this regard, we provided evidence in an in vitro cell culture system that the S2-Bim conjugate substantially augmented standard of care treatment regiments currently used in clinical settings. More specifically, combination with chemotherapy and, more profoundly, radiation therapy increased target cell death by nearly 100% with an overall killing of more than 60%. While radiation therapy has its intrinsic caveats, combination of S2-Bim with gemcitabine resulted in a much improved killing response. Therefore, we are currently performing preclinical studies using the latter combination therapy to assess its efficacy in an orthotopic mouse model of pancreatic cancer.
In summary, we present a platform concept for the cancer-selective, SV119-mediated (σ-2 receptor dependent) co-delivery of apoptosis-inducing cargo (peptides or small molecules) to the target cells. Once inside the cell, these bifunctional conjugates mediate enhanced killing via the combined activities of both moieties. As a result, the baseline cytotoxic activity of the delivery agent is augmented following the signaling pathway of its cargo. This new concept represents an exciting opportunity for the development of future small molecule drugs with dual functionality combined in a single reagent. In fact, we have recently developed another sigma-2-specific ligand, the SV119 homolog SW43, which turned out to be twice as cytotoxic by itself (24) and could represent the building block for the next generation of bifunctional (e.g. SW43-Bim) therapeutics to treat patients with pancreas cancer. Even though our study was focused on pancreatic cancer, we are developing evidence that this concept could also be applied to other types of malignancies including ovarian and breast cancers.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. David Linehan for expert advice along the preparation of the manuscript and Stacey Plambeck-Suess for excellent technical assistance.
This work was supported by the American Cancer Society (MRSG08019-01CDD), a Veterans Administration Merit Award (#1136919) (WGH), the Doug Phillips Research Fund #4125-35604) (DS), a Surgical Oncology Training Grant (#5T32CA009621-22) (POS and JRH) and a Siteman Cancer Center Pilot Feasibility Grant (#5P30CA09184208) (WGH). It was further supported by the NIH (CA102869) and the US Army Medical Research and Material Command under DAMD (17-01-1-0446) (RHM).
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Sierzega M, Popiela T, Kulig J, Nowak K. The ratio of metastatic/resected lymph nodes is an independent prognostic factor in patients with node-positive pancreatic head cancer. Pancreas. 2006;33:240–5. doi: 10.1097/01.mpa.0000235306.96486.2a. [DOI] [PubMed] [Google Scholar]
- 3.Linehan DC, Tan MC, Strasberg SM, Drebin JA, Hawkins WG, Picus J, et al. Adjuvant interferon-based chemoradiation followed by gemcitabine for resected pancreatic adenocarcinoma: a single-institution phase II study. Ann Surg. 2008;248:145–51. doi: 10.1097/SLA.0b013e318181e4e9. [DOI] [PubMed] [Google Scholar]
- 4.Karasek P, Skacel T, Kocakova I, Bednarik O, Petruzelka L, Melichar B, et al. Gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer: a prospective observational study. Expert Opin Pharmacother. 2003;4:581–6. doi: 10.1517/14656566.4.4.581. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler KT, Wang LM, Wallen CA, Childers SR, Cline JM, Keng PC, et al. Sigma-2 receptors as a biomarker of proliferation in solid tumours. Br J Cancer. 2000;82:1223–32. doi: 10.1054/bjoc.1999.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi SR, Yang B, Plossl K, Chumpradit S, Wey SP, Acton PD, et al. Development of a Tc-99m labeled sigma-2 receptor-specific ligand as a potential breast tumor imaging agent. Nucl Med Biol. 2001;28:657–66. doi: 10.1016/s0969-8051(01)00234-7. [DOI] [PubMed] [Google Scholar]
- 7.Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J Am Coll Surg. 2004;198:591–9. doi: 10.1016/j.jamcollsurg.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Flick MB, O'Malley D, Rutherford T, Rodov S, Kamsteeg M, Hao XY, et al. Apoptosis-based evaluation of chemosensitivity in ovarian cancer patients. J Soc Gynecol Investig. 2004;11:252–9. doi: 10.1016/j.jsgi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine-and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–32. [PubMed] [Google Scholar]
- 10.Hou C, Tu Z, Mach R, Kung HF, Kung MP. Characterization of a novel iodinated sigma-2 receptor ligand as a cell proliferation marker. Nucl Med Biol. 2006;33:203–9. doi: 10.1016/j.nucmedbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Zeng C, Vangveravong S, Xu J, Chang KC, Hotchkiss RS, Wheeler KT, et al. Subcellular localization of sigma-2 receptors in breast cancer cells using two-photon and confocal microscopy. Cancer Res. 2007;67:6708–16. doi: 10.1158/0008-5472.CAN-06-3803. [DOI] [PubMed] [Google Scholar]
- 12.Kashiwagi H, McDunn JE, Simon PO, Jr., Goedegebuure PS, Xu J, Jones L, et al. Selective sigma-2 ligands preferentially bind to pancreatic adenocarcinomas: applications in diagnostic imaging and therapy. Mol Cancer. 2007;6:48. doi: 10.1186/1476-4598-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford KW, Bowen WD. Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res. 2002;62:313–22. [PubMed] [Google Scholar]
- 14.Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, et al. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, et al. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–61. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Moudgil T, Ross HJ, Hu HM. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein Bim. Cell Death Differ. 2005;12:292–303. doi: 10.1038/sj.cdd.4401554. [DOI] [PubMed] [Google Scholar]
- 17.Garber K. New apoptosis drugs face critical test. Nat Biotechnol. 2005;23:409–11. doi: 10.1038/nbt0405-409. [DOI] [PubMed] [Google Scholar]
- 18.Kashiwagi H, McDunn JE, Goedegebuure PS, Gaffney MC, Chang K, Trinkaus K, et al. TAT-Bim Induces Extensive Apoptosis in Cancer Cells. Ann Surg Oncol. 2007;14:1763–71. doi: 10.1245/s10434-006-9298-z. [DOI] [PubMed] [Google Scholar]
- 19.Knobbe CB, Reifenberger J, Blaschke B, Reifenberger G. Hypermethylation and transcriptional downregulation of the carboxyl-terminal modulator protein gene in glioblastomas. J Natl Cancer Inst. 2004;96:483–6. doi: 10.1093/jnci/djh064. [DOI] [PubMed] [Google Scholar]
- 20.Chae KS, Martin-Caraballo M, Anderson M, Dryer SE. Akt activation is necessary for growth factor-induced trafficking of functional K(Ca) channels in developing parasympathetic neurons. J Neurophysiol. 2005;93:1174–82. doi: 10.1152/jn.00796.2004. [DOI] [PubMed] [Google Scholar]
- 21.Simon PO, Jr., McDunn JE, Kashiwagi H, Chang K, Goedegebuure PS, Hotchkiss RS, et al. Targeting AKT with the proapoptotic peptide, TAT-CTMP: a novel strategy for the treatment of human pancreatic adenocarcinoma. Int J Cancer. 2009;125:942–51. doi: 10.1002/ijc.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibbons JJ, Abraham RT, Yu K. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol. 2009;36(Suppl 3):S3–S17. doi: 10.1053/j.seminoncol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Rowinsky EK. Targeting the molecular target of rapamycin (mTOR) Curr Opin Oncol. 2004;16:564–75. doi: 10.1097/01.cco.0000143964.74936.d1. [DOI] [PubMed] [Google Scholar]
- 24.Hornick JR, Xu J, Vangveravong S, Tu Z, Mitchem JB, Spitzer D, et al. The novel sigma-2 receptor ligand SW43 stabilizes pancreas cancer progression in combination with gemcitabine. Mol Cancer. 2010;9:298. doi: 10.1186/1476-4598-9-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Tu Z, Jones LA, Vangveravong S, Wheeler KT, Mach RH. [3H]N-[4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl]-2-methoxy-5 -methylbenzamide: a novel sigma-2 receptor probe. Eur J Pharmacol. 2005;525:8–17. doi: 10.1016/j.ejphar.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 26.Vangveravong S, Xu J, Zeng C, Mach RH. Synthesis of N-substituted 9-azabicyclo[3.3.1]nonan-3alpha-yl carbamate analogs as sigma2 receptor ligands. Bioorg Med Chem. 2006;14:6988–97. doi: 10.1016/j.bmc.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, Jaattela M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65:8975–83. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- 28.Zeng C, Vangveravong S, Jones LA, Hyrc K, Chang KC, Xu J, et al. Characterization and Evaluation of Two Novel Fluorescent Sigma-2 Receptor Ligands as Proliferation Probes. Mol Imaging. 2011 Apr 27; [PMC free article] [PubMed] [Google Scholar]
- 29.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP, Jr., et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–26. [PubMed] [Google Scholar]
- 30.High LM, Szymanska B, Wilczynska-Kalak U, Barber N, O'Brien R, Khaw SL, et al. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol Pharmacol. 2010;77:483–94. doi: 10.1124/mol.109.060780. [DOI] [PubMed] [Google Scholar]
- 31.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 32.Chonghaile TN, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008;27(Suppl 1):S149–S157. doi: 10.1038/onc.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.