

Abstract
Renal cell carcinoma is increasing in incidence but the molecular mechanisms regulating its growth remain elusive. Co-expression of the monocytic growth factor CSF-1 and its receptor CSF-1R on renal tubular epithelial cells (TEC) will promote proliferation and anti-apoptosis during regeneration of renal tubules. Here we show that a CSF-1-dependent autocrine pathway is also responsible for the growth of renal cell carcinoma (RCC). CSF-1 and CSF-1R were co-expressed in RCC and TEC proximally adjacent to RCC. CSF-1 engagement of CSF-1R promoted RCC survival and proliferation and reduced apoptosis, in support of the likelihood that CSF-1R effector signals mediate RCC growth. In vivo CSF-1R blockade using a CSF-1R tyrosine kinase inhibitor decreased RCC proliferation and macrophage infiltration in a manner associated with a dramatic reduction in tumor mass. Further mechanistic investigations linked CSF-1 and EGF signaling in RCC. Taken together, our results suggest that budding RCC stimulates the proximal adjacent microenvironment in the kidney to release mediators of CSF-1, CSF-1R and EGF expression in RCC. Further, our findings imply that targeting CSF-1/CSF-1R signaling may be therapeutically effective in RCC.
Keywords: Tubular epithelial cells, Macrophages, Renal carcinoma, CSF-1R, CSF-1
Introduction
Renal cell carcinoma has been steadily on the rise for several decades. Renal clear cell carcinoma (RCC) is derived from proximal tubule epithelial cells and is by far the most common (70–80%) form of kidney carcinomas (1, 2). However, despite the increased incidence of RCC, the molecular mechanisms that regulate the growth of this tumor remain elusive (3).
CSF-1 and CSF-1R are instrumental during the progression of epithelial tumors of the female reproductive tract and prostate (4–7). For example, CSF-1 and CSF-1R are co-expressed in >50% of mammary tumors and elevated circulating CSF-1 levels are an indicator of early metastatic relapse in patients with breast cancer (8, 9). In addition, CSF-1 expression in primary breast carcinoma and RCC correlates with infiltration of inflammatory cells and, in turn, poor prognosis of this tumor (10). In fact, CSF-1 generated by mammary tumors and RCC recruit and activate large numbers of tumor associated Mø (TAMS) that release trophic cytokines and other growth factors thereby, enhances mammary and renal carcinoma growth and facilitate tumor metastases (10–14). Moreover, an autocrine loop in breast cancer cell lines expressing both CSF-1 and the CSF-1R, may contribute to tumor invasion and metastasis (4,9,15). Furthermore, recent findings indicate that CSF-1R is expressed on tubular epithelial cells (TEC) in RCC, however the relevance to this tumor's development and progression was not elucidated (5,16). Thus, signaling thru the CSF-1R may promote the progression of renal epithelial cell tumors.
Inflammation is meant to set the stage for repair. We recently uncovered a CSF-1 dependent mechanism of renal tubular repair (17). After transient renal ischemia, CSF-1 and CSF-1R are co-expressed on tubular epithelial cells (TEC), including those in the proximal tubule, in mice and humans. CSF-1 engaging with the CSF-1R induces TEC to proliferate and inhibits further apoptosis leading to the replenishment of injured TEC. CSF-1 is integral in the healing process as CSF-1 injected into mice after ischemia/reperfusion (I/R) hastens tubular healing, while blocking the CSF-1R prevents renal tubular regeneration (17,18). However, CSF-1 was originally identified as the principle Mø developmental molecule that stimulates survival, differentiation, proliferation and activation of Mø (19, 20). And CSF-1 has a sole receptor, the c-fms tyrosine kinase proto-oncogene that is expressed on cells of the monocyte lineage (Mø, dendritic cells) (21) As Mø are implicated in the repair of numerous tissues (22), we probed for the contribution of a CSF-1 dependent Mø along with autocrine TEC renal repair after I/R. We determined that CSF-1 mediated tubular repair is dependent on TEC autocrine Mø-independent and Mø-dependent mechanisms following transient injury (17). Thus, signaling via the CSF-1R on TEC and Mø are intended to protect the kidney by mediating tubular repair.
