

Abstract
Major histocompatibility complex class I (MHCI) molecules modulate activity-dependent refinement and plasticity. Here, we show that MHCI also negatively regulates the density and function of cortical synapses during their initial establishment both in vitro and in vivo. MHCI molecules are expressed in cortical neurons before and during synaptogenesis. In vitro, decreasing surface MHCI (sMHCI) on neurons increases glutamatergic and GABAergic synapse density, while overexpression decreases it. In vivo, synapse density is higher throughout development in β2m−/ − mice. MHCI also negatively regulates mEPSC, but not mIPSC, amplitude and controls the balance of excitation and inhibition onto cortical neurons. sMHCI levels are modulated by activity and are necessary for activity to negatively regulate glutamatergic synapse density. Finally, acute changes in sMHCI and activity alter synapse density exclusively in early postnatal development. These results identify a novel function for immune proteins in negatively regulating the initial establishment and function of cortical connections.
The initial formation of connections in the mammalian central nervous system (CNS) occurs when axons reach their proper target and is guided largely by molecules that promote synaptogenesis1–2. In the cerebral cortex, this initial phase of synapse formation leads to an exuberant number and pattern of connections that are later sculpted by molecules that either strengthen or eliminate synapses in an activity-dependent manner1. Recent evidence suggests that the initial establishment of cortical connections is also regulated by molecules that negatively affect connectivity. MEF2 transcription factors, semaphorin-5B, and ephexin5 limit the early establishment of glutamatergic synapses in young neuronal cultures3–4–5 and RSY-1 antagonizes presynaptic assembly in C. elegans6. Another molecular family that could negatively regulate the initial establishment of cortical connections is major histocompatibility complex I (MHCI) because these molecules are present in early postnatal cortex and regulate refinement of connections during later stages of development7–9.
MHCI molecules are trimeric proteins comprised of a transmembrane heavy chain, a soluble β2-microglobulin (β2m) light chain, and a peptide bound to the heavy chain. Complete trimers are transported to the cell surface; in the absence of β2m, MHCI fails to be trafficked to the plasma membrane10–12. In the immune system, MHCI molecules are the primary mediators of the adaptive immune response. In the CNS, MHCI functions in activity-dependent plasticity of the developing visual system7–9,13. Retinal axons fail to segregate into eye-specific layers in the LGN in transgenic mice in which β2m and TAP1, a protein required for peptide loading, are knocked-out (β2m/TAP1−/−)9. Similarly, in mice lacking two MHCI genes, H2-Kb and H2-Db, retinogeniculate refinement is impaired and ocular dominance plasticity is enhanced 13. Finally, hippocampal long-term potentiation is enhanced and long-term depression is eliminated in β2m/TAP1−/− mice9. Although synapse density is not changed in the hippocampus of β2m/TAP1−/− mice, mEPSC frequency is increased in β2m/TAP1−/− hippocampal cultures14, consistent with the reported changes in long-term synaptic plasticity. Interestingly, mEPSC frequency in β2m/TAP1−/− cortical slices is increased much more dramatically14, suggesting that MHCI molecules may play a greater role in the establishment of cortical versus hippocampal connectivity.
The function of MHCI molecules during the initial establishment of connections in the CNS remains unknown. Here, we show that MHCI proteins are well-positioned to influence the initial establishment of cortical connectivity; they are present on the surface of neurons (surface MHCI; sMHCI) at the time of, and in the structures that participate in, synapse formation. Through manipulating neuronal sMHCI levels, we found that MHCI proteins negatively regulate the establishment of cortical connections. In cultured neurons, β2m knockdown (KD) increased glutamatergic synapse density, while overexpression (OE) of a GFP-tagged MHCI subtype, H2-Kb, decreased it. Similarly, glutamatergic synapse density was higher in neurons from β2m−/− mice both in vitro and in vivo throughout development. MHCI negatively regulated both mEPSC frequency and amplitude. MHCI KD also increased GABAergic synapse density and mIPSC frequency, but to a lesser extent, with no affect on mIPSC amplitude. This differential effect of MHCI on glutamatergic and GABAergic synapses dramatically altered the balance of cortical excitation and inhibition. In addition, sMHCI levels were modulated by neuronal activity and were necessary for activity to negatively regulate glutamatergic synapse density. Finally, these effects of MHCI and neural activity occurred exclusively during the initial establishment of connections.
RESULTS
MHCI is present on the neuronal surface during development
MHCI protein has been found in synaptosomes of the adult CNS9, is colocalized with PSD-95 in cultured hippocampal neurons13–14, and is present in both axon terminals and postsynaptic densities of synapses in the visual cortex at multiple ages10. If MHCI molecules regulate the initial establishment of cortical connectivity, then they must be present in the plasma membrane of neurons before and during synaptogenesis. To test this hypothesis, the localization of MHCI proteins relative to synaptic proteins was examined in low-density dissociated rat cortical neurons, allowing unambiguous detection of neuronal MHCI independent of glial MHCI expression. A pan-specific antibody recognizing the conserved, extracellular α-3 domain of the MHCI heavy chain was used to label MHCI protein (OX-18, Serotec; Suppl. Fig. 1). The specificity of OX-18 for MHCI was confirmed by three experiments. First, two antibodies raised against different epitopes of MHCI molecules—OX-18 and a rabbit monoclonal antibody (ab52922; AbCAM)—produced similar patterns of staining of P3 rat cortical membranes in Western blot (Suppl. Fig. 1a). Second, two antibodies raised against different epitopes of MHCI molecules—OX-18 and F16-4-4—produced similar staining patterns in immunocytochemistry (ICC) of 8 day in vitro (d.i.v.) cortical neurons (Suppl. Fig. 1b–c). Finally, there was little-to-no sMHCI staining on 8 d.i.v. neurons made from β2m−/− mice (Suppl. Fig. 1d).
MHCI proteins were found in dendrites of all examined cortical neurons in small clusters before (3 d.i.v.), during (8 d.i.v.), and after (14 d.i.v.) the peak of synaptogenesis (Fig. 1a and b). The density of dendritic MHCI clusters detected after membrane permeabilization (Fig. 1a) did not vary with age in culture (Fig. 1c). For all experiments, “n” equals 30 dendrites from 10 neurons for each condition. Most values reported here were normalized to age and culture-matched controls unless otherwise specified, to control for inter-experiment variability inherent in young cortical cultures.
Fig. 1. MHCI is present at the surface of cortical neurons before, during, and after synapse formation.

(a–b) Images of dendrites from cortical neurons immunostained for MHCI proteins using the OX-18 antibody at 3, 8, and 14 d.i.v. (top to bottom) are shown proximal to distal from soma (left to right in each image). (a) MHCI is present in clusters throughout all dendrites at all ages examined. (b) Surface MHCI (sMHCI) is also present on dendrites of all cells at all ages examined (n=540 dendrites, 180 cells). (c) The density of MHCI detected after permeabilization does not vary over time (normalized to 8 d.i.v.: 3 d.i.v., 1.06 ± 0.05, p = 0.48; 8 d.i.v., 1.00 ± 0.05; 14 d.i.v., 1.07 ± 0.05, p = 0.45). (d) sMHCI clusters are present in isolated axons at 3 and 8 d.i.v. (e) sMHCI clusters on dendrites increase in density with age. (normalized to 8 d.i.v.: 3 d.i.v., 0.70 ± 0. 04, p < 0.001; 8 d.i.v., 1.00 ± 0.07; 14 d.i.v., 1.98 ± 0.09, p < 0.001). (f) At 3 d.i.v., sMHCI clusters are also present on axonal and dendritic growth cones (n = 57 growth cones, 20 neurons). (g) At 3 and 8 d.i.v., sMHCI density is higher in proximal dendrites and lower in distal portions of the same dendrites (normalized to 8 d.i.v. proximal: 3 d.i.v., proximal, 0.58 ± 0.03, distal, 0.38 ± 0.03, p < 0.001; 8 d.i.v. proximal, 1.00 ± 0.07, distal, 0.50 ± 0.03, p < 0.001; *, significant difference from 8 d.i.v. proximal density values; Ξ, significant difference between distal and same age proximal densities). At 14 d.i.v., this uneven distribution is not observed (sMHCI normalized to 8 d.i.v. proximal: 14 d.i.v proximal, 1.22±0.08; distal, 1.12±0.06, p=0.07). (h) Glutamatergic synapses (excitatory synapses; ES) are also unevenly distributed along dendrites. At 8 d.i.v., ES density is lower in proximal dendrites and higher in distal dendritic regions (normalized to proximal: proximal, 1.00 ± 0.13; distal, 1.78 ± 0.16, p <0.001; *, significant difference from same age proximal density values). At 14 d.i.v., synapses are evenly distributed across dendrites (synapse values normalized to 14 d.i.v. proximal: proximal, 1.00 ± 0.07; distal, 0.83 ± 0.06, p = 0.07). *, p<0.05; Scale bars = 5 μm.
To label MHCI proteins on the neuronal surface, neurons were fixed and immunolabeled for both sMHCI and microtubule-associated protein-2 (MAP2) under nonpermeabilizing conditions. To control for fixation-induced permeabilization15, sMHCI was analyzed exclusively in MAP2-negative dendrites. MHCI proteins were present on the surface of all examined neurons in clusters before, during, and after the peak of synaptogenesis (Fig. 1b). MHCI clusters were also found on the surface of axons at 3 and 8 d.i.v. (Fig. 1d)16. Whereas MHCI remained at a constant density over development after permeabilization, the density of sMHCI in dendrites increased 2-fold from 3 to 14 d.i.v. (Fig. 1e). Similar results were obtained in neurons that were labeled for sMHCI prior to fixation, to control for possible fixation-induced permeabilization not detected by the MAP-2 control (Suppl. Fig. 2a). In addition, MHCI clusters were present on the surface of axonal and dendritic growth cones before contact with other neurons (Fig. 1f). At this early age, MHCI molecules were also highly colocalized with the synaptic vesicle (SV) protein synapsin in isolated axons (83.80±0.58%, n=18). Thus, MHCI proteins are present at the right time and the right place in the plasma membrane to influence the initial establishment of cortical connections.
Consistent with this hypothesis, MHCI is present at synapses during (8 d.i.v.) and after (14 d.i.v.) the period of maximal synaptogenesis (Suppl. Fig. 3). However, these data reflect mostly internal MHCI since the permeabilization required to label synaptic proteins disrupted the majority of sMHCI staining (Suppl. Fig. 1e). Thus, we were unable to visualize synaptic sMHCI or determine the percentage of total sMHCI that was synaptic. Importantly, it is the expression of sMHCI, but not internal MHCI, that changes so dramatically over development (Fig. 1c,e).
As an alternative metric, the ratio between sMHCI and glutamatergic synapses was quantified in separate regions of dendrites from neurons in sister cultures over development. Proximal dendrites were defined as the initial 25μm of dendrite from the cell soma and distal regions as the remainder (up to 200μm) of the same primary dendrite. Neither sMHCI nor glutamatergic synapses were evenly distributed along dendrites. At 3 and 8 d.i.v., sMHCI clusters were significantly more dense in proximal than in distal dendrites (Fig. 1g and Suppl. Fig. 2), quantified using either density per length or area of dendrite (Suppl. Fig. 2b–c). Conversely, glutamatergic synapses (excitatory synapses; ES) were rarely formed by 3 d.i.v., but by 8 d.i.v. their distribution was low proximally and high distally as determined by colocalized presynaptic (vGlut1) and postsynaptic (NR2A/B) proteins (Fig. 1h). By 14 d.i.v., both sMHCI and glutamatergic synapses were distributed equally along dendrites (Fig. 1g and h). This inverse correlation between the density of sMHCI and glutamatergic synapses suggests that if sMHCI regulates the initial formation of cortical connections, it should negatively regulate glutamatergic synapse density.
MHCI molecules negatively regulate synapse density
To test whether sMHCI negatively regulates the initial establishment of glutamatergic connections, sMHCI levels were decreased using both KD and knockout approaches and the resulting effects on synapse density were quantified. First, sMHCI levels were decreased specifically during the initial establishment of connections in cortical cultures (5–8 d.i.v.) using acute RNA interference (Fig. 2). To knockdown most sMHCI isoforms, an siRNA was designed to target β2m, the light chain required for trafficking MHCI to the plasma membrane10–12. Neurons were co-transfected at the start of synaptogenesis at 5 d.i.v. with either anti-β2m siRNA or a non-targeting sequence siRNA (NTS; control), plus a RISC-free Cy3-tagged siRNA for visual identification of transfected cells. To determine the time-course and magnitude of KD, β2m and sMHCI densities were quantified at 6, 7, and 8 d.i.v.. The density of β2m clusters decreased to approximately 50% of control levels by 8 d.i.v. (Fig. 2a–b). As expected, sMHCI also decreased to approximately 50% with a similar time-course (Fig. 2a–b). This reduction in sMHCI levels increased glutamatergic synapse density on transfected neurons by approximately 70% (Fig. 2c–d). These effects were replicated with a separate siRNA targeting a distinct region of β2m (8 d.i.v., 1.71±0.13, p<0.001).
Fig. 2. Acute β2m knockdown decreases sMHCI and increases glutamatergic and GABAergic synapse density.

(a) β2m siRNA knocks down endogenous β2m and sMHCI protein. Neurons were transfected with β2m siRNA at 5 d.i.v., resulting in a significant decrease in β2m cluster density (green, solid line) within 24h and reaching a maximum of 51% KD by 8 d.i.v. (6 d.i.v., 0.83 ± 0.03, p < 0.001; 7 d.i.v., 0.51 ± 0.02, p < 0.001; 8 d.i.v., 0.49 ± 0.02, p < 0.001). β2m KD decreases sMHCI density (blue, dashed line) within 48h, reaching a maximum KD of 46% also by 8 d.i.v. (6 d.i.v., 0.96 ± 0.05, p = 0.8; 7 d.i.v., 0.69 ± 0.02, p < 0.001; 8 d.i.v., 0.54 ± 0.02, p < 0.001). All values are normalized to age-matched non-targeting sequence (NTS) controls. (b) Images showing the distribution of β2m (top two images) and sMHCI (lower two images) at 8 d.i.v. Dendrites are oriented proximal to distal, left to right. (c–d) Decreasing sMHCI via β2m KD increases glutamatergic synapse (excitatory synapse; ES) density (red, solid line) within 48h of transfection, reaching a maximum 72% increase by 8 d.i.v. (6 d.i.v., 1.08 ± 0.07, p = 0.34; 7 d.i.v., 1.23 ± 0.06, p < 0.01; 8 d.i.v., 1.72 ± 0.08, p < 0.001). (d) Images of dendrites immunostained for vGlut1 (left) and NR2A/B (middle) from neurons transfected with NTS control (left panels) or β2m siRNA (right panels). Yellow in the overlay image indicates synapses. Dendrites are oriented proximal to distal, top to bottom. (e) The inversely related proximal-distal distributions of sMHCI and glutamatergic synapses are preserved after β 2m KD (sMHCI density: proximal, 0.60 ± 0.03, p < 0.001; distal, 0.48 ± 0.05, p < 0.001; ES density: proximal, 1.43 ± 0.1, p < 0.01; distal, 2.00 ± 0.14, p < 0.001). (f) β2m KD increases the percentage of vGlut1 clusters at synapses by almost 30% (26±0.03%, p<0.001), with no effect on NR2A/B enrichment at synapses (1.07 ± 0.03, p = 0.14). (g) Addition of exogenous β2m (eβ2m) does not change the effect of decreasing sMHCI on ES (normalized to β2m siRNA: 0.93 ± 0.04, p = 0.29). (h) Images of dendrites immunostained for synapsin (syn; left) and GABA receptor subunits (GABAR; middle) from neurons transfected with NTS control (left panels) or β2m siRNA (right panels). Yellow in the overlay image indicates synapses. (i) Decreasing sMHCI via β2m KD increases GABAergic synapse (inhibitory synapse; IS) density by almost 30% (1.27 ± 0.10, p < 0.05, n=20 dendrites). Scale bars = 5 μm.
Although sMHCI density was reduced by β2m KD, the differential distribution of sMHCI and glutamatergic synapses along dendrites was maintained. β2m KD decreased sMHCI and increased synapse density both proximal and distal to the soma by 8 d.i.v. (Fig. 2e; sMHCI). In proximal regions where sMHCI levels were higher, synapse density was lower, and vice versa for distal regions. This local inverse relationship suggests that sMHCI density may be differentially regulated within dendritic regions, which could influence the local density of glutamatergic synapses.
The β2m KD-induced increase in glutamatergic synapse density resulted from both an increase in density and colocalization of new pre- and postsynaptic protein clusters, and an increase in synaptic recruitment of SVs. Decreased sMHCI increased the densities of both SV and NMDAR clusters by 39±0.07 and 63±0.07%, respectively (vGlut1, p<0.001; NR2A/B, p<0.001). vGlut1 also became enriched at synapses by 26±0.03% (Fig. 2f), while there was no significant change in enrichment of NMDARs at synapses (Fig. 2f). These results suggest that sMHCI prevents recruitment of vGlut1 to young synapses and downregulates the abundance of both pre- and postsynaptic glutamatergic protein clusters.
Since extracellular surface β2m can be released from the MHCI complex and potentially signal independent of the heavy chain17–18, the increase in glutamatergic synapse density after β2m KD could result either from a decrease in free β2m signaling or decreased sMHCI. If the increase in synapse density was caused by a lack of free β2m signaling, then adding exogenous β2m (eβ2m) should rescue the effect of β2m KD on synapse density. However, the density of glutamatergic synapses on β2m KD neurons was unaltered by addition of eβ2m (Fig. 2g), suggesting that free β2m does not regulate synaptogenesis independent of sMHCI and supporting the interpretation that MHCI is instrumental in limiting the initial establishment of cortical connections.
Finally, to determine if sMHCI also affects the establishment of inhibitory synapses, neurons were transfected with β2m siRNA and immunostained to label GABAergic synapses, defined as colocalized synapsin I and GABAR α2/γ2 subunit clusters (GABARs; Fig. 2h)19. By 8 d.i.v., β2m KD significantly increased GABAergic synapse density by almost 30% (Fig. 2h–i). This increase in GABAergic synapse density resulted primarily from an increase in density of SV clusters (36±0.09%; p<0.001) and a trend toward synaptic enrichment of GABARs (1.17±0.09, p=0.1) with no change in GABAR density (1.1±0.1, p= 0.44) or synaptic enrichment of synapsin (0.94±0.07, p=0.52, n=20 dendrites). Thus, sMHCI molecules normally downregulate the abundance of SV clusters and glutamatergic and GABAergic connectivity.
To rule out the possibility of off-target effects caused by acute β2m KD, glutamatergic synapse density was quantified using neurons from mice with a targeted deletion of the β2m gene (β2m−/−). Mice with this mutation appear to develop normally and have almost no dendritic sMHCI10 (Suppl. Fig. 1d). Cortical neurons from P0 β2m−/− and wild-type mice (WT) were cultured and immunostained at 8 d.i.v. for glutamatergic synapses, defined as colocalized PSD-95 and vGlut1 puncta. Synapse density was increased in neurons from β2m−/− mice compared to WT (Fig. 3a–c), similar to the effects of β2m KD, supporting the interpretation that the KD results are not due to off-target effects. The increase in synapse density was less in β2m−/− neurons compared to KD neurons, suggesting that there may be some compensation for the loss of β2m on synapse density in the knockout. Together, results from both acute KD and chronic knockout approaches indicate that MHCI functions to limit the initial establishment of cortical connections.
Fig. 3. Synapse density is increased between visual cortical neurons from β2m−/− mice both in vitro and in vivo throughout development.
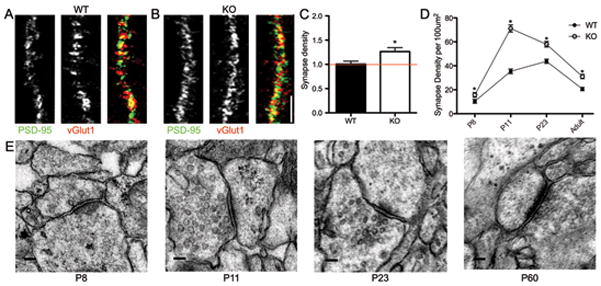
Images of neurons from 8 d.i.v. WT (a) and β2m−/− (b) mouse cultures immunostained for vGlut1 (green) and PSD-95 (red). Yellow indicates glutamatergic synapses. Scale bar = 5μm. (c) Glutamatergic synapse density is increased by 26% in cultured β2m−/− neurons (normalized to WT, 1.26±0.08, p<0.01). (d) Total synapse density in β2m−/− mice is greater at all ages examined compared to WT controls (P8: WT, 10.35 ± 0.74, β2m−/−, 15.76 ± 1.13; n = 40 sections each, p < 0.001; P11: WT, 35.42 ± 1.85, β2m−/−, 71.18 ±2. 96, n =48 and 54 sections, respectively, p < 0.001; P23: WT, 43.80 ± 1.70, β2m−/−, 58.05 ± 2.27, n = 39 sections each, p < 0.001; P60: WT, 20.65 ± 1.16, β2m−/−, 31.12 ± 1.23, n = 39 sections each, p < 0.001. (e) Transmission electron micrographs of synapses from P8, P11, P23, and P60 (adult) sections. Sections were blinded and analyzed independently by two different researchers. Only those synapses that were confirmed by both were included in the quantification. Total synapse density was calculated as the number of synapses per 100μm2 of neuropil. * = p<0.05. Scale bars = 0.5 μm.
MHCI limits synapse density in vivo in visual cortex
To determine whether MHCI regulates synapse density in the developing cortex in vivo, synapse density was quantified from electron micrographs of layer 5 of primary visual cortex from β2m−/− and WT mice at four ages—the start of synaptogenesis (P8), the peak of synaptogenesis (P11), the peak of the critical period for visual cortical plasticity (P23), and in the adult (P60). Synapses were identified based on standard criteria20 and their density was quantified from a large area of neuropil (Fig. 3d–e). Symmetric and asymmetric synapses were not differentiated due to the difficulty of discriminating them in young cortical tissue.
At all ages examined, β2m−/− mice had significantly higher synapse density per unit area of neuropil (Fig. 3d). Total synapse density in β2m−/− mice was 52% greater at P8, 2-fold greater at P11, 33% greater at P23, and 50% greater in the adult compared to WT controls. Thus, MHCI molecules also limit the establishment of cortical synapses in vivo during the initial formation of connections, consistent with our data from neuronal cultures. Moreover, MHCI does not simply delay the establishment of connections but rather limits synapse density at all ages.
MHCI bidirectionally regulates synapse density
If sMHCI negatively regulates cortical connectivity, then MHCI overexpression (OE) should decrease glutamatergic synapse density. To test this hypothesis, a single MHCI gene (H2-Kb-CFP) was transfected into neurons at 6 d.i.v. and glutamatergic synapse density was assessed at 8 d.i.v. H2-Kb is a mouse isoform of MHCI expressed in the cortex early in development8–9,13. MHCI OE increased sMHCI levels in cultured rat neurons by about 75% by 8 d.i.v. (Fig. 4a–b) and decreased synapse density by a third (Fig. 4b–c). Remarkably, MHCI OE decreased synapse density to levels less than those at 5 d.i.v. (normalized to 5 d.i.v.: 0.76 ± 0.06, p < 0.05, n =15 dendrites, 5 cells), suggesting that MHCI may actively eliminate synapses as well as limit their formation. MHCI OE and KD elicit opposite effects on glutamatergic synapse density, demonstrating that MHCI bidirectionally regulates the initial establishment of cortical connectivity.
Fig. 4. MHCI overexpression decreases glutamatergic synapse density.

(a) Neurons were transfected with H2-Kb-CFP at 6 d.i.v. Within 48h of transfection, MHCI was overexpressed in both proximal and distal dendrites. sMHCI density in dendrites (high magnification images in middle and right panels) of H2-Kb-CFP expressing cells (right) was qualitatively greater than in control cells (middle). Dendrites are oriented proximal to distal, top to bottom. Scale bars = 20 μm, left panel; 5 μm right panels. (b) H2-Kb-CFP OE increases sMHCI density by 76 ± 0.12% (p < 0.001) and decreases glutamatergic synapse (ES) density by 33 ± 0.03% (p < 0.001). (c) Neurons transfected with H2-Kb-CFP (right panels) or their non-transfected neighbors (left panels) were immunostained for vGlut1 (left) and NR2A/B (NR2; middle). Synapses are yellow in the overlay images (right). (d) MHCI OE ablates the proximal-distal distribution of both sMHCI (normalized to same region control: proximal: 1.70 ± 0.14, p < 0.001; distal: 2.13 ± 0.18, p < 0.001, n = 17 dendrites, 8 neurons each) and excitatory synapses (ES; normalized to same region control: proximal: 0.73 ± 0.08, p < 0.05; distal: 0.66 ± 0.03, p < 0.001, n = 19 dendrites, 8 neurons each). (e) Neurons transfected with H2-Kb-CFP (right panels) or their non-transfected neighbors (left panels) were immunostained for synapsin (syn: left) and GABAR subunits (GABAR; middle). Synapses are yellow in the overlay images (right). (f) H2-Kb-CFP OE decreases GABAergic synapse density (IS) by a little over 25% (0.74 ± 0.08, n = 20 dendrites, 10 neurons each, p < 0.05). Scale bars = 5μm. * = p<0.05.
The MHCI OE-induced reduction in synapse density resulted from both a decrease in synaptic proteins and in synaptic recruitment of SVs. MHCI OE decreased both vGlut1 and NMDAR densities by 25–30% (vGlut1: 0.74±0.03, p<0.001; NR2A/B: 0.72±0.03, p<0.001). In addition, vGlut1 enrichment at synapses decreased by 15±0.03% (p<0.05), with no change in synaptic enrichment of NR2A/B (0.93±0.03, p=0.09). Thus, both KD and OE experiments indicate that sMHCI regulates the abundance of both glutamatergic SVs and NMDARs, and the recruitment of SVs to new synapses.
The effects of MHCI OE also support the conclusion that sMHCI levels regulate synapse density locally on dendrites. MHCI OE increased sMHCI both proximal and distal to the soma, ablating the proximal-distal distribution (Fig. 4d). The distribution of glutamatergic synapse density was similarly ablated (Fig. 4d). These results support the interpretation that local concentrations of sMHCI regulate the local density of glutamatergic synapses.
Finally, MHCI OE also decreased the density of GABAergic synapses by 26% (Fig. 4e–f). In these neurons, there was no significant change in the density of pre- and postsynaptic proteins (synapsin; 1.0±0.07, p=0.86; GABAR subunits: 0.85±0.07, p=0.18), nor was there a change in synaptic enrichment of GABARs (0.86±0.07, p=0.15). However, there was a significant decrease in synapsin enrichment at inhibitory synapses (0.76±0.06, p<0.01), suggesting that MHCI may limit the accumulation of SVs at nascent GABAergic synapses similar to its proposed role at glutamatergic synapses.
MHCI negatively regulates synaptic transmission
To determine if MHCI alters synaptic transmission, two approaches were utilized—FM dye labeling and whole-cell patch-clamp recording. First, FM1-43 and FM4-64 were used to determine the density of presynaptic terminals that were capable of depolarization-dependent vesicle cycling 21. β2m KD increased the density of functional presynaptic terminals labeled by FM1-43 by 55% (Fig. 5a), implying that many of the new synapses formed during sMHCI suppression are capable of SV release. Conversely, MHCI OE decreased the density of functional, FM4-64-labeled presynaptic terminals by approximately 35% (Fig. 5b).
Fig. 5. MHCI bidirectionally regulates glutamatergic and GABAergic synaptic transmission.

(a) β2m KD increases the density of FM1-43 labeled puncta by 55±0.12% (values normalized to NTS; p < 0.01, n = 15 dendrites, 5 cells each) (b) whereas MHCI OE decreases the density of FM4-64 staining by 35% (values normalized to GFP-transfected controls; 0.63 ± 0.06, p < 0.05, n = 6 dendrites, 3 neurons each). (c) Representative traces from whole-cell patch-clamp recordings of mEPSCs from 8–10 d.i.v. control cultured cortical neurons or neurons transfected with either β2m siRNA or H2-Kb-CFP. Qualitatively, β2m KD dramatically increases, and H2-Kb OE strongly decreases, glutamatergic synaptic transmission. (d) Representative traces from whole-cell patch-clamp recordings of mIPSCs from 8–10 d.i.v. control cultured cortical neurons or neurons transfected with either β2m siRNA or H2-Kb-CFP. Qualitatively, β2m KD increases, and H2-Kb OE decreases, GABAergic synaptic transmission. (e) β2m KD significantly increases both mEPSC frequency (2.74 ± 0.60, p < 0.001, n = 8) and amplitude (1.63 ± 0.22, p < 0.05, n = 8), while H2-Kb-CFP OE decreases both measures (freq: 0.45 ± 0.04, p < 0.001, n = 8; amp: 0.72 ± 0.10, p < 0.05, n = 8). (f) In contrast, β2m KD significantly increases mIPSC frequency (1.77 ± 0.12, p < 0.05, n = 12), while H2-Kb-CFP OE decreases it (0.68 ± 0.10, p < 0.05, n = 12), but neither manipulation changes mIPSC amplitude (KD: 0.96 ± 0.03, p = 0.78; OE: 1.01 ± 0.03, p = 0.36).
Manipulation of sMHCI levels also changed glutamatergic and GABAergic synaptic transmission. β2m KD more than doubled the frequency of miniature excitatory postsynaptic currents (mEPSCs; Fig. 5c,e), while MHCI OE decreased mEPSC frequency by more than half (Fig. 5c,e). These data are consistent with the direction of changes in synapse density measured using ICC (Figs. 2 and 4). Because mEPSC frequency was increased to a much greater magnitude than synapse density, MHCI effects on presynaptic release probably also contribute to changes in mEPSC frequency. Surprisingly, mEPSC amplitude also changed in neurons with altered sMHCI levels. β2m KD increased mEPSC amplitude, while MHCI OE decreased it (Fig. 5c,e). Although the effects on GABAergic transmission were generally smaller in magnitude, β2m KD also increased mIPSC frequency, while MHCI OE decreased it (Fig. 5d,f). Similar results were obtained using two distinct recording paradigms for mIPSCs (Suppl. Fig. 4). These results are similar to those measured using ICC both in direction and magnitude. However, unlike the changes in strength of glutamatergic synapses, there was no change in mIPSC amplitude following MHCI manipulations (Fig. 5f). These data suggest that MHCI negatively regulates glutamatergic synaptic strength, in addition to controlling both glutamatergic and GABAergic synapse density.
Because the changes in glutamatergic and GABAergic transmission were different in magnitude, we next examined if MHCI levels dictate the balance of excitation and inhibition onto young cortical neurons. Following MHCI manipulations, the amount of charge transferred during excitatory neurotransmission (Fig. 5c, mEPSCs) was divided by that transferred during inhibitory neurotransmission (mIPSCs; Fig. 5d). Charge was calculated as the average integrated area of each event multiplied by the average frequency. The excitation/inhibition ratio doubled as a consequence of β2m KD and decreased by about 60% from MHCI OE (KD: 3.56/1.75 = 2.03, OE: 0.25/0.78 = 0.32). Thus, MHCI controls the ratio of excitation to inhibition on cortical neurons; altering sMHCI expression can profoundly change that balance during the initial establishment of cortical connections.
Homologous MHCI negatively regulates synapse density
Although trafficking of MHCI out of the ER to the cell surface requires association between the MHCI heavy chain and β2m10–12, this complex (heterologous sMHCI) can dissociate once inserted into the plasma membrane, resulting in release of β2m and generation of ‘free’ heavy chain (homologous sMHCI)17–18,22. Homologous sMHCI self associates23 and enables binding to other proteins24. The ratio of homologous to heterologous sMHCI is dynamic since homologous sMHCI can also re-associate with β2m23,25–26.
To test if the homologous to heterologous ratio of sMHCI regulates glutamatergic synapse density, neurons were treated with eβ2m for 36 hours and the ratio was assessed by comparing the density of sMHCI without and with sβ2m. Treatment with eβ2m decreased the percentage of homologous sMHCI by 45±0.05% (Fig. 6a), revealing that both heterologous and homologous MHCI are normally present on neurons. Decreasing the density of homologous sMHCI clusters increased the density of glutamatergic synapses by about 50% (Fig. 6b–c). Thus, it is not just the presence of sMHCI, but specifically free MHCI heavy chain that is critical for negatively regulating synapse density in early cortical development.
Fig. 6. Homologous MHCI clusters negatively regulate glutamatergic synapse density.

(a) Exposure of neurons to eβ2m for 36h decreases the homologous (MHCI alone) to heterologous (MHCI plus β2m) ratio of sMHCI clusters by 45% at 8 d.i.v. (p < 0.001). (b) ICC staining for vGlut1 (top) and NR2A/B (middle) qualitatively shows the increase in ES density (yellow in overlay, bottom) caused by decreasing homologous sMHCI. (c) Decreasing the homologous to heterologous ratio of sMHCI clusters increases ES density by 53 ± 0.05% (p < 0.001). All values are normalized to the culture-matched controls. Scale bar = 5 μm.
MHCI modulates activity-dependent synaptogenesis
Although MHCI mRNA expression is regulated by neuronal activity8,27, the effect of synaptic activity on sMHCI protein levels remains unknown. To determine if, and in what direction, synaptic activity regulates sMHCI in young cortical cultures, neural activity was acutely blocked by suppressing sodium-dependent action potentials using 1μM TTX for 24hr between 7–8 d.i.v. TTX treatment decreased sMHCI levels 25% by 8 d.i.v. (Fig. 7a) without shifting the homologous to heterologous ratio (0.91±0.08, p=0.47, n=15 dendrites). Interestingly, TTX had the opposite effect on MHCI levels detected after permeabilization (41.2% increase in MHCI levels; Suppl. Fig. 5a–b), providing more evidence that sMHCI is highly regulated and the distribution of MHCI after permeabilization does not necessarily reflect sMHCI.
Fig. 7. Changes in MHCI levels are necessary for activity-dependent changes in synapse density.
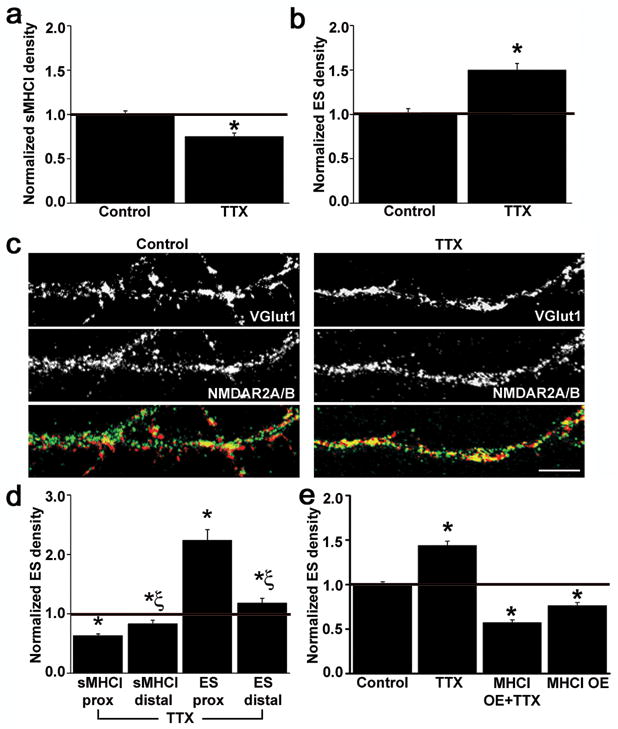
(a) Exposure to TTX (1μM, 24h) decreases sMHCI density at 8 d.i.v. by 25 ± 0.04% (p < 0.001). (b) TTX treatment increases glutamatergic synapse (ES) density, as defined by co-localized vGlut1 and NR2A/B, by 44 ± 0.07% (p<0.001). (c) Images of neurons treated with vehicle (left panels) or TTX (right panels) and immunostained for VGlut1 (top) and NR2A/B (middle). Synapses are visible as yellow puncta in the overlay (bottom). Dendrites are oriented proximal to distal, left to right. (d) TTX inverts the proximal-distal distribution of sMHCI (normalized to same region control: proximal: 0.63 ± 0.03, p < 0.001; distal: 0.83 ± 0.06, p < 0.05) and glutamatergic synapses (ES; proximal: 1.89 ± 0.15, p < 0.001; distal: 1.22 ± 0.06, p < 0.05), but maintains the inverse relationship between local levels of sMHCI and ES (*, significant difference from control; Ξ, significant difference of distal density from same age proximal density values). (e) The TTX-induced increase in ES density is completely prevented by MHCI OE (TTX: 1.44 ± 0.05, n = 25 dendrites, p < 0.001; MHC OE: 0.77 ± 0.03, n =71, p < 0.001; MHC OE + TTX: 0.57 ± 0.03, n = 48, p < 0.001). Scale bar = 5 μm.
Because neural activity regulates sMHCI levels and sMHCI regulates glutamatergic synapse density, activity may regulate glutamatergic synapse density through altering sMHCI in early cortical development. To test this hypothesis, cultures were treated with TTX (1μM) for 24 hr and glutamatergic synapse density was assessed at 8 d.i.v. At this age, TTX treatment increased synapse density by 45% (Fig. 7b–c). This increase was obtained not only by quantifying NR2A/B-vGlut1 positive synapses (Fig. 7b–c), but also by quantifying synapsin-PSD-95 positive synapses (1.35±0.09, p<0.01, n=8 dendrites, 4 neurons each) and vGlut1-GluR1/2 positive synapses (1.52±0.12, p<0.01, n=6 dendrites, 3 neurons each). Thus, the new synapses formed during activity blockade were not “silent” synapses. Like sMHCI, neural activity acts to limit the density of glutamatergic synapses during the initial establishment of cortical connectivity.
While TTX treatment decreased sMHCI and increased glutamatergic synapse density both proximal and distal to the soma, it inverted their proximal-distal distribution (Fig. 7d). sMHCI levels were decreased in proximal regions more dramatically than in distal regions. Remarkably, as the local sMHCI distribution shifted, the distribution of glutamatergic synapses also changed, maintaining the inverse relationship between sMHCI and synapse levels and inverting the differential distribution found in control neurons (Fig. 7d). These data support the conclusion that local dendritic sMHCI levels regulate the density of local innervation.
The increase in glutamatergic synapse density following activity blockade resulted from both an increase in density of pre- and postsynaptic proteins and an increase in their synaptic recruitment. Similar to the effects of decreasing sMHCI, TTX increased the densities of SVs, NMDARs, and PSD-95 (vGlut1: 1.33±0.06, p<0.001, n=47 dendrites, 19 neurons; NR2A/B: 1.22±0.04, p<0.001, n=29 dendrites, 10 neurons; PSD-95: 1.34±0.08, p<0.01, n=6 dendrites, 3 neurons) and modestly increased the synaptic recruitment of vGlut1 (vGlut1: 1.12±0.04, p<0.001). However, unlike the effects of decreasing sMHCI, TTX also increased the synaptic recruitment of glutamate receptors by 20–30% (% synaptic NR2A/B: 1.18±0.04, p<0.001, n=29 dendrites, 10 neurons; % synaptic GluR1: 1.32±0.07, p<0.05, n=6 dendrites, 3 neurons; % synaptic GluR2: 1.27±0.03, p<0.05, n=6 dendrites, 3 neurons). Thus, blockade of neural activity and suppression of sMHCI increase glutamatergic synapse density via overlapping mechanisms.
To establish if TTX requires changes in sMHCI levels to increase glutamatergic synapse density, sMHCI was overexpressed during TTX treatment to attempt to rescue the effect. Consistent with this hypothesis, MHCI OE completely prevented the TTX-induced increase in synapse density (Fig. 7e). Moreover, MHCI and TTX appear to function in overlapping pathways since decreasing sMHCI in the presence of TTX did not increase glutamatergic synapse density above the increase elicited by either treatment alone (TTX: 1.44±0.07, p<0.001, TTX + β2m KD: 1.43±0.05, p<0.001; β2m KD: 1.69±0.05, p<0.001, n = 24 dendrites from 8 neurons each). These data indicate that the ability of neural activity to influence glutamatergic synapse density in early postnatal development depends, at least in part, on MHCI.
MHCI and activity regulate synapse density only in young cortex
To test whether the ability of neural activity and sMHCI to negatively regulate glutamatergic synapse density is developmentally regulated, older cortical neurons (13d.i.v.) were treated with TTX or e β 2m, or were transfected with H2-Kb-CFP, as above. Neither activity blockade nor changing sMHCI levels altered glutamatergic synapse density in older neurons (TTX: 0.89±0.05, p=0.21, n=15 dendrites, 5 cells; H2-Kb-CFP: 0.97±0.05, p=0.60, n=11 dendrites, 5 cells; e β 2m, 1.07±0.06, p=0.41, n= 5 dendrites, 5 cells). Therefore, the ability of both neural activity and sMHCI to regulate glutamatergic synapse density is specific to the period of the initial establishment of cortical connections.
Discussion
Although MHCI molecules regulate activity-dependent refinement during late postnatal development7–9,13, the function of these molecules in the initial establishment of connections in the CNS remains unknown. Here, we show that MHCI molecules are present in the plasma membrane of both axons and dendrites of cortical neurons prior to and during synapse formation. MHCI negatively regulates the density and function of glutamatergic and GABAergic connections and controls the balance of cortical excitation and inhibition. In addition, neural activity regulates glutamatergic synapse density in part through MHCI. Finally, the ability of MHCI molecules and neural activity to modulate cortical glutamatergic connectivity occurs selectively during the early postnatal period.
There are relatively few reports of molecules that limit the initial establishment of cortical connectivity. Recently, evidence demonstrates that MEF2 transcription factors, semaphorin-5B, and ephexin5 negatively regulate the early establishment of glutamatergic connections in mammalian cultures3–5 and RSY-1 antagonizes presynaptic assembly in C. elegans 6. Our results identify MHCI as a new class of molecules that limit early postnatal connectivity in the CNS. These findings counter the prevailing assumption that the initial establishment of connectivity in the CNS is guided primarily by molecules that promote synaptogenesis1,28 and instead support an alternative hypothesis—that synapse formation is bidirectionally controlled.
Our observation that MHCI negatively regulates glutamatergic synapse formation in cortical neurons differs from a recent report showing that there is no change in synapse density in older postnatal hippocampal cultures or in vivo in the hippocampus of β2m/TAP1−/− mice14. While the increase in mEPSC frequency in hippocampal neurons in these knockout mice is caused by a modest increase in number of SVs per synapse14, the reported doubling of mEPSC frequency between cortical neurons in these mice14 must be caused, at least in part, by an increase in glutamatergic synapse density—a hypothesis consistent with our results. Changes in sMHCI levels altered synapse density by every measure used in our study. First, acute β 2m KD increased glutamatergic and GABAergic synapse density in vitro, measured by ICC and electrophysiology. Second, glutamatergic synapse density was also increased in cultured neurons from β2m−/− mice. Third, MHCI OE caused the predicted, opposite results from β2m KD or knockout. Fourth, synapse density was increased in layer 5 of visual cortex in β2m−/− mice at every age examined, showing that the changes observed from acute KD and OE do not simply reflect temporal shifts in synapse formation. Moreover, because acute manipulations of sMHCI affected synapse density only during the initial establishment of connections, our results suggest that changes in sMHCI in early postnatal development may have lasting effects on cortical circuitry. Finally, predicted changes in synapse density were obtained following alterations of sMHCI by eβ2m and TTX treatment, neither of which involved directly manipulating MHC gene expression. Together, these results provide strong support for the conclusion that sMHCI is integral to limiting synapse density during the initial establishment of cortical connections and that chronic decreases in sMHCI levels, as in the β2m −/− mouse, would cause dramatically increased cortical connections especially during two critical periods of development—the initial formation of connections and activity-dependent refinement of those connections.
In addition to its ability to control both glutamatergic and GABAergic synapse density, sMHCI regulates the function of glutamatergic synapses. β2m KD increased both mEPSC frequency and amplitude, while MHCI OE decreased both measures. These changes in mEPSC frequency are consistent with changes in synapse density as well as changes in presynaptic release, as previously shown in mature hippocampal neurons from β2m/TAP1−/− mice14. However, the effect of sMHCI on mEPSC amplitude is surprising, given the lack of change in mEPSC amplitude in cortical slices from β 2m/TAP1−/− mice14. Perhaps this difference is due to homeostatic compensation in the function of glutamatergic synapses in the knockout mouse with age. Regardless, sMHCI clearly controls the function of young glutamatergic synapses through altering the quantity of functional AMPA receptors at synapses, which mediate mEPSC amplitude. In contrast to glutamatergic synapses, sMHCI appears to affect only mIPSC frequency and not amplitude, suggesting that sMHCI alters GABAergic synapse density and possibly also presynaptic release at GABAergic synapses. Finally, although sMHCI alters glutamatergic and GABAergic synapse density and function in the same direction, the magnitude of change is less for GABAergic synapses, leading to dramatic changes in the excitation/inhibition (E/I) balance onto young cortical neurons. Future work is needed to clarify how sMHCI controls synaptic AMPAR dynamics, whether (and how) it controls presynaptic release, and the ramifications of changes in E/I balance for cortical network formation and plasticity.
Although neural activity is not required for synapses to form in the mammalian CNS29–32, synaptic transmission may modulate the number of synapses that a neuron forms 33. Our data demonstrates that suppression of action potentials for just 24h increases glutamatergic synapse density at 8d.i.v. Consistent with this result, increases in the density of synapses and pre- and postsynaptic proteins and synaptic recruitment of glutamate receptors have been described in older hippocampal and cortical neurons following either chronic or acute TTX treatment34–36. However, in younger neurons (7–10d.i.v.), 48hr of TTX has been reported to change only the glutamate receptor content of existing synapses, with no change in presynaptic properties or synapse number36. It is possible that our experiments have revealed a new effect of activity blockade during the initial establishment of cortical connections that changes as cortical networks mature. Indeed, the high percentage of SVs found at synapses in the previous report36 indicate that those cultures are more mature than ours. In sum, our results are consistent with reports that suggest different rules of homeostasis apply in young versus older neurons36–40, but extend those models by providing evidence that neurons respond to altered network activity by changing synapse density during the initial establishment of cortical connectivity.
Manipulation of both neural activity and sMHCI levels revealed that neural activity regulates glutamatergic synapse density at least in part through its effects on sMHCI levels. This conclusion is consistent with recent reports showing that MHCI may be required for synaptic scaling induced by TTX-treatment in older hippocampal neurons14. In our experiments, MHCI OE prevented the TTX-induced increase in synapse density. However, the degree of increase in synapse density observed with TTX is greater than would be expected based on the sMHCI decrease, suggesting that additional pathways mediate these effects together with MHCI. Similarly, the TTX-specific enrichment of both pre- and postsynaptic proteins at new synapses and the sMHCI-specific synaptic enrichment of only glutamatergic SVs indicate that their underlying mechanisms are overlapping, but not identical.
Although most of the effects described here are due to changing postsynaptic sMHCI levels, the mechanisms by which MHCI proteins regulate the initial establishment of cortical connections remain unknown. Because synapse density at any age is the net result of synapse formation and elimination, it is possible that MHCI controls cortical connectivity through altering either, or both, of these processes. MHCI may regulate trafficking of SV precursors to, or their stabilization at, nascent synapses. SV precursors are highly colocalized with sMHCI in axons prior to synaptogenesis, SVs at mature synapses are colocalized and co-fractionate with MHCI10, and both gain-of-function and loss-of-function studies revealed that sMHCI regulates the density and synaptic enrichment of glutamatergic and GABAergic SVs. These data are consistent with the increase in SV density at hippocampal synapses in adult β2m/TAP1−/− mice 14. sMHCI may also regulate the transcription of synaptic proteins, leading to changes in their density and availability during synaptogenesis, since ligation of MHCI alters gene expression in non-neuronal cells47. In addition to functioning across the synapse in trans through binding to an as yet unknown MHCI receptor, sMHCI could also limit glutamatergic synapse density indirectly by interacting with other proteins in cis on the same cell24,41–43. In other cell types, increasing the homologous to heterologous sMHCI ratio increases binding of MHCI with other proteins in cis44, changing their surface expression and altering their sensitivity to ligand. Here, we report that homologous, but not heterologous MHCI, limits synapse density. Thus, the homologous to heterologous sMHCI ratio is critical for sMHCI signaling and function in neurons, as in other cell types. Defining which proteins interact with MHCI in trans and/or in cis will be an important future step in identifying how MHCI regulates synapse density.
The inverse, non-uniform distributions of sMHCI and glutamatergic synapses on dendrites of young cortical neurons suggest that local regulation of sMHCI density may serve as a mechanism for cortical neurons to locally regulate glutamatergic synapse density. Consistent with this idea, this inverse relationship was maintained after all manipulations in our study, even those that inverted the original distribution. This is one of the first demonstrations of a protein whose irregular distribution appears to regulate the dendritic location of synapses in an activity-dependent manner. It is also possible that local control of sMHCI and synapse density might be instructive for the elaboration of dendritic arbors. Limiting the formation of too many synapses in proximal dendrites while permitting synapse formation in more distal growing dendritic regions could maximize the number of neurons with which a given neuron connects and allow for extended dendritic arbors to develop. In this way, sMHCI may help to determine whether and where synapses form on young neurons during the initial establishment of cortical connectivity.
Since MHCI molecules mediate the immune response45 and regulate cortical connectivity9,14 as well as the balance of excitation and inhibition in the brain, it is possible that an abnormal systemic immune response during early cortical development could alter neuronal connectivity and change cognition through changing neuronal MHCI levels. This hypothesis is receiving increased attention due to mounting evidence for immune dysregulation in a number of neurodevelopmental disorders, including autism and schizophrenia46–48. Results presented here are consistent with the idea that altering MHCI proteins during early cortical development could alter cortical connectivity and the balance of excitation and inhibition, pathologies characteristic of these disorders. To determine the relevance of our results to disease, it will be critical to define whether, how, and when systemic immune dysregulation might alter neuronal MHCI to cause changes in cortical connectivity.
Methods
Rat cortical neuron culture
All protocols were approved by the Institutional Animal Care and Use Committee. Neurons from the occipital cortex of newborn (P0-3) Long-Evans rats were dissociated with papain in HEPES-buffered EBSS with kynurenate (1mM), plated on poly-L-lysine coated glass coverslips at densities of 22K/ 24mm slip, and inverted over cortical astrocyte monolayers into N2 medium16.
Mouse cultures
Neurons from the occipital cortex of newborn (P0-3) C57BL/6J mice or mice lacking β2m (Jackson Labs, B6.129P2- β2m<tm1Unc>/J) were dissociated with papain in HEPES-buffered EBSS with kynurenate (1mM), plated on poly-L-lysine coated glass coverslips at densities of 35,000/ 24mm slip, and inverted over cortical astrocyte monolayers as above.
Immunoblot analysis
Total cellular membranes were isolated from P3 primary visual cortex and processed as previously reported 8. Briefly, tissue was homogenized with a motorized glass-teflon homogenizer in cold 10mM HEPES (pH 7.4), 0.32M sucrose, and protease inhibitors (Complete Mini with EDTA, Roche). Protein was centrifuged for 10 min at 1000xg. The resulting supernatant was centrifuged at 16,000xg for 35 min. The resulting pellet was dissolved for 15 min in cold RIPA with inhibitors and centrifuged for 15 min at 14,000 RPM at 4°C in a desk-top centrifuge. Protein in SDS sample buffer was separated in 8% SDS-PAGE, transferred to PVDF, and incubated with anti-MHCI antibodies OX-18 (mouse monoclonal, Serotec) and ab52922 (rabbit monoclonal, AbCAM). Odyssey secondaries were utilized with the Odyssey Infrared Imaging System for protein detection (LI-COR).
Transfections
siRNA transfections were optimized for concentration and time interval of maximum protein KD. 25nM siRNA targeting β2m (Antisense: 5′-PUAACUCUGCAAGCAUAUACUU, Dharmacon) were transfected into neurons at 5 d.i.v. with Lipofectamine 2000. 25nM of a Risc-free siRNA conjugated to Cy3 (siGLO RISC-free siRNA, Dharmacon) was cotransfected in liposomes carrying β 2m siRNA to enable visual identification of transfected neurons. 25nM siRNA of non-targeting sequence (NTS; siCONTROL siRNA #1, Dharmacon) co-transfected in liposomes with the fluorophore-bound RISC-free sequence defined the control. Co-transfection efficiencies were consistently greater than 90% as measured by the correlation between the Cy3 and sMHCI immunostaining. sMHCI densities were assessed on culture-matched coverslips for each experimental trial assessing synapse density to confirm suppression of sMHCI by β 2m KD. H2-Kb-CFP (a kind gift of Dr.M. Edidin, Johns Hopkins University) was transfected into neurons at 6 d.i.v. with Lipofectamine 2000. Non-transfected neurons from the H2-Kb transfected coverslip expressed densities of sMHCI and glutamatergic synapses that were not significantly different from cells transfected with an eGFP-N1 construct (Clontech).
Exogenous β 2m and Activity Blockers
5mM exogenous human β2m (e β2m; Serotec) was added to cultures at 6.5 d.i.v. (36h). 1 μM TTX (Sigma) was added to cultures at 7d.i.v. (24h). All neurons were fixed and immunostained at 8d.i.v.
Immunocytochemistry
Total protein labeling: Coverslips were fixed in 4% paraformaldehyde in 0.01M PBS with 4% sucrose for 10 min. Cells were washed with 0.1M glycine in PBS for 5min, permeabilized with 0.25% Triton-X 100 for 5 min, and washed with PBS. Neurons were then blocked for 30 min with 10% BSA in PBS and incubated with primary antibodies in 3% BSA in PBS for 1–1.5 h. Following primary binding, cells were washed with 3% BSA in PBS. Then, neurons were incubated with secondary antibodies in 3% BSA in PBS for 45 min. Following secondary binding, cells were washed with 3% BSA in PBS, rinsed with ddH2O, and mounted on slides in Fluoromount-G (EMS). Surface labeling: Coverslips were fixed, blocked, and incubated with primary antibodies to surface proteins as above, with omission of the permeabilization step. Controls for membrane integrity were conducted with every surface experiment by staining for the internal protein MAP2. The absence of MAP2 staining confirmed intact membrane integrity; dendritic regions with positive MAP2 staining were excluded from analysis49. Primary antibodies: mouse anti-MHCI heavy chain (1:100, OX-18 clone, Serotec), mouse anti- β2m (1:100, Serotec), human anti-β2m (1:100, Abcam), chicken anti-MAP2 (1:1500, Abcam), guinea pig anti-vGlutI (1:2000, Chemicon), rabbit anti-NR2A (1:200, Upstate), rabbit anti-NR2B (1:200, Chemicon), synapsin (1:1000; Chemicon), mouse anti-PSD95 (1:1000, NeuroMab, UC Davis), mouse or chicken anti-GFP (1:200, 1:800; Chemicon), guinea pig anti-GABAR α2/ γ2 (1:1000; gift from Dr. JM Fritschy). Secondary antibodies: anti-mouse-488 and -647, anti-chicken-488 and -647, anti-rabbit-488, -568, and -647 (1:400, Molecular Probes) and anti-guinea pig-Cy3 (1:400, Jackson). For Suppl. Fig. 1, neurons were incubated with mouse anti-MHCI (1:50, OX-18, Serotec) and rabbit anti-MAP2 (1:2000 Chemicon) in 3% BSA in PBS for 90 min, washed 3×5min in PBS, incubated with biotin anti-mouse (1:100, Vector) for 45 min, washed 3×5min in PBS, then incubated with Streptavidin-FITC (1:200, Vector) and Alexa 568 anti-rabbit (1:400, Molecular Probes) in 3% BSA in PBS, washed 3×5min in PBS, rinsed with ddH2O, and mounted on slides in Fluoromount-G (EMS).
Animals and tissue samples for EM
WT mice and β2m−/− mice at P8, P11, P23 and P60 were anesthetized with pentobarbital and perfused intracardially with saline, followed by a mixture of 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer. 60–80 μm coronal sections were cut on a vibratome (Leica VT1000) and collected in cold 0.1M phosphate buffer.
Electron microscopy
Vibratome sections were osmicated, dehydrated in ethanol and acetone and flat embedded in Araldite. Embedded sections containing primary visual cortex were identified by light microscopy and photographed at X10. Approximately 1–2 mm wide slabs of the visual cortex (from pia matter to white matter) were cut in a dissecting microscope and glued to blank resin blocks. Semithin sections (2–4 μm thickness) were cut on an ultramicrotome (Leica Ultracut), mounted on gelatinized glass slides and stained with 1% toluidine blue. Sections were photographed and Layer V was identified and used as the guide for trimming the blocks for ultrathin sectioning. Seventy nanometer thin sections were cut on the ultramicrotome and collected on Formvar-coated single slot copper grids. The grids were stained with uranyl acetate and lead citrate and examined in a Philips CM 120 electron microscope at 80 kV. To obtain images of synapses in layer V, thin neuropil regions (avoiding the neuronal and glial somata, blood vessels, large dendrites and myelinated axons) 20 were selected by low magnification scanning and multiple regions separated by at least 100 mm were photographed at X 8400 or X 11000 using a 2k X 2k CCD camera (Gatan). Images were collected and processed in the computer using DigitalMicrograph software (Gatan).
EM Quantification
At least 19 single section transmission electron micrographs were randomly taken from dense neuropil of layer 5 primary visual cortex for each condition and age, from at least 2 animals. This approach permitted analysis of a large area of neuropil20. The area of each image taken at X 8400 was approximately 34.9 μm2; the area of each image taken at X 11000 was approximately 17.4 μm2. For P8, the whole area for synaptic counting per genotype was 1396 μm2; for P11, the area was 934 μm2; for P23, the area was 1361 μμm2; for P60, the area was 1361 μm2. Synapses were identified based on generally accepted criteria20; three-criteria synapses included an unambiguous post-synaptic density (PSD), a clear synaptic cleft, and three or more SVs while two-criteria synapses included a PSD and SVs but no discernable cleft. These two classes were analyzed separately; however, since the same magnitude changes in synapse density were found using either criterion, the numbers were combined to give total synapse density. Two animals were analyzed for each genotype at each age. Since the samples from each animal were processed together and the ultrastructural features were similar, the data were pooled for blinded quantification.
FM Staining
Coverslips were washed with warm ACSF (120mM NaCl, 3mM KCl, 2mM CaCl2, 2mM MgCl2, 30mM dextrose, 0.2% sorbitol, 20mM HEPES, ph 7.3) and transferred to imaging chambers. Cells were bathed in 15 μm FM1-43 (Molecular Probes), for siRNA experiments, or FM4-64 (Molecular Probes) for overexpression experiments, in high K+ ACSF (38mM NaCl, 85mM KCl, 2mM CaCl2, 2mM MgCl2, 30mM dextrose, 0.2% sorbitol, 20mM HEPES, ph 7.3) at room temperature for 1 min. Cells were then washed via constant perfusion of ACSF for 15 min and an image of the neuron was taken; this is the “stained” image. Cells were then constantly perfused with high K+ ACSF for 3 min and a second image of the same neuron was taken; this is the “destained” image. Subtraction of the destained from the stained image provided the data for quantification. Only one neuron was imaged per coverslip. Images were acquired on a Zeiss Pascal confocal system with a 63x oil objective (1.4 NA). Laser power was low and scan times were between 3–4 seconds to avoid phototoxicity. Images were analyzed with ImagePro software as described49. Dendrites of lengths 50–150 μm were analyzed for each neuron. Measures were pooled across a given set of neurons for statistical analysis.
Image acquisition and analysis
Growth cones were imaged on an Olympus Fluoview 2.1 laser scanning confocal system with a 60X PlanApo oil immersion objective (1.4 NA) on an IX70 inverted microscope using 2X zoom. Images for β2m synapse quantification from culture were collected on an Olympus Fluoview 300 confocal microscope with a 60x oil objective using 2.5X zoom. All other images were collected using sequential triple-channel confocal fluorescence imaging on a Zeiss LSM 510 Meta with 63x oil immersion objective (1.4 NA). PMT levels were set based on a no-primary control for non-specific secondary antibody labeling; the highest PMT employed to capture images from samples was lower than any PMT registering positive signal from the no-primary control. PMTs were also set below saturation for images intended for quantification. Neurons were selected for imaging and analysis from networks of comparable complexity.
Images were analyzed with ImagePro software (Media Cybernetics) as described49. Briefly, background was defined manually for each neurite and each channel as 2-times the standard deviation plus the average intensity value of 10 regions of neighboring diffuse fluorescence. Protein clusters were selected manually on images from each of the channels independently to create a template of points that include all stained clusters in all fluorescent channels. The presence of signal in each fluorescent channel was then assessed and colocalization determined. Synapses were defined as sites of colocalized pre- and postsynaptic proteins. Unless otherwise specified, 10 cells from 2 separate cultures were analyzed at each developmental time point. 3 dendrites of lengths 50–150 μm were analyzed for each parameter on each neuron. Dendrites were selected for analysis that appeared uniform in diameter along their initial 100 μm length. 1–3 regions of axon were analyzed over 50–150 μm. 10 neurons were selected as a representative sample size because values calculated from their dendrites generated reproducible results and small standard errors. Images for quantification of synapse density in β2m−/− mouse cultures was performed using custom-written journals in Metamorph software (Molecular Devices). Data for most experiments was normalized to the indicated values to control for any contribution from inter-experiment variability.
Electrophysiology
Whole-cell patch-clamp recordings were made from rat cortical neurons at 8–10 d.i.v.. Neurons were co-transfected (at 5 d.i.v.) with eGFP and a control siRNA (siNTS) or siRNA targeting β2m (si β2M) and recorded at 8–10 d.i.v. Neurons were transfected with H2-Kb-YFP for OE and recorded 48hours later at 8–10 d.i.v. To isolate mEPSCs, whole-cell patch-clamp recordings were made at −65mV in the presence of 500 nM TTX and 100 mM picrotoxin. The extracellular solution consisted of, in mM: 110 NaCl, 3 KCl, 10 Hepes, 10 D-glucose, 10 glycine, 2 CaCl2, 2 MgCl2 at pH 7.3. The intracellular solution consisted of, in mM, 130 K-gluconate, 10 KCl, 10 Hepes, 10 EGTA, 1 CaCl2, 5 ATP-Mg2+, 5 GTP-Li2+, pH 7.31. To isolate mIPSCs, two distinct recording paradigms were used. First, recordings were made from cells voltage-clamped at −40 and −20mV in the presence of 500 nM TTX. The extracellular solution consisted of, in mM: 119 NaCl, 5 KCl, 20 Hepes, 30 D-glucose, 10 μM glycine, 1 CaCl2, 1 MgCl2 at pH 7.3. The internal solution consisted of, in mM: 148.5 K-gluconate, 9 KCl, 1 MgCl2, 10 Hepes, 0.2 EGTA, pH 7.3. Using these solutions, mIPSCs reverse at −65 mV. Results from mIPSCs recorded at −40mV and −20mV were not significantly different, so they were normalized to the appropriate control and pooled. Second, recordings were made from cells voltage-clamped at −70mV in the presence of 500 nM TTX, 10 μM CNQX, 50 μM APV, as previously described in ref. 50. The extracellular solution consisted of, in mM: 136 NaCl, 2.5 KCl, 10 HEPES, 10 D-glucose, 2 CaCl2, 1.3 MgCl2 at pH 7.3. The internal solution consisted of, in mM: 68 K-gluconate, 68 KCl, 0.2 EGTA, 2 MgSO4, 2 HEPES, 3 ATP-Mg2+, 0.2 GTP-Li2+, pH 7.3. For this set of Cl− solutions, the reversal potential for Cl− was -18mV and thus yielded larger mIPSCs. Recordings were filtered at 2 kHz using an Axopatch 200B amplifier and digitized at 5 kHZ using Clampex 8 (Axon Instruments). Events were detected with MiniAnalysis software (Synaptosoft), at a threshold 4x the RMS noise and confirmed by visual inspection. Statistical analysis of average amplitude and frequency was performed using Prism software (Graphpad).
Statistics
Because of variability in sMHCI and glutamatergic synapse density on neurons from different culture preparations, controls were generated and analyzed for every experiment and values from manipulated or treated neurons were normalized to those controls as indicated in the text. Unpaired t-tests were used to determine statistical significance (* = p< 0.05) when there were two data sets to compare and ANOVAs were performed when there were multiple treatment groups from one data set to compare. T-test and ANOVA are the most accurate statistical tests to use for our data since our data sets are usually normally-distributed. Normality was determined using the D’Agostino-Pearson omnibus test in GraphPad Prism software (GraphPad). T-tests and ANOVA are also safe statistical tests to use for our analysis since small deviations from a normal distribution do not usually affect the results.
Supplementary Material
(a) Western blot immunoprobed with two anti-Rt1A MHCI antibodies (OX-18 and ab52922) show similar band patterns. (b–c) Two distinct anti-MHCI antibodies (OX-18 and F16-4-4) show similar immunostaining patterns in cultured 7 d.i.v. rat visual cortical neurons. (d) OX-18 was used to stain non-permeabilized 8 d.i.v visual cortical cultures from rat (top), wild type mice (middle), and β2m−/− mice (bottom). The images show dendritic sMHCI. All images were taken at the same confocal settings for equivalent comparison. The intensity of OX-18 surface staining is drastically decreased in dendrites from β2m−/− mice compared to WT mouse and rat cultures. Scale bar = 5μm. (e) sMHCI staining of a 7 d.i.v. neuron stained without permeabilization (left) and after permeabilization (right). Both the pattern and intensity of sMHCI staining decreases after permeabilization.
To control for possible fixation-induced permeabilization in the experiments shown in Fig. 1, neurons were exposed to primary antibody prior to fixation and then stained as described in Methods in the absence of detergent. (a) Images of dendrites from cortical neurons immunostained for surface MHCI protein at 4, 7, 9 and 14 d.i.v. (top to bottom) are shown proximal to distal from soma (left to right in each image). sMHCI is present on dendrites of all cells at all ages examined. (b) sMHCI clusters on dendrites increase in density with age. (*, p<0.05). At 4, 7 and 10 d.i.v., sMHCI density is higher in proximal dendrites and lower in distal portions of the same dendrites. At 14 d.i.v., this uneven distribution is not observed. (c) The uneven distribution of sMHCI on dendrites is maintained even when quantified as density by area of dendrite (*, significant difference from 4 and 7 d.i.v. density; Ξ, significant difference between distal and same age proximal densities).
Internal MHCI colocalizes with synaptic proteins during the peak of synaptogenesis at 8 d.i.v. (a) and after synapse density has stabilized by 14 d.i.v. (b) in culture. Following permeabilization, neurons were immunostained for MHCI (green), vGlut1 (blue), and the NMDAR subunits, NR2A/B (red). Low magnification image of neurons at each age are shown to the left and then higher magnification images of dendrites from these cells are shown to the right to illustrate the punctate distribution of these proteins. The overlays (bottom higher mag images) illustrate the amount of MHCI that is found at synapses (indicated by arrows in each panel). (c) Approximately 65% of glutamatergic synapses (ES) at 8 d.i.v. contain MHCI, while a little over 50% of these synapses contain MHCI at 14 d.i.v. (d) The percentage of total MHCI clusters found at synapses is about 37% at 8 d.i.v. and 41% at 14 d.i.v. While these data clearly show that MHCI is present at glutamatergic synapses in cultured cortical neurons, it should be noted that the MHCI detected after permeabilization, as in this figure, represents mostly internal MHCI with some residual sMHCI. As shown in Suppl. Fig. 1e, most of the sMHCI is removed from neurons following permeabilization.
Because the mIPSCs recorded −40 and −20mV in Fig. 5 were small and noisy, we performed these experiments again under conditions that cause mIPSCs to be larger in amplitude. Internal and external solutions with close to equal chloride solutions were used to cause chloride to reverse at −18mV and the neurons were voltage-clamped at −70mV for recording. mIPSCs were isolated pharmacologically by including 500 nM TTX, 10 μM CNQX, 50 μM APV in the bath. (a) Representative traces from whole-cell patch-clamp recordings of mIPSCs from 8–10 d.i.v. control cultured cortical neurons or neurons transfected with either β2m siRNA or H2-Kb-CFP. Qualitatively, β2m KD increases, and H2-Kb OE decreases, GABAergic synaptic transmission. (b) Quantitatively, β2m KD significantly increases mIPSC frequency, while H2-Kb-CFP OE decreases it. Neither manipulation changes mIPSC amplitude. These results are similar to those obtained using the distinct recording paradigm described in Fig. 5. n=9 and 7 for β2m siRNA and NTS, respectively and n=3 each for H2-Kb-CFP OE and GFP controls, * = p<0.05.
Although the expression of MHCI protein on the surface of neurons is decreased after 24hr of TTX treatment (Fig. 7), the density of MHCI clusters detected in permeabilized neurons following TTX treatment is increased by about 41% (a), and is readily apparent qualitatively (b). Taken together with data from Fig. 7, these results suggest that TTX causes a rapid internalization of sMHCI to increase the density of MHCI inside the neurons.
Acknowledgments
We are grateful to Dr. M. Edidin (Johns Hopkins University) for the H2-Kb-CFP construct, Dr. J. Fritschy (University of Zurich) for GABA receptor antibodies, and Dr. J. Trimmer (UC Davis) for the PSD-95 antibody. Thanks also to Dr. J. Trimmer and Dr. E. Diaz (UC Davis) for advice and support of the project. This project was funded by a National Institute for Environmental Health Sciences Training Grant (M.W.G.), the John Merck Fund (A.K.M.), the March of Dimes (A.K.M.), Cure Autism Now (A.K.M.), Autism Speaks (A.K.M.) and NINDS R01NS060125 (A.K.M.). We are especially grateful to the Higgins and Gassin Family Foundations for their invaluable support of basic science research and our work.
Footnotes
Author Contributions:
M.W. Glynn initiated this project, conducted most of the experiments using ICC to measure glutamatergic synapse density, and wrote a draft of the manuscript. B.M.E. worked with X.-B. Liu to generate and quantify the electron microscopy images. P.A.G. established whole-cell patch-clamp recording and performed all of the electrophysiology experiments. L.A.N. performed essential control experiments for antibody specificity. F. E-S. performed all of the ICC experiments on GABAergic synapses and most of the TTX/MHC experiments. All authors edited the manuscript. A.K.M. supported all aspects of this project, designed and helped to analyze all experiments, and wrote the manuscript.
References
- 1.Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annual review of neuroscience. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- 2.McAllister AK. Dynamic aspects of CNS synapse formation. Annual review of neuroscience. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flavell SW, et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 4.Margolis SS, et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442–455. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connor TP, et al. Semaphorin 5B mediates synapse elimination in hippocampal neurons. Neural Dev. 2009;4:18. doi: 10.1186/1749-8104-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel MR, Shen K. RSY-1 is a local inhibitor of presynaptic assembly in C. elegans. Science. 2009;323:1500–1503. doi: 10.1126/science.1169025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulanger LM, Shatz CJ. Immune signalling in neural development, synaptic plasticity and disease. Nature reviews. 2004;5:521–531. doi: 10.1038/nrn1428. [DOI] [PubMed] [Google Scholar]
- 8.Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 9.Huh GS, et al. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Needleman LA, Liu XB, El-Sabeawy F, Jones EG, McAllister AK. MHC class I molecules are present both pre- and postsynaptically in the visual cortex during postnatal development and in adulthood. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16999–17004. doi: 10.1073/pnas.1006087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiliotis ET, Pentcheva T, Edidin M. Probing for membrane domains in the endoplasmic reticulum: retention and degradation of unassembled MHC class I molecules. Molecular biology of the cell. 2002;13:1566–1581. doi: 10.1091/mbc.01-07-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams DB, Barber BH, Flavell RA, Allen H. Role of beta 2-microglobulin in the intracellular transport and surface expression of murine class I histocompatibility molecules. J Immunol. 1989;142:2796–2806. [PubMed] [Google Scholar]
- 13.Datwani A, et al. Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron. 2009;64:463–470. doi: 10.1016/j.neuron.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- 16.Sabo SL, McAllister AK. Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nature neuroscience. 2003;6:1264–1269. doi: 10.1038/nn1149. [DOI] [PubMed] [Google Scholar]
- 17.Schnabl E, et al. Activated human T lymphocytes express MHC class I heavy chains not associated with beta 2-microglobulin. The Journal of experimental medicine. 1990;171:1431–1442. doi: 10.1084/jem.171.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madrigal JA, et al. Molecular definition of a polymorphic antigen (LA45) of free HLA-A and -B heavy chains found on the surfaces of activated B and T cells. The Journal of experimental medicine. 1991;174:1085–1095. doi: 10.1084/jem.174.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sassoe-Pognetto M, Panzanelli P, Sieghart W, Fritschy JM. Colocalization of multiple GABA(A) receptor subtypes with gephyrin at postsynaptic sites. J Comp Neurol. 2000;420:481–498. doi: 10.1002/(sici)1096-9861(20000515)420:4<481::aid-cne6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 20.DeFelipe J, Marco P, Busturia I, Merchan-Perez A. Estimation of the number of synapses in the cerebral cortex: methodological considerations. Cereb Cortex. 1999;9:722–732. doi: 10.1093/cercor/9.7.722. [DOI] [PubMed] [Google Scholar]
- 21.Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Demaria S, Schwab R, Bushkin Y. The origin and fate of beta2m-free MHC class I molecules induced on activated T cells. Cellular Immunology. 1992;142 doi: 10.1016/0008-8749(92)90272-q. [DOI] [PubMed] [Google Scholar]
- 23.Chakrabarti A, Matko J, Rahman NA, Barisas BG, Edidin M. Self-association of class I major histocompatibility complex molecules in liposome and cell surface membranes. Biochemistry. 1992;31:7182–7189. doi: 10.1021/bi00146a022. [DOI] [PubMed] [Google Scholar]
- 24.Santos SG, Powis SJ, Arosa FA. Misfolding of major histocompatibility complex class I molecules in activated T cells allows cis-interactions with receptors and signaling molecules and is associated with tyrosine phosphorylation. The Journal of biological chemistry. 2004;279:53062–53070. doi: 10.1074/jbc.M408794200. [DOI] [PubMed] [Google Scholar]
- 25.Bodnar A, et al. Class I HLA oligomerization at the surface of B cells is controlled by exogenous beta(2)-microglobulin: implications in activation of cytotoxic T lymphocytes. International immunology. 2003;15:331–339. doi: 10.1093/intimm/dxg042. [DOI] [PubMed] [Google Scholar]
- 26.Hochman JH, Shimizu Y, DeMars R, Edidin M. Specific associations of fluorescent beta-2-microglobulin with cell surfaces. The affinity of different H-2 and HLA antigens for beta-2-microglobulin. J Immunol. 1988;140:2322–2329. [PubMed] [Google Scholar]
- 27.Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. The Journal of experimental medicine. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAllister AK. Dynamic aspects of cortical synaptogenesis. Annual review of neuroscience. 2007 doi: 10.1146/annurev.neuro.29.051605.112830. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craig AM, Blackstone CD, Huganir RL, Banker G. Selective clustering of glutamate and gamma-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12373–12377. doi: 10.1073/pnas.91.26.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harms KJ, Craig AM. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. The Journal of comparative neurology. 2005;490:72–84. doi: 10.1002/cne.20635. [DOI] [PubMed] [Google Scholar]
- 31.Varoqueaux F, et al. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verhage M, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 33.Bouwman J, et al. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience. 2004;126:115–126. doi: 10.1016/j.neuroscience.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 34.Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 35.Lauri SE, et al. Activity blockade increases the number of functional synapses in the hippocampus of newborn rats. Mol Cell Neurosci. 2003;22:107–117. doi: 10.1016/s1044-7431(02)00012-x. [DOI] [PubMed] [Google Scholar]
- 36.Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- 37.Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- 38.Burrone J, Murthy VN. Synaptic gain control and homeostasis. Current opinion in neurobiology. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Huupponen J, Molchanova SM, Taira T, Lauri SE. Susceptibility for homeostatic plasticity is down-regulated in parallel with maturation of the rat hippocampal synaptic circuitry. J Physiol. 2007;581:505–514. doi: 10.1113/jphysiol.2007.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirov SA, Goddard CA, Harris KM. Age-dependence in the homeostatic upregulation of hippocampal dendritic spine number during blocked synaptic transmission. Neuropharmacology. 2004;47:640–648. doi: 10.1016/j.neuropharm.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 41.Naranda T, Goldstein A, Olsson L. A peptide derived from an extracellular domain selectively inhibits receptor internalization: target sequences on insulin and insulin-like growth factor 1 receptors. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11692–11697. doi: 10.1073/pnas.94.21.11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsson L, Goldstein A, Stagsted J. Regulation of receptor internalization by the major histocompatibility complex class I molecule. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9086–9090. doi: 10.1073/pnas.91.19.9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stagsted J. Journey beyond immunology. Regulation of receptor internalization by major histocompatibility complex class I (MHC-I) and effect of peptides derived from MHC-I. Apmis. 1998;85:1–40. [PubMed] [Google Scholar]
- 44.Fishman D, Elhyany S, Segal S. Non-immune functions of MHC class I glycoproteins in normal and malignant cells. Folia biologica. 2004;50:35–42. [PubMed] [Google Scholar]
- 45.Abbas A, Lichtman AH, Pober J. Cellular and Molecular Immunology. W.E. Saunders Company; Philadelphia, PA: 2000. [Google Scholar]
- 46.Nawa H, Takahashi M, Patterson PH. Cytokine and growth factor involvement in schizophrenia--support for the developmental model. Molecular psychiatry. 2000;5:594–603. doi: 10.1038/sj.mp.4000730. [DOI] [PubMed] [Google Scholar]
- 47.Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Current opinion in neurobiology. 2002;12:115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- 48.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glynn MS, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nature Protocols. 2006 doi: 10.1038/nprot.2006.220. in press. [DOI] [PubMed] [Google Scholar]
- 50.Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nature neuroscience. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Western blot immunoprobed with two anti-Rt1A MHCI antibodies (OX-18 and ab52922) show similar band patterns. (b–c) Two distinct anti-MHCI antibodies (OX-18 and F16-4-4) show similar immunostaining patterns in cultured 7 d.i.v. rat visual cortical neurons. (d) OX-18 was used to stain non-permeabilized 8 d.i.v visual cortical cultures from rat (top), wild type mice (middle), and β2m−/− mice (bottom). The images show dendritic sMHCI. All images were taken at the same confocal settings for equivalent comparison. The intensity of OX-18 surface staining is drastically decreased in dendrites from β2m−/− mice compared to WT mouse and rat cultures. Scale bar = 5μm. (e) sMHCI staining of a 7 d.i.v. neuron stained without permeabilization (left) and after permeabilization (right). Both the pattern and intensity of sMHCI staining decreases after permeabilization.
To control for possible fixation-induced permeabilization in the experiments shown in Fig. 1, neurons were exposed to primary antibody prior to fixation and then stained as described in Methods in the absence of detergent. (a) Images of dendrites from cortical neurons immunostained for surface MHCI protein at 4, 7, 9 and 14 d.i.v. (top to bottom) are shown proximal to distal from soma (left to right in each image). sMHCI is present on dendrites of all cells at all ages examined. (b) sMHCI clusters on dendrites increase in density with age. (*, p<0.05). At 4, 7 and 10 d.i.v., sMHCI density is higher in proximal dendrites and lower in distal portions of the same dendrites. At 14 d.i.v., this uneven distribution is not observed. (c) The uneven distribution of sMHCI on dendrites is maintained even when quantified as density by area of dendrite (*, significant difference from 4 and 7 d.i.v. density; Ξ, significant difference between distal and same age proximal densities).
Internal MHCI colocalizes with synaptic proteins during the peak of synaptogenesis at 8 d.i.v. (a) and after synapse density has stabilized by 14 d.i.v. (b) in culture. Following permeabilization, neurons were immunostained for MHCI (green), vGlut1 (blue), and the NMDAR subunits, NR2A/B (red). Low magnification image of neurons at each age are shown to the left and then higher magnification images of dendrites from these cells are shown to the right to illustrate the punctate distribution of these proteins. The overlays (bottom higher mag images) illustrate the amount of MHCI that is found at synapses (indicated by arrows in each panel). (c) Approximately 65% of glutamatergic synapses (ES) at 8 d.i.v. contain MHCI, while a little over 50% of these synapses contain MHCI at 14 d.i.v. (d) The percentage of total MHCI clusters found at synapses is about 37% at 8 d.i.v. and 41% at 14 d.i.v. While these data clearly show that MHCI is present at glutamatergic synapses in cultured cortical neurons, it should be noted that the MHCI detected after permeabilization, as in this figure, represents mostly internal MHCI with some residual sMHCI. As shown in Suppl. Fig. 1e, most of the sMHCI is removed from neurons following permeabilization.
Because the mIPSCs recorded −40 and −20mV in Fig. 5 were small and noisy, we performed these experiments again under conditions that cause mIPSCs to be larger in amplitude. Internal and external solutions with close to equal chloride solutions were used to cause chloride to reverse at −18mV and the neurons were voltage-clamped at −70mV for recording. mIPSCs were isolated pharmacologically by including 500 nM TTX, 10 μM CNQX, 50 μM APV in the bath. (a) Representative traces from whole-cell patch-clamp recordings of mIPSCs from 8–10 d.i.v. control cultured cortical neurons or neurons transfected with either β2m siRNA or H2-Kb-CFP. Qualitatively, β2m KD increases, and H2-Kb OE decreases, GABAergic synaptic transmission. (b) Quantitatively, β2m KD significantly increases mIPSC frequency, while H2-Kb-CFP OE decreases it. Neither manipulation changes mIPSC amplitude. These results are similar to those obtained using the distinct recording paradigm described in Fig. 5. n=9 and 7 for β2m siRNA and NTS, respectively and n=3 each for H2-Kb-CFP OE and GFP controls, * = p<0.05.
Although the expression of MHCI protein on the surface of neurons is decreased after 24hr of TTX treatment (Fig. 7), the density of MHCI clusters detected in permeabilized neurons following TTX treatment is increased by about 41% (a), and is readily apparent qualitatively (b). Taken together with data from Fig. 7, these results suggest that TTX causes a rapid internalization of sMHCI to increase the density of MHCI inside the neurons.