

Abstract
Purpose
The inhibition of c-Src results in a striking reduction in cancer cell invasion, but the effect on cell survival is modest. Defining mechanisms that limit apoptosis following c-Src inhibition could result in an ideal therapeutic approach that both inhibits invasion and leads to apoptosis. In this regard, we discovered a novel feedback loop that results in STAT3 reactivation following sustained c-Src inhibition. Here we define the mechanism underlying this feedback loop and examine the effect of inhibiting it in vivo.
Methods
We measured levels and activity of pathway components using PCR, Western blotting, and kinase assays following their manipulation using both molecular and pharmacologic approaches. We utilized a heterotransplant animal model in which human oral squamous cancer is maintained exclusively in vivo.
Results
Following c-Src inhibition, STAT5 is durably inhibited. The inhibition of STAT5A, but not STAT5B, subsequently reduces the expression of suppressors of cytokine signaling 2 (SOCS2). SOCS2 inhibits Janus kinase 2 (Jak2) activity and Jak2-STAT3 binding. SOCS2 expression is necessary for STAT3 inhibition by c-Src inhibitors. Overexpression of SOCS2 is adequate to prevent STAT3 reactivation and to enhance the cytotoxic effects of c-Src inhibition. Likewise, the combination of Jak and c-Src inhibitors led to significantly more apoptosis than either agent alone in vivo.
Conclusions
To our knowledge, ours is the first study that fully defines the mechanism underlying this feedback loop, in which sustained c-Src inhibition leads to diminished SOCS2 expression via sustained inhibition of STAT5A, allowing activation of Jak2 and STAT3, Jak2-STAT3 binding, and survival signals.
Keywords: STAT3, Src, JAK, SOCS2, STAT5
Introduction
One potential and promising therapeutic cancer target is c-Src, given its well-defined roles in promoting cell migration and metastasis as well as regulating proliferation, survival, and angiogenesis. The Src family kinases (SFKs) are nonreceptor tyrosine kinases involved in signal transduction in both normal and cancer cells (1). c-Src is the SFK that is most often implicated in cancer progression. Inhibition of c-Src results in a nearly universal reduction in invasion of cancers in vitro and in vivo (2, 3). However, despite c-Src expression and activation in epithelial tumors and c-Src’s robust inhibition by clinically relevant agents, the effect of c-Src inhibition on epithelial cancer cell survival and proliferation has been modest (3). A clinical trial of the SFK inhibitor dasatinib as a single agent in head and neck squamous cell carcinoma (HNSCC) did not demonstrate significant activity (4). Current treatment for HNSCC includes a combination of cytotoxic chemotherapy, radiotherapy, and surgery. Cetuximab enhances the efficacy of chemotherapy and radiotherapy, but no kinase inhibitors are currently a standard of care for HNSCC. Although invasion is important in the pathophysiology of many cancers, local invasion is a critical determinant of both morbidity and mortality for HNSCC and is associated with worse locoregional control and decreased survival. There is a great need to improve systemic therapy to treat both local recurrence and distant metastatic disease. Thus, defining mechanisms that limit the pro-apoptotic effects of c-Src inhibitors could result in an ideal combination of therapeutic agents that both inhibit local invasion and lead to significant cytotoxicity.
Because signal transducers and activators of transcription (STATs) are known to be c-Src substrates and can mediate c-Src’s biologic effects (5), we explored the potential role of STATs in modulating the biologic effects of c-Src inhibition. The STAT family of transcription factors, especially STAT3 and STAT5, regulates oncogenic signaling in many different tumor types. In HNSCC cells, c-Src’s inhibition results in reduced STAT3 and STAT5 activation and reduced cell proliferation (6). Correspondingly, inhibition of STAT3 in HNSCC leads to increased apoptosis, decreased proliferation, and decreased tumor size (7, 8). However, we found that whereas inhibition of c-Src led to durable inhibition of STAT5, c-Src’s inhibition of STAT3 was only transient, with levels of phosphoSTAT3 (pSTAT3, Y705) returning to baseline or above by 7 hours. We confirmed this finding by reducing c-Src specifically with small interfering RNAs (siRNAs) and by measuring STAT3 activity using DNA binding and transcriptional activity assays (9). We also established the biologic importance of this feedback loop by demonstrating that abrogation of STAT3 reactivation enhanced the cytotoxicity, cell cycle arrest, and apoptosis caused by c-Src inhibition in vitro. These findings established that the STAT3 compensatory pathway is important for maintaining cancer cell proliferation and survival after sustained c-Src inhibition. Furthermore, the depletion of STAT3 by an siRNA reduced the 50% inhibitory concentration (IC50) of the c-Src inhibitor dasatinib from 23 nM to 4 nM, increasing sensitivity to levels comparable with those observed after inhibition of Bcr-Abl in leukemia.
In addition to regulation by c-Src, STAT3 can be activated by the nonreceptor tyrosine kinases Jaks. Following activation, Jak molecules phosphorylate cytokine receptors, thus allowing the binding of the monomeric inactive STATs present in the cytoplasm. STATs then become Jak substrates and the pSTATs undergo dimerization and nuclear translocation. In HNSCC cells, Jak inhibition or knockdown completely and durably blocked both basal activation of STAT3 and subsequent reactivation of STAT3 following c-Src inhibition (9, 10). Consistent with the effects of c-Src inhibition on STAT3 activity, c-Src inhibition resulted in initial inhibition and then recovery of Jak2 kinase activity, confirming that the reactivation of STAT3 is mediated by Jak reactivation.
Although there are no known positive feedback loops leading to Jak activation after its inhibition, loss of a negative feedback loop could play such a role. There are three canonical negative feedback loops that regulate Jak/STAT function after cytokine signaling: SH-2– containing phosphatases (SHPs), which inactivate Jak by dephosphorylation; protein inhibitors of activated STAT (PIAS), which are negative regulators of STAT transcription downstream; and SOCS, which inhibit Jak kinase activity, facilitate proteasomal degradation of Jak, and reduce STATs binding to cytokine receptors (5).
The mechanism by which sustained c-Src inhibition allows Jak reactivation is unknown. We observed changes in Jak activity and Jak-STAT binding following c-Src inhibition that suggest SOCS proteins to be the most likely candidates for regulating Jak/STAT function in this setting. Our hypothesis is that the inactivation of STAT5 caused by sustained c-Src inhibition suppresses the expression of one or more of the SOCS proteins. This loss allows recovery of Jak2-STAT3 binding and Jak2 kinase activity and relieves STAT3 inhibition, thereby reactivating proliferative signals through Jak2 and STAT3. Additionally, the two STAT5 isoforms (A and B) are known to have distinct roles in cancer and in embryonic development, but the roles of these isoforms in this feedback loop have never been explored (5, 11). Understanding the basis for STAT3 reactivation is essential to maximizing the anti-apoptotic effect of c-Src inhibitors.
To test our hypothesis, we measured the levels of all known SOCS family members following c-Src knockdown or inhibition with the ATP-competitive SFK inhibitor, dasatinib, and found that SOCS2 expression was consistently decreased. To further define this novel feedback loop, we manipulated the levels of SOCS2, STAT3, STAT5A, and STAT5B to demonstrate that c-Src inhibition leads to STAT5 inactivation, that STAT5A drives SOCS2 protein expression, and that SOCS2 inhibits Jak2-STAT3 binding, Jak activity, and STAT3 activation. We previously demonstrated that c-Src inhibition did not affect total levels of Jak2 protein (9). Moreover, SOCS2 loss caused increased resistance to dasatinib, and SOCS2 overexpression led to increased sensitivity to c-Src inhibitors. We confirmed the biological importance of this feedback pathway using a heterotransplant model of HNSCC and clinically relevant inhibitors of Jak and c-Src.
Materials and Methods
Cells and reagents
Dasatinib was purchased from Selleck Chemicals and the clinical pharmacy. INCB016562 was provided by Incyte Corporation. Both were prepared as 10 mmol/L stock solutions in DMSO. Antibodies used included c-Src, pSFK (Y416), pSTAT3 (Y705), pJak2 (Y221), pJak2 (Y1007/1008), pSTAT5 (Y694) XP, and SOCS2 (Cell Signaling Technology); total phosphotyrosine and total STAT5B (Upstate Biotechnology); SOCS1 and total Jak2 (BD Biosciences, Franklin Lakes, NJ); total STAT5A (Abcam); and β-actin (Sigma Chemical).
Human HNSCC cell lines were obtained from Dr. Jeffrey Myers and maintained as described previously (9). All cell lines were validated by cross-comparing their allelic short tandem repeat profiling (Johns Hopkins Fragment Analysis Core Facility) and patterns generated with the PowerPlex 1.2 platform (Promega) to those from the American Type Culture Collection repository database.
Western blot analysis and immunoprecipitation
Western blot analysis and immunoprecipitation were performed as previously described (3, 9). Briefly, for immunoprecipitation, cells were lysed and equal amounts of protein cell lysates (800 μg) were precleared with protein A-G-sepharose beads (Invitrogen) for 1 hour. The precleared lysate was incubated with 5 μg agarose-conjugated primary antibody overnight. The immunocomplexes were washed and resolved by SDS-PAGE. Following transfer to nitrocellulose membranes, immunoblots were probed with primary antibody and proteins detected with horseradish peroxidase-conjugated secondary antibody (Bio-Rad Laboratories) and enhanced chemiluminescent reagent (Pierce Biotechnology).
Cytotoxicity assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess cytotoxicity as previously described (3). Eight wells were treated for each experimental condition.
Transfection with siRNA and recombinant plasmids
siRNAs were predesigned sets of four independent sequences (siGENOME SMARTpool, Dharmacon). Controls included cells that were mock transfected (no siRNA) and those transfected with a nontargeting (scrambled) siRNA. The pUSE STAT5A 1*6, pUSE STAT5B 1*6 recombinant plasmids and pMet7-FLAG-mSOCS2 constructs were used to achieve overexpression of STAT5A and/or STAT5B and mouse full-length SOCS2, respectively, in cells. Mouse SOCS2 shows 94% identity and 95% amino acid sequence similarity with human SOCS2. Cells were harvested, washed, and suspended (106/100μL) in Nucleofector V solution (Lonza Group). siRNA (200 pmol/100μL), DNA (5 μg/100μL), or controls were added and electroporated using the U-31 Nucleofector program (Lonza) as described previously (9).
Quantitative PCR (qPCR)
Total RNA was isolated from cells that had been either transfected with siRNAs or incubated with dasatinib (as indicated in the figure legends) by using an RNeasy mini kit (Qiagen). Total RNA (2 μg) was converted into cDNA using 1× MMLV buffer, 1 μL RNasin, 10 μM random hexamer, 500 μM deoxyribonucleotide triphosphates, 100 mg/mL BSA, and 1.5μL MMLV reverse transcriptase enzyme. The final reaction volume was 20 μL. The reaction mixture was incubated at 42°C for 2 hours, and the reaction was terminated by heating the mixture at 99°C for 5 minutes and cooling it at 5°C for 5 minutes.
The level of mRNA for the SOCS genes was measured with SYBR green-based real-time PCR in triplicate. The primers were designed by using Primer Express (Applied Biosystems) (Supplemental Table 1). Each cDNA sample was amplified by using SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's suggested protocol. The PCR products and their dissociation curves were detected using the ABI Prism 7500 fast real-time PCR system. The level of the housekeeping gene L32 ribosomal gene (Rpl32) was used as an internal control. Individual data sets were normalized with control vehicle-treated cells; absolute quantities were normalized with L32 as internal control.
In vitro kinase assay
Purified recombinant Jak2 (Abcam) and SOCS2 (Abnova) proteins were incubated at a 1:1 molar stoichiometric ratio with 15 μCi [γ-32P]ATP (3,000 Ci/mmol), and kinase activity was assayed as described previously (9).
Xenograft nude mouse models
All animal procedures were in accordance with the policies of MD Anderson's Institutional Animal Care and Use Committee. For the orthotopic models, the tongues of five 6-week-old female Swiss nu/nu mice were injected with 5 × 105 Osc19 cells. For the heterotransplant studies, residual tumor from a patient with untreated oral squamous carcinoma was identified by a head-and-neck pathologist (AE) at the time of surgical resection and implanted directly into the flank of a nude mouse. The resulting tumor was divided and transplanted into subsequent mice until 40 fifth-generation tumors were produced. The heterotransplant tumors were never cultured in vitro. Dasatinib (20 mg/kg), INCB016562 (60 mg/kg), both, or vehicle was administered by oral gavage daily for 7 days (orthotopic) or 17 days (heterotransplant). Mice were killed 2 hours after the last drug dose, tumors were dissected, and the mice were examined for distant metastases. The tumors were homogenized and subjected to Western blot analysis as described previously (12).
Immunohistochemistry analysis
Immunohistochemical staining was performed as previously described using the following specific conditions: antigen retrieval was performed using a Dako-Target retrieval at pH 6.0 for PCNA, CD31 (Abcam), and pSFK (Y416) (Cell Signaling Technology). Peroxide blocking was performed using 3% methanol and hydrogen peroxide (PCNA and CD31) or 3% water and hydrogen peroxide (pSFK Y416) (13). Primary antibody dilutions were: PCNA (1:100), CD31 (1:50), and pSFK (1:50). Slides were examined by a blinded observer for the intensity and extent of immunostaining by light microscopy using a ×20 magnification objective. Nuclear PCNA expression was quantified using a three-value intensity: 0, none; low (1+, weak and 2+, moderate); and high (3+, strong and +4 very strong). CD31-positive vessels were counted in five high-powered fields by a blinded observer.
TUNEL assay
TUNEL staining was performed using the DeadEnd Colorimetric TUNEL system from Promega per the manufacturer’s instructions as previously described (13). TUNEL-positive nuclei were counted for each representative treatment group.
Statistical Methods
All experiments in which error bars and p values are provided were done in at least triplicate. The Student’s T test was used to determine if the mean values of these continuous variables were different in the various treatment groups.
Results
c-Src inhibition leads to decreased SOCS2 expression and STAT5 inactivation
We postulated that the loss of one of the SOCS proteins could contribute to STAT3 reactivation after sustained c-Src inhibition. To test this hypothesis, we determined the expression level of all members of the SOCS family after 7 hours of c-Src inhibition with dasatinib using qPCR analysis in a panel of six different HNSCC cell lines (Fig. 1A). Among the eight family members of SOCS proteins, only SOCS2 showed consistent downregulation in all six cell lines. We also measured the expression of the four PIAS family members but found no significant alteration in PIAS expression following dasatinib treatment (representative data, Fig. S1). STAT3 reactivation was not mediated by an autocrine mechanism such as cytokine release (Fig. S2).
Figure 1.



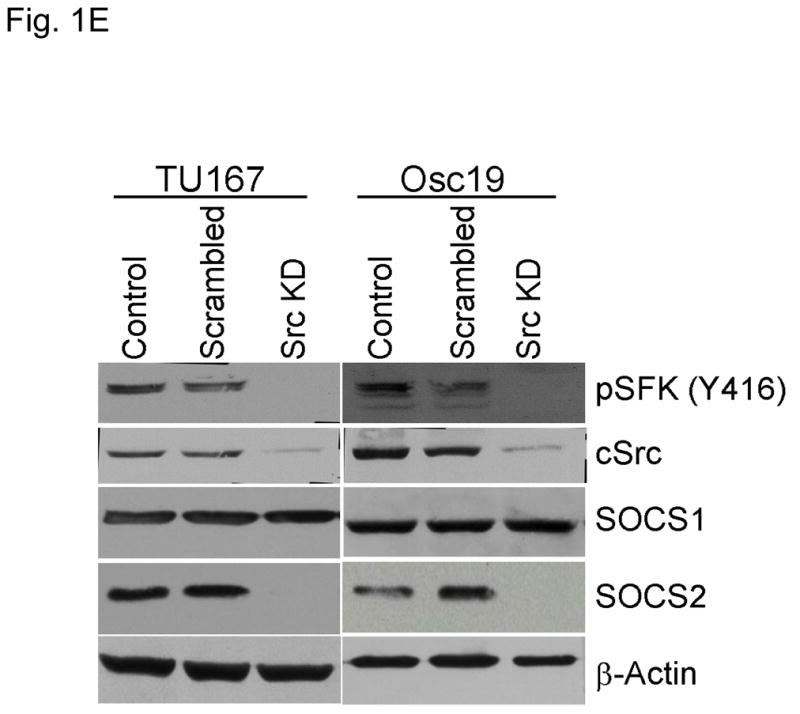
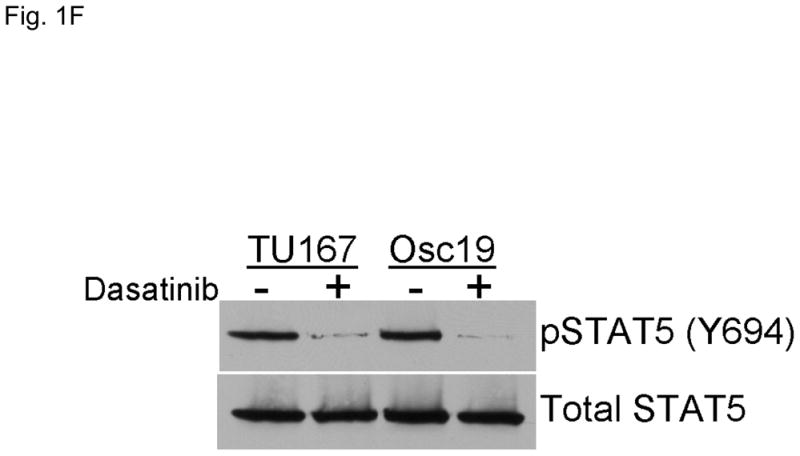
c-Src inhibition leads to decreased SOCS2 expression and STAT5 activation. (A) Six HNSCC cell lines were incubated with 100 nM dasatinib for 7 hours, and mRNA levels of all the known SOCS molecules were measured by qPCR and expressed as fold control (vehicle treatment). # 6.2-fold control. † SOCS5 was not detectable in four cell lines. *P<0.05. (B) TU167 and Osc19 cell lines were incubated with 100 nM dasatinib for the indicated times, lysed, and analyzed by Western blot analysis with the indicated antibodies. (C–E) TU167 and Osc19 cells were transfected with nontargeting siRNA or c-Src–specific siRNA. The mRNA levels of all the SOCS molecules were measured by qPCR analysis (C–D) or Western blot analysis (E). (F) TU167 and Osc19 cell lines were treated with 100 nM dasatinib for 7 hours, and lysates were subjected to immunoprecipitation with the total STAT5 antibody. Immunoprecipitates were resolved on 8% SDS-PAGE and blotted with the pSTAT5 (Y694) monoclonal antibody. The data in A, C, and D are presented as an average of three replicates; bars indicate the standard deviation. *P<0.05.
To characterize the effect of c-Src inhibition on SOCS2 protein expression, we examined the effect of dasatinib in two representative HNSCC cell lines, that grow well both in vitro and in vivo, using Western blot analysis (Fig. 1B). As expected, c-Src phosphorylation was rapidly and durably inhibited at a site associated with its activation (pSFK, Y416). SOCS2 protein expression was significantly downregulated after sustained c-Src inhibition.
To determine whether SOCS2 expression is downstream of c-Src specifically, we transfected HNSCC cells with siRNAs specific to c-Src and examined the effect on SOCS family members’ mRNA (Fig. 1C–D) and protein (Fig. 1E) expression. Upon c-Src depletion, the levels of SOCS2 mRNA and protein decreased significantly. In addition to SOCS2, CIS1 expression was decreased following c-Src knockdown (Fig. 1C–D), but CIS1 was not consistently affected by incubation with dasatinib (Fig. 1A). These experiments demonstrate that c-Src activation is upstream of SOCS2 transcription.
Given that STAT5 can regulate SOCS2 expression, we investigated whether c-Src could regulate STAT5 activation in HNSCC cell lines. We incubated cells with dasatinib for 7 hours and measured pSTAT5 (Y694). c-Src inhibition rendered STAT5 durably inactive which is consistent with our previous results demonstrating STAT5 inhibition from 2 – 24 h following dasatinib treatment (Fig. 1F) (3).
SOCS2 expression is regulated by STAT5A but not STAT3 or STAT5B
Previous reports showed that STAT5 can act as a transcriptional regulator for SOCS family proteins in hematopoietic cells (14, 15). We sought to determine whether the modulation of STAT5 activity regulates SOCS2 expression in HNSCC cells. HNSCC cell lines express both isoforms of STAT5 (STAT5A and STAT5B) and their roles may be distinct (11). Likewise, we found that selective STAT5A knockdown using siRNA led to a considerable decrease in SOCS2 expression, whereas STAT5B depletion alone had little effect on SOCS2 expression (Fig. 2A). In contrast, selective STAT3 depletion with siRNA did not affect SOCS2 expression (Fig. 2B).
Figure 2.

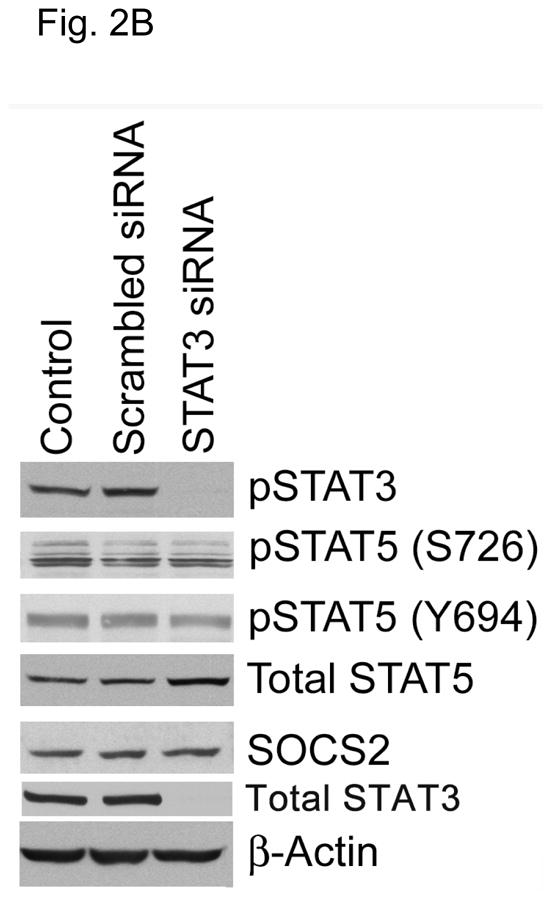

STAT5A regulates SOCS2 expression in HNSCC cells. (A) TU167 and Osc19 cells were transfected with nontargeting scrambled siRNA or STAT5A- or STAT5B-specific siRNA either alone or in combination (KD, knockdown). Cells were harvested and lysed after 72 hours of transfection, and the indicated molecules were visualized using Western blot analysis. SOCS1 and SOCS2 expression was normalized with that of β-actin (bar graph). (B) TU167 cells were transfected with STAT3-specific siRNA and analyzed by Western blot analysis 72 hours later. (C) TU167 and Osc19 cells were transiently transfected with empty vector, CA-STAT5A or CA-STAT5B (OE, overexpression), or both. Cells were harvested and lysed after 72 hours of transfection, and the indicated molecules were visualized using Western blot analysis. The terms lighter and darker refer to film exposure. SOCS1 and SOCS2 expression was normalized with that of β-actin (bar graph).
To further elucidate the function of the STAT5 isoforms in the regulation of SOCS2 expression and STAT3 activation, we selectively overexpressed constitutively active forms of both STAT5 isoforms. STAT5A activation led to increased expression of SOCS2 but not SOCS1 (Fig. 2C). Likewise, STAT5A overexpression resulted in decreased activation of STAT3, thus supporting our hypothesis that STAT5A regulates SOCS2 expression, which subsequently acts as a negative regulator of STAT3 activation. In contrast, STAT5B overexpression alone did not significantly alter basal SOCS2 protein levels or pSTAT3 (Y705) expression.
Selective knockdown of SOCS2 leads to STAT3 activation
To determine whether SOCS2 downregulation could lead to STAT3 activation, we selectively decreased SOCS2 expression in HNSCC cell lines using siRNA. Upon SOCS2 knockdown, STAT3 phosphorylation increased markedly by 4.6- and 4.8-fold in TU167 and Osc19 cell lines, respectively, over that in control cells (Fig. 3A). This result supports our hypothesis that SOCS2 has a negative regulatory role in the Jak2-STAT3 signaling pathway. Total Jak2 protein levels were also increased by SOCS2 knockdown, a result consistent with the known role of SOCS in promoting Jak protein degradation. In our previous work, however, we did not observe changes in total Jak2 levels following dasatinib treatment or c-Src knockdown (9).
Figure 3.





SOCS2 expression negatively regulates STAT3 activation. (A–B) TU167 and Osc19 cells were mock-transfected with no siRNA, nontargeting (scrambled), or SOCS2-specific siRNA (KD, knockdown). Cells were subjected to no further treatment (A) or incubated with 100 nM dasatinib for 30 minutes before lysis (B) and then harvested and lysed. The indicated molecules were visualized using Western blot analysis. (C) TU167 and Osc19 cell lines were transiently transfected with the pMet7-FLAG -mSOCS2 overexpression construct (SOCS2 OE) or vector control. Cells were harvested and lysed 72 hours after transfection, and the indicated molecules were analyzed by Western blot analysis. TU167 (D) and Osc19 (E) cells were transfected with SOCS2 OE. Cells were treated with 100 nM dasatinib for the indicated times starting 48 hours after transfection, lysed, and analyzed by Western blot analysis.
SOCS2 depletion results in sustained STAT3 activation despite acute c-Src inhibition
Our previous experiments have demonstrated that acute c-Src inhibition results in transient STAT3 inactivation (10). We hypothesized that early SOCS2 depletion would allow STAT3 to remain activated despite acute c-Src inhibition. To test this hypothesis, we examined the effect of dasatinib on STAT3 reactivation in cells with depleted SOCS2. As we showed previously, TU167 cells incubated with dasatinib showed significant downregulation of STAT3 phosphorylation 30 minutes after treatment (SOCS2 levels were not affected at this early timepoint). In contrast, SOCS2-depleted TU167 cells had incomplete inhibition of STAT3 phosphorylation at 30 minutes after dasatinib treatment (Fig. 3B). This result demonstrates that SOCS2 expression is required for STAT3 inhibition by c-Src. In contrast, STAT5 was inhibited by dasatinib independently of SOCS2 expression.
SOCS2 overexpression leads to STAT3 inhibition
To further explore the role of SOCS2 as a negative regulator of STAT3, we transiently overexpressed SOCS2, which resulted in significant sustained decreases in both STAT3 and Jak2 activation while leaving total STAT3, SOCS1, and pSFK levels unchanged (Fig. 3C). To determine the effect of forced SOCS2 expression following sustained c-Src inhibition, we transfected Osc19 and TU167 cells with either SOCS2 or empty vector and exposed them to dasatinib for 30 minutes to 7 hours (Fig. 3D and E). The overexpression of SOCS2 significantly diminished the basal activation and reactivation of STAT3 compared with controls.
SOCS2 expression mediates sensitivity and resistance to c-Src inhibition
To determine the biological significance of SOCS2 in this feedback loop, we transiently overexpressed or knocked down SOCS2 and estimated cytotoxicity in the presence of the c-Src inhibitor dasatinib (Fig. 4). SOCS2 knockdown led to increased resistance to dasatinib in both HNSCC cell lines compared with results in controls (Fig. 4A and C). In contrast, overexpression of SOCS2 in either line led to increased sensitivity to c-Src inhibition (Fig. 4B and D). The basal differences in dasatinib sensitivity between Osc-19 and TU167 cells are likely due to distinct interactions between c-Src and c-Met (16).
Figure 4.

SOCS2 expression levels affect sensitivity to c-Src inhibition. TU167 (A, B) and Osc19 (C, D) cells were mock-transfected (no siRNA) or transfected with either nontargeting siRNA or SOCS2-specific siRNA (A, C), empty vector, or pMet7-FLAG-mSOCS2 overexpression construct (B, D). Twenty-four hours after transfection, cells were seeded in 96-well plates for the MTT assay. Cell viability was assayed in the presence of increasing concentrations of dasatinib for 48 hours (B, D) or 72 hours (A, C). Data are presented as the average of eight replicates; bars indicate the standard deviation.
Although the manipulation of SOCS2 expression affected sensitivity to c-Src inhibition in a predictable manner, we were concerned that the biologic effects of STAT5 modulation might not parallel what we observed with direct SOCS2 manipulation, because STAT5 itself can promote cancer cell survival and proliferation in HNSCC (17). We transfected cells with constitutively active STAT5A or B or both and then measured cytotoxicity in the presence of dasatinib. HNSCC cells that overexpressed STAT5A (and had increased SOCS2 and decreased STAT3 activation, Fig. 2C) were slightly more sensitive to dasatinib. However, those cells overexpressing STAT5B (no change in SOCS2) or both isoforms (increased SOCS2) were more resistant to dasatinib (Fig. S3A), suggesting that STAT5B promotes cancer survival through an independent mechanism. In TU167 cells, STAT5A and B knockdown led to a modest increase in sensitivity to dasatinib, whereas in Osc19 cells, this observation was reversed (Fig. S3B). Because dasatinib causes STAT5 inhibition, it is not surprising that STAT5 knockdown does not have a striking effect on dasatinib-induced cytotoxicity.
SOCS2 inhibits Jak2-STAT3 binding and Jak2 kinase activity
Previous reports have demonstrated that SOCS family members bind to Jaks and inhibit their kinase activity, as well as compete with STAT molecules for recruitment to the receptor complex (18). To determine whether SOCS2 affects Jak2-STAT3 binding in HNSCC cells, we overexpressed SOCS2 in TU167 cells and immunoprecipitated total Jak2; immunocomplexes were analyzed by immunoblotting (Fig. 5A and S4). When SOCS2 was overexpressed, Jak2-STAT3 binding was significantly decreased.
Figure 5.

SOCS2 regulates Jak2-STAT3 binding and Jak2 kinase activity. (A) Lysates from control and SOCS2-overexpressing cells were immunoprecipitated with Jak2 antibodies, and immunoprecipitated complexes were resolved on SDS-PAGE. The indicated molecules were visualized using Western blot analysis. (B) Purified recombinant SOCS2 and Jak2 proteins were incubated at room temperature in the presence of 15 μCi γ32-ATP and the exogenous substrate enolase for 30 minutes. Reactions were terminated with the addition of sample buffer, boiled for 5 minutes, and separated on an 8% SDS-PAGE. Radiolabeled proteins were detected by autoradiography.
To determine whether SOCS2 can directly affect Jak2 activity, we performed an in vitro kinase assay in which purified Jak2 and SOCS2 proteins were incubated together at a 1:1 molar stoichiometric ratio with ATP; we detected phosphorylated molecules by autoradiography (Fig. 5B). In the presence of SOCS2, Jak2 autophosphorylation and activity toward an exogenous substrate (enolase) were significantly inhibited. As expected, SOCS2 alone showed no kinase activity. These observations confirm that SOCS2 acts as a negative regulator of Jak2-STAT3 signaling by inhibiting Jak2 activity as well as Jak2-STAT3 binding.
Jak inhibition enhances the anti-tumor effects of c-Src inhibition in vivo
To determine whether the reactivation of STAT3 is biologically significant in vivo, we utilized a heterotransplant model of HNSCC in which an oral squamous carcinoma tumor was transplanted directly into a mouse. The resulting tumor was divided and serially passaged into mice; the tumors were never cultured in vitro. The resulting tumors maintained the histological characteristics of the primary tumor from which they were derived (Fig. S5). Heterotransplants maintain the gene expression profiles of the original tumors and their pattern of response to chemotherapy resembles those observed in the clinic (19), suggesting that this model may be superior to other xenograft approaches for therapeutic studies. Both dasatinib and the Jak inhibitor INCB16562 modestly inhibited tumor growth; the combination was significantly more effective than the single agents (Fig. 6A).Likewise, the tumors treated with the combination had significantly more apoptosis (Fig. 6B) and less proliferation (Fig. 6C, Table S2). Consistent with our in vitro results, c-Src inhibition did not result in STAT3 inhibition, but Jak inhibition abrogated STAT3 activation (Fig. 6D); c-Src was inhibited in vivo by dasatinib (Fig. 6E). Tumor microvessels were stained with CD31 and counted; the tumors from mice treated with dasatinib, INCB16562, and the combination had lower microvessel density compared with controls (0.68-, 0.65-, and 0.64-fold control levels, respectively), but the differences were not statistically significant (P values of 0.12, 0.11, and 0.06, respectively).
Figure 6.




The combination of SFK and Jak inhibition resulted in increased cytotoxic effects in vivo. Mice bearing heterotransplanted oral squamous carcinoma were treated with vehicle alone, dasatinib alone, INCB16562 alone, or the two agents combined, and tumor size was measured on the indicated days (A). (B) TUNEL assay was performed with heterotransplanted tumor tissues, and TUNEL-positive nuclei were counted and calculated as the percentage of total nuclei. (C) Tumor tissues were stained for PCNA. Nuclear staining was classified as low, intermediate, or high grade. (D–E) Tumors from treated mice were subjected to Western blotting with the indicated antibodies (D) or immunohistochemical staining with an anti-pSFK (Y416) Ab (E) (*P < 0.05).
We also utilized an orthotopic HNSCC model in which Osc19 cells were implanted into the tongue. Mice were treated with dasatinib or INCB016562 or the combination for 7 days. Tumors consisted primarily of HNSCC cells (>90%) with no distant metastases. As expected, dasatinib treatment inhibited c-Src, and STAT3 remained activated (1.7-fold) over the control level. In the presence of INCB016562, pSTAT3 reactivation upon dasatinib treatment was significantly reduced to 0.2-fold (Fig. S6).
Discussion
Our current findings define the mechanism underlying a novel feedback loop in which sustained c-Src inhibition or knockdown leads to diminished SOCS2 expression via the sustained inhibition of STAT5A. This relieves the negative constitutive inhibition of SOCS2 on the Jak2-STAT3 pathway, specifically allowing the activation of Jak2 kinase activity, Jak2-STAT3 binding, and STAT3 activation. Although SOCS2 can affect Jak2 protein levels by promoting protein degradation, in our previous studies we observed no changes in total Jak2 expression following c-Src inhibition or knockdown. Ultimately, the loss of SOCS2 expression leads to the reactivation of proliferative signals through STAT3 despite sustained c-Src inhibition (Fig. S7).
Although it is well established that SOCS proteins can inhibit Jak/STAT function, we are aware of only one other study in which altered signaling led to the loss of SOCS function with subsequent Jak/STAT activation and cancer promotion (20). Jak1 activation is important for v-Abl-induced transformation of pre-B cells. In nontransformed cells, the induction of SOCS1 acts as a negative feedback loop to suppress Jak/STAT function, but v-Abl phosphorylates SOCS1 and inhibits its targeting of Jak1 for degradation. Thus, v-Abl’s inhibition of SOCS1 allows sustained Jak1 and STAT5 activation, contributing to cytokine independence in the transformed cells. Our study showed a distinct role for a SOCS protein in regulating Jak/STAT function; in HNSCC, SOCS2 was regulated at the transcriptional level and not by post-translational modification and degradation.
SOCS proteins have been most extensively studied in normal immune function and hematologic malignancies, where they function as classic mediators of a negative feedback loop downstream of cytokine receptors (21). The roles of SOCS proteins in epithelial cancers are not as well known, although studies support a tumor-suppressor role for SOCS proteins via Jak/STAT suppression in nonhematologic malignancies. In this context, SOCS1 and SOCS3 are the most extensively studied, although the loss of SOCS2 can promote intestinal growth, polyp formation, and colon cancer progression (22–24). The expression of SOCS1, which is downregulated via methylation in about a third of HNSCC tumors, can inhibit STAT3 activation by Jak in HNSCC cell lines (25). In those cell lines with SOCS1 expression, STAT3 was shown to be activated via EGFR; in those lines lacking SOCS1, STAT3 was activated via IL6 and Jak. The effects of SOCS1 on STAT5 were not examined (25). SOCS3 is commonly hypermethylated and downregulated in HNSCC tumors; its overexpression in HNSCC cell lines leads to apoptosis (26). SOCS3 is also hypermethylated in lung cancer cell lines and tissues (27). In melanoma, the SOCS1 expression was decreased and STAT3 and Jak2 expression increased compared with primary tumor cells. Restoration of SOCS1 expression leads to STAT3 inactivation and inhibition of brain metastasis (28). Similarly, exogenous expression of SOCS1, SOCS3, or SOCS5 in thyroid cancer cells reduces STAT3 phosphorylation and sensitizes cells to chemotherapy in vitro and in vivo (29).
In our experiments, SOCS2 had a function distinct from its classically understood role described in hematopoietic cells (21). SOCS2 did inhibit Jak2 kinase activity but does not contain the classic kinase inhibitory region that SOCS1 and SOCS3 proteins possess (21). However, our study was limited in that we used isolated recombinant proteins that may function differently from native proteins in an intact cell. SOCS2 also is classically understood to promote the degradation of Jak2, yet we did not observe changes in total STAT3 or Jak2 levels in HNSCC cells following prolonged c-Src inhibition or knockdown (9). However, we did observe that SOCS2 knockdown led to increased Jak2 expression, demonstrating that SOCS2 is capable of this classical function in HNSCC cells.
SOCS2 expression is dependent upon STAT5 (30). There are at least 5 STAT5A binding sites in the SOCS2 promoter (intron 1)1 (31). STAT5A and STAT5B share similar binding sequences (31). Given the high level of homology between STAT5A and STAT5B, it is not clear how the two could be differentially regulating SOCS2 expression based exclusively on sequence data. Another layer of complexity in the regulation of SOCS function is that SOCS2 may compete with or regulate other SOCS proteins. SOCS2 can lead to proteasome-dependent SOCS3 degradation (32). Such a complex system of inter-regulation may explain why we observed diverse effects on the levels of multiple SOCS proteins in HNSCC cell lines following c-Src inhibition.
Although STAT5A and STAT5B may possess some functional redundancy, their roles in both normal physiology and cancer biology are distinct. Their separate roles in normal physiology are demonstrated by discrete tissue expression patterns, distinct phenotypes of the knockout mice, and different roles in cell signaling [reviewed in (5)]. STAT5 has been studied in multiple cancer types, but the distinction between STAT5A and STAT5B has been examined only infrequently in epithelial tumors (11, 33, 34). STAT5A and STAT5B have differential regulatory roles in HNSCC, breast cancer, glioblastoma, and hepatocellular carcinoma (35–39). In HNSCC, STAT5 activation led to increased cell and tumor growth and increased invasion and induced epithelial-to-mesenchymal transition (17). Activated and total STAT5B, but not STAT5A, was found to increase in HNSCC tumors compared with normal-appearing mucosa. Likewise, in a xenograft model of HNSCC, STAT5B antisense was found to inhibit tumor growth in mice, whereas STAT5A antisense did not affect tumor size (40). Cells containing a dominant-negative STAT5B construct fail to proliferate in vitro (11). Erythropoietin mediates invasion in HNSCC through the activation of STAT5A; STAT5A did not promote tumor proliferation (41). These studies support a role for STAT5B, but not STAT5A, in the progression of HNSCC. Although we did not study the differential roles of STAT5A and STAT5B in HNSCC cells with unperturbed c-Src, our model would support a role for STAT5A as a tumor suppressor (by driving SOCS2 expression and subsequent suppression of Jak2-STAT3 activation). Also consistent with the finding that STAT5B promotes HNSCC cancer progression, we found that activation of STAT5B resulted in resistance to c-Src inhibition (Fig. S3A). Although STAT5 contributes to the progression of HNSCC, activation of STAT5 correlates with improved survival in breast cancer, where it may promote differentiation rather than progression (36, 42).
Our study has demonstrated that STAT3 and STAT5 are regulated independently. STAT5 activity was predominantly dependent upon c-Src, as the reactivation of Jak activity did not result in STAT5 reactivation. In contrast, STAT3 activation was predominantly Jak dependent, as STAT3 was reactivated in the presence of c-Src inhibition (9). Moreover, acute c-Src inhibition alone did not result in complete STAT3 inhibition unless SOCS2 was present (Fig. 3B). Jaks are the classic regulators of STAT5 and STAT3, but they are not the only kinases that can do so. ErbB receptor-induced activation of STAT1, STAT3, and STAT5 was found to be mediated by c-Src and independent of Jak (43). Likewise, c-Src can directly phosphorylate STAT5A (Y694) and activate STAT3 (44–46). c-Src can activate STAT5B directly by phosphorylation or indirectly by phosphorylating EGFR (Y845) (47). In HNSCC specifically, c-Src inhibition using both molecular and pharmacologic agents leads to STAT3 and STAT5 inhibition downstream of EGFR (6). EGFR possesses a STAT-binding capacity and can activate STATs in a Jak-independent manner (48, 49). EGFR, though an important mediator of both c-Src and STAT3 activation in HNSCC, does not function in STAT3 reactivation following sustained c-Src inhibition (10). The functions of Jaks, c-Srcs, and growth factor receptors are not independent, as they can cooperate to enhance STAT3 activation during oncogenesis (47, 50).
One unanswered question is what mechanism leads to Jak kinase inhibition. Our previous studies demonstrated that c-Src inhibition led to a rapid and significant inhibition of Jak kinase activity (9). However, Jak is not a known c-Src substrate. Another unresolved issue is the potential role for a cytokine or growth factor receptor as a scaffold for the Jak2/STAT3/SOCS2 complex. Although there is no role for a soluble growth factor or cytokine in this feedback loop [(10) and Fig. S2] and our previous work did not support the role for the kinase activity of a growth factor receptor (10), these experiments do not preclude the role of such a receptor as a scaffold for the complex. Future studies will be needed to address these issues.
Our study could have a direct clinical application. We have found STAT3 reactivation in cell lines from lung cancer, mesothelioma, and squamous carcinoma of the skin (10, 51). We have also observed STAT3 reactivation in vivo, after specific c-Src knockdown and using three different pharmacologic inhibitors (9, 10, 12); the combination of c-Src and Jak inhibitors leads to significant cancer cell apoptosis in vivo. The reciprocal regulation of c-Src and STAT3 activation in tumors from lung cancer patients suggests that this pathway functions in human tumors (12). These results demonstrate that STAT3 reactivation is likely to occur in patients with a broad range of cancers that are treated with any c-Src inhibitor. Specific and potent kinase inhibitors of c-Src and Jak are well tolerated in humans (1, 5). Specific SOCS mimetics are being developed and may be more specific and presumably less toxic than Jak inhibitors (52). STAT3 inhibitors also are being developed, but none have completed clinical trials (5).
Despite the finding of c-Src expression in epithelial tumors and the availability of agents to sustain its inhibition, the effects of c-Src inhibition on cell survival and proliferation have been moderate and inconsistent. c-Src mediates its effects on cancer cell survival and proliferation via diverse substrates including STATs. We have discovered a heretofore unknown compensatory pathway culminating in STAT3 reactivation and cancer cell survival. Our long-term goal is to use these results to design clinical trials combining these or other more specific c-Src inhibitors with Jak2 or STAT3 inhibitors or SOCS mimetics to improve the survival of patients with HNSCC and other cancers.
Supplementary Material
Translational Relevance.
Despite c-Src expression in epithelial tumors and the availability of potent inhibitors, the effects of c-Src inhibition on cancer cell survival have been modest. Our current study defines the molecular mechanism that underlies a compensatory pathway that ultimately leads to STAT3 activation and survival despite sustained c-Src inhibition; the combination of c-Src and Jak inhibitors leads to significant cancer cell apoptosis in vivo. Our study could have a direct clinical application because previous results demonstrate that STAT3 reactivation is likely to occur in a broad range of cancers and inhibitors of c-Src and Jak are well tolerated in humans. Specific STAT3 inhibitors are being developed and may be more specific and less toxic than Jak inhibitors. Our long-term goal is to use these results to design clinical trials combining c-Src inhibitors with Jak2 or STAT3 inhibitors to improve the survival of cancer patients.
Acknowledgments
The vector control plasmid, pUSEamp(+), and plasmids carrying the activated mutants of STAT5A and STAT5B (pUSE STAT5A 1*6 and pUSE STAT5B 1*6 plasmids) were kind gifts from Dr. Jennifer Grandis. Mouse full-length SOCS2 construct (pMet7-FLAG-mSOCS2) was a kind gift from Dr. Julie Piessevaux. Drs. Jeffrey Myers and Gary Gallick provided technical assistance with cell line authentication and in vitro kinase assays, respectively. We thank Brenda Robinson and Michael Worley for editorial assistance.
Grant Support
This work was supported by R01-CA143369-01 (FMJ). SYL was supported by NIH Mentored Career Development Award K08 DE018061.
This work was supported by R01-CA143369-01 (FMJ).
Footnotes
The authors declare that they have no conflict of interest.
References
- 1.Johnson FM, Gallick G. Src Family Kinase Inhibitors in Cancer Therapy. Hauppauge NY: Nova Science Publishers Inc; 2009. [Google Scholar]
- 2.Ammer AG, Kelley LC, Hayes KE, Evans JV, Lopez-Skinner LA, Martin KH, et al. Saracatinib Impairs Head and Neck Squamous Cell Carcinoma Invasion by Disrupting Invadopodia Function. J Cancer Sci Ther. 2009;1:52–61. doi: 10.4172/1948-5956.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 4.Brooks HD, Glisson BS, Bekele BN, Johnson FM, Ginsberg LE, El-Naggar A, et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer. 2011;117:2112–9. doi: 10.1002/cncr.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai SY, Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resist Updat. 2010;13:67–78. doi: 10.1016/j.drup.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Xi S, Zhang Q, Dyer KF, Lerner EC, Smithgall TE, Gooding WE, et al. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278:31574–83. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 7.Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19:2489–95. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 8.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–35. [PubMed] [Google Scholar]
- 9.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–65. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson FM, Saigal B, Tran H, Donato NJ. Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin Cancer Res. 2007;13:4233–44. doi: 10.1158/1078-0432.CCR-06-2981. [DOI] [PubMed] [Google Scholar]
- 11.Leong PL, Xi S, Drenning SD, Dyer KF, Wentzel AL, Lerner EC, et al. Differential function of STAT5 isoforms in head and neck cancer growth control. Oncogene. 2002;21:2846–53. doi: 10.1038/sj.onc.1205385. [DOI] [PubMed] [Google Scholar]
- 12.Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009;15:6852–61. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang X, Kadara H, Behrens C, Liu D, Xiao Y, Rice DC, et al. Abnormalities of the TITF-1 lineage-specific oncogene in NSCLC: Implications in lung cancer pathogenesis and prognosis. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-10-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, Misawa H, et al. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood. 1997;89:3148–54. [PubMed] [Google Scholar]
- 15.Takatori H, Nakajima H, Kagami S, Hirose K, Suto A, Suzuki K, et al. Stat5a inhibits IL-12-induced Th1 cell differentiation through the induction of suppressor of cytokine signaling 3 expression. J Immunol. 2005;174:4105–12. doi: 10.4049/jimmunol.174.7.4105. [DOI] [PubMed] [Google Scholar]
- 16.Sen B, Peng S, Saigal B, Williams MD, Johnson FM. Distinct interactions between c-Src and c-Met in mediating resistance to c-Src inhibition in head and neck cancer. Clin Cancer Res. 2011;17:514–24. doi: 10.1158/1078-0432.CCR-10-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppikar P, Lui VW, Man D, Xi S, Chai RL, Nelson E, et al. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin Cancer Res. 2008;14:7682–90. doi: 10.1158/1078-0432.CCR-08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14:6456–68. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 20.Limnander A, Danial NN, Rothman PB. v-Abl signaling disrupts SOCS-1 function in transformed pre-B cells. Mol Cell. 2004;15:329–41. doi: 10.1016/j.molcel.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 22.Newton VA, Ramocki NM, Scull BP, Simmons JG, McNaughton K, Lund PK. Suppressor of cytokine signaling-2 gene disruption promotes Apc(Min/+) tumorigenesis and activator protein-1 activation. Am J Pathol. 2010;176:2320–32. doi: 10.2353/ajpath.2010.090684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaylira CZ, Simmons JG, Ramocki NM, Scull BP, McNaughton KK, Fuller CR, et al. Suppressor of cytokine signaling-2 limits intestinal growth and enterotrophic actions of IGF-I in vivo. Am J Physiol Gastrointest Liver Physiol. 2006;291:G472–81. doi: 10.1152/ajpgi.00218.2005. [DOI] [PubMed] [Google Scholar]
- 24.Michaylira CZ, Ramocki NM, Simmons JG, Tanner CK, McNaughton KK, Woosley JT, et al. Haplotype insufficiency for suppressor of cytokine signaling-2 enhances intestinal growth and promotes polyp formation in growth hormone-transgenic mice. Endocrinology. 2006;147:1632–41. doi: 10.1210/en.2005-1241. [DOI] [PubMed] [Google Scholar]
- 25.Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- 26.Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, et al. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24:6699–708. doi: 10.1038/sj.onc.1208818. [DOI] [PubMed] [Google Scholar]
- 27.He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–8. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang FJ, Steeg PS, Price JE, Chiu WT, Chou PC, Xie K, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. 2008;68:9634–42. doi: 10.1158/0008-5472.CAN-08-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francipane MG, Eterno V, Spina V, Bini M, Scerrino G, Buscemi G, et al. Suppressor of cytokine signaling 3 sensitizes anaplastic thyroid cancer to standard chemotherapy. Cancer Res. 2009;69:6141–8. doi: 10.1158/0008-5472.CAN-09-0994. [DOI] [PubMed] [Google Scholar]
- 30.Vidal OM, Merino R, Rico-Bautista E, Fernandez-Perez L, Chia DJ, Woelfle J, et al. In vivo transcript profiling and phylogenetic analysis identifies suppressor of cytokine signaling 2 as a direct signal transducer and activator of transcription 5b target in liver. Mol Endocrinol. 2007;21:293–311. doi: 10.1210/me.2006-0096. [DOI] [PubMed] [Google Scholar]
- 31.Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000;20:389–401. doi: 10.1128/mcb.20.1.389-401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25:9115–26. doi: 10.1128/MCB.25.20.9115-9126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan SH, Nevalainen MT. Signal transducer and activator of transcription 5A/B in prostate and breast cancers. Endocr Relat Cancer. 2008;15:367–90. doi: 10.1677/ERC-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol. 2010;176:1959–72. doi: 10.2353/ajpath.2010.090653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–9. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- 36.Tran TH, Utama FE, Lin J, Yang N, Sjolund AB, Ryder A, et al. Prolactin inhibits BCL6 expression in breast cancer through a Stat5a-dependent mechanism. Cancer Res. 2010;70:1711–21. doi: 10.1158/0008-5472.CAN-09-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang JZ, Zuo ZH, Kong XJ, Steiner M, Yin Z, Perry JK, et al. Signal transducer and activator of transcription (STAT)-5A and STAT5B differentially regulate human mammary carcinoma cell behavior. Endocrinology. 2010;151:43–55. doi: 10.1210/en.2009-0651. [DOI] [PubMed] [Google Scholar]
- 38.Liang QC, Xiong H, Zhao ZW, Jia D, Li WX, Qin HZ, et al. Inhibition of transcription factor STAT5b suppresses proliferation, induces G1 cell cycle arrest and reduces tumor cell invasion in human glioblastoma multiforme cells. Cancer Lett. 2009;273:164–71. doi: 10.1016/j.canlet.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng IO, et al. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res. 2006;66:9948–56. doi: 10.1158/0008-5472.CAN-06-1092. [DOI] [PubMed] [Google Scholar]
- 40.Xi S, Zhang Q, Gooding WE, Smithgall TE, Grandis JR. Constitutive activation of Stat5b contributes to carcinogenesis in vivo. Cancer Res. 2003;63:6763–71. [PubMed] [Google Scholar]
- 41.Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A, et al. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene. 2005;24:4442–9. doi: 10.1038/sj.onc.1208635. [DOI] [PubMed] [Google Scholar]
- 42.Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, et al. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–60. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 43.Olayioye MA, Beuvink I, Horsch K, Daly JM, Hynes NE. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J Biol Chem. 1999;274:17209–18. doi: 10.1074/jbc.274.24.17209. [DOI] [PubMed] [Google Scholar]
- 44.Okutani Y, Kitanaka A, Tanaka T, Kamano H, Ohnishi H, Kubota Y, et al. Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene. 2001;20:6643–50. doi: 10.1038/sj.onc.1204807. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, et al. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J Biol Chem. 2000;275:24935–44. doi: 10.1074/jbc.M002383200. [DOI] [PubMed] [Google Scholar]
- 46.Chaturvedi P, Reddy MV, Reddy EP. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene. 1998;16:1749–58. doi: 10.1038/sj.onc.1201972. [DOI] [PubMed] [Google Scholar]
- 47.Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–23. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- 48.Xia L, Wang L, Chung AS, Ivanov SS, Ling MY, Dragoi AM, et al. Identification of both positive and negative domains within the epidermal growth factor receptor COOH-terminal region for signal transducer and activator of transcription (STAT) activation. J Biol Chem. 2002;277:30716–23. doi: 10.1074/jbc.M202823200. [DOI] [PubMed] [Google Scholar]
- 49.Leaman DW, Pisharody S, Flickinger TW, Commane MA, Schlessinger J, Kerr IM, et al. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol Cell Biol. 1996;16:369–75. doi: 10.1128/mcb.16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 51.Tsao AS, He D, Saigal B, Liu S, Lee JJ, Bakkannagari S, et al. Inhibition of c-Src expression and activation in malignant pleural mesothelioma tissues leads to apoptosis, cell cycle arrest, and decreased migration and invasion. Mol Cancer Ther. 2007;6:1962–72. doi: 10.1158/1535-7163.MCT-07-0052. [DOI] [PubMed] [Google Scholar]
- 52.Flowers LO, Subramaniam PS, Johnson HM. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene. 2005;24:2114–20. doi: 10.1038/sj.onc.1208437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.