To probe for the mechanisms that promote RCC growth, we hypothesized that: 1) RCC co-express CSF-1 and the CSF-1R and 2) the CSF-1 mediated autocrine feedback loop, intended to promote tubular repair in normal kidneys is “hijacked” by the RCC and instead triggers tumor cell proliferation and inhibits tumor cell apoptosis, thereby promoting tumor growth.
Materials and Methods
Renal biopsy specimens
Renal carcinomas tissue (discarded tissues) with a confirmed pathological diagnosis were provided by the Department of Pathology, Rush University Medical Center, Chicago, IL and the Department of Pathology Johannes-Gutenberg University, Mainz, Germany. RCC samples were analysed using the morphological classification of the carcinomas according to World Health Organization (WHO) specifications to evaluate clinical outcome.
CSF-1R and CSF-1 expression
Immunofluorescence
In vitro
We cultured RCC (786-0 and Caki), HK2 and MCF-7 lines according to the manufactures instructions (Cell Line Services, Eppelheim, Germany). Cells (1×104 well) were stimulated with TPA (Phorbol 12-Myristate 13 Acetate) for 48 h. We fixed cells with methanol for 5 min on coverslips and incubated them with rabbit-anti-mouse CSF-1R Ab or rabbit IgG (Santa Cruz) for 1 h at RT. We detected CSF-1R by incubating cells with anti-rabbit Cy3 Ab (Vector, Burlingame, CA) for 30 min. We mounted the coverslips with Vectoshield with Dapi (Vector) and analysed them using a Nikon Eclipse E1000 upright fluorescence microscope.
Immunohistochemistry
A) Paraffin sections
We determined CSF-1 (CSF-1-Ab, N-16; Santa Cruz), phospho-M-CSFR (rabbit anti-human phospho-M-CSFR Tyr 723 Ab, 49C10; Cell signalling), CSF-1R (CSF-1R-Ab; Santa Cruz) as previously described (23). We determined the number of positive cells in 10 randomly selected high-power fields and evaluated the corresponding areas within serial sections for the correlation analysis.
CSF-1/EGF ELISA
To quantify the levels of CSF-1 in supernatants, we evaluated samples using an ELISA for each as previously described (23). We analysed EGF in supernatants using an ELISA (R&D systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Human Cell lines
The following cell lines were used and cultured as previously described: immortalized RCC lines (786-0 and Caki), immortalized proximal TEC line (HK2) (24), promyelocytic leukemia cell line (HL60) (25) and immortalized T cell line (JURKAT). Carcinoma cells were purchased from the Cell Line Services, Germany.
TEC stimulated with RCC and Mø supernatant
To determine CSF-1R and CSF-1 expression on TEC induced by mediators released by RCC and Mø we stimulated RCC cell lines and Mø with TNF-α (6 ng/ml), TNF-α/LPS (6ng/ml, 12 ng/ml) and PMA for 24h. Then the supernatant was removed and fresh media was added. After an additional 24h the supernatant (undiluted and diluted: 1:1, 1:4 and 1:8 with media) was added to cultured TEC (HK2). After 48h CSF-1 in the cultured TEC supernatant was evaluated (ELISA assay) and CSF-1 and CSF-1R transcript expression on cultured TEC was evaluated by RTQ-PCR. We measured CSF-1 concentrations in the supernatant of RCC and Mø before co-incubation with TEC to determine the baseline expression of CSF-1 in these cells. CSF-1 expression in TEC that were not stimulated served as controls.
CSF-1R and CSF-1 transcript expression
We analyzed the CSF-1R and CSF-1 expression in human cell lines using real-time, two-step, quantitative PCR as previously described (RTQ-PCR) (26). The mRNA levels were normalized to those of GAPDH. We used the following PCR primers: GAPDH; sense, 5′-CCC TCA ACG ACC ACT TTG TCA-3′, anti-sense, 5′-TTC CTC TTG TGC TCT TGC TGG-3′; CSF-1R; sense 5′-TGA GCA AGA CCT GGA CAA GGA-3′, anti-sense 5′-CCA TTG GTC AAC AGC ACG TTA-3′. CSF-1; sense 5′-CCC ATA TTG CGA CAC CGA A-3′; antisense 5′-AAG CAG TAA CTG AGC AAC GGG-3′.
CSF-1R RNA interference
For transient inhibition of CSF-1R mRNA production, 786-0 and HK2 cell lines were transfected with a commercially available pool of 4 target-specific 20–25-nucleotide-long small interfering RNA (siRNA) designed to “knock down” CSF-1R expression and with a control siRNA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For stable inhibition of CSF-1R expression, 786-0 and HK2 cell lines were transfected using a shRNA Plasmid (pool of 4 target-specific lentiviral vector plasmids each encoding 19–25 nucleotide-long (plus hairpin) shRNAs are designed to target the CSF-1R knockdown or control shRNA Plasmid, Santa Cruz, Biotechnology. Each plasmid contains a puromycin resistance gene to select cells stably expressing shRNA. These transfections were conducted with shRNA Plasmid Transfection reagent according to the manufacturer's instructions, Santa Cruz Biotechnology.
Preparation of VHL in 786-0+ cell line
Using a retroviral gene transfer approach 293T cells were transfected with pBABe-puro-VHL DNA and a control DNA as previous described (27). 786-0 cells expressing the VHL gene were selected with puromycin (1μg/ml).
Proliferation
1) Immunohistochemistry
We stained paraffin sections using a primary Ab against rabbit anti-human/mouse Ki67 (SP6; Lab Vision) to identify proliferating TEC.
2) MTT assay
We cultured RCC (786-0 and Caki-1 (Caki), Caki-2 and human TEC (HK2) and human breast carcinoma (MCF-7) lines in 96-well plates (5×103 cells/well) for 12 h, and stimulated these cells for 72h with human recombinant CSF-1 or EGF (Sigma-Aldrich, St Louis, MO) at various concentrations (5, 20, 40 and 80 ng/ml). To verify specificity by blocking the CSF-1R, we stimulated cells with CSF-1 in combination with CSF-1R Ab (25 ng/ml, Santa Cruz) or CSF-1R tyrosine kinase inhibitor (5-(3-Methoxy-4-((4-methoxybenzyl)oxy)benzyl)-pyrimidine-2-4-diamine, Calbiochem, Darmstadt, Germany)), or with EGF and anti-EGFR Ab for 72h. We used control antibodies for CSF-1R Ab (rabbit IgG) and EGFR Ab (mouse IgG2B) in all experiments. Furthermore, we stimulated cultured RCC, HK2 and MCF-7 lines with varying concentrations of TNF-α (3, 6, 15 and 30 ng/ml), LPS (6, 12, 25 and 50ng/ml) (Sigma-Aldrich) in combination with CSF-1R Ab (25 ng/ml, Santa Cruz), or anti-EGFR Ab. We analysed proliferation using the MTT colorimetric assay (Roche, Palo Alto, CA) according to the manufacturer's instructions.
Apoptosis
We cultured RCC, HK2 and MCF-7 lines in 6-well (apoptosis-assay) or 96-well (proliferation assay) plates (5×104/well) for 12 h and stimulated these cells with varying concentrations of human recombinant CSF-1 (5-, 20-,39-, and 78 ng/ml) in combination with TNF-α (3, 6, 15 and 30 ng/ml), LPS (6, 12, 25 and 50ng/ml). After 72 h we assessed apoptosis by flow cytometry using an Annexin-V-FITC-PI kit (BD Bioscience, San Jose, CA) according to the manufacturer's instructions.
Mice
We purchased athymic nu/nu BALB/c mice from The Jackson Laboratory (Bar Harbor, ME). Mice were housed at Johannes-Gutenberg University in Mainz, Germany. The use of mice in this study was reviewed and approved by the Standing Committee on Animals at the University of Mainz
Implanting TEC under the renal capsule
We implanted RCC (786-0) or HK2 cells under the renal capsule of athymic nu/nu BALB/c mice at 3 mo of age as previously described (27).
Treatment with CSF-1R tyrosine kinase inhibitor
We injected mice (i.p.) every 24 h with the CSF-1R tyrosine kinase inhibitor (Calbiochem, Darmstadt, Germany), (25mg/kg/body wt) beginning at 1.0d after surgery and ending at 21d after surgery. The CSF-1R inhibitor is a cell-permeable diaminopyrimidine compound that acts as a potent, selective, and ATP-competitive inhibitor of cFMS kinase activity (IC50 = 30 nM) with minimal inhibition towards a panel of 26 other kinases (IC50 >5 μM). It is shown to selectively inhibit cFMS-mediated cellular functions in vitro as well as CSF-1-dependent tumor growth in vivo (28).
Statistical Analysis
The data represent the mean ± SEM and were prepared using GraphPad PRISM version 4.0. We used the nonparametric Mann-Whitney U test to evaluate p values. For correlation analysis, we used the Spearman correlation calculation.
Results
CSF-1R and CSF-1 are expressed in Renal Clear Cell Carcinoma (RCC), but not Papillary Carcinoma (PC)
CSF-1 and the CSF-1R are co-expressed on TEC following a transient ischemic injury and via an autocrine/paracrine mechanism mediate renal tubular repair (17). Moreover, the CSF-1R is expressed on epithelial cells of carcinomas including breast and prostate (6,29). Thus, we hypothesized that CSF-1 and the CSF-1R are co-expressed on renal cell carcinoma. To test this hypothesis we probed for CSF-1 and CSF-1R protein expression in human kidney carcinomas, RCC and PC specimens in comparison to normal kidney by immunostaining. We detected robust CSF-1R expression in RCC as compared with normal kidney that lacked CSF-1R expression (Figure 1A). Since the CSF-1R is internalized following activation, CSF-1R is expressed on the membrane and in the cytoplasm (30,31). To determine if CSF-1R expression is restricted to the RCC, we examined the TEC adjacent and distant to the RCC. We detected ubiquitous expression of CSF-1R on TEC adjacent to RCC, while CSF-1R expression was limited to far fewer TEC distant to RCC (Figure 1A). In fact, there was a CSF-1R expression gradient emanating from the RCC and declining in proportion to the distance from the tumor. To determine if CSF-1 is expressed along with CSF-1R in RCC, we stained for the presence of CSF-1 in RCC compared with normal kidney. We detected CSF-1 expression along with CSF-1R expression in RCC. Moreover, the CSF-1 expression was most pronounced in the TEC contiguous to the tumor, diminished with distance from RCC, and was not detectable in the areas most distant to the tumor. Furthermore, we detected a correlation in the expression of CSF-1R and CSF-1 in RCC and the TEC adjacent, but not distant, to RCC (Figure 1B). In contrast, we rarely detected CSF-1R and CSF-1 expression in PC (Figure 1A). Taken together, CSF-1 and CSF-1R are ubiquitously co-expressed in RCC and TEC closest to the RCC and declines with distance from the tumor.
Figure 1. CSF-1R and CSF-1 are expressed in RCC, but not PC.

We detected robust CSF-1R and CSF-1 expression in RCC and TEC adjacent to RCC as compared with areas distant to RCC and normal human kidney. We did not detect a rise in PC. CSF-1R and CSF-1 Ab specificity was verified u s i n g p e p t i d e p r e-absorption. Representative photomicrographs (magnification 20×) are shown. B. CSF-1R expression in TEC correlates with CSF-1 expression in RCC and TEC adjacent to RCC, but not distant to RCC. Values are the mean ± SEM.
CSF-1R and CSF-1 expression in RCC and TEC adjacent to RCC correlates with infiltrating Mø, but not T cells
Increased numbers of Mø in human breast cancer is an index of poor prognosis (32,33). Moreover, mammary tumor metastasis is attenuated in mice with reduced numbers of Mø (34). To determine whether enhanced expression of CSF-1R and CSF-1 on TEC in RCC fosters Mø and T cell incited inflammation, we probed for Mø and T cells in RCC and the area adjacent and distant to the tumor. We detected a greater magnitude of Mø in RCC as compared to normal kidneys (Figure 2A). Moreover, Mø were more abundant in the RCC than in the area adjacent and distant to RCC. In fact, the magnitude of CSF-1R and CSF-1 expression correlated with CD68+ Mø in RCC and TEC, adjacent but not distant, to RCC (Figure 2B). Of note, we did not detect an increase in CD3+ T cells in RCC, nor in TEC adjacent and distant to RCC (Figure 2A). And CSF-1R and CSF-1 expression did not correlate with the number of T cells in the RCC and the adjacent TEC (Figure 2B). And we did not detect an increase in T cells in PC (Figure 2A). Taken together, our findings suggest that the magnitude of CSF-1R and CSF-1 is an index of the extent of Mø, but not T cells, in RCC and the adjacent TEC.
Figure 2. CSF-1R and CSF-1 expression on RCC and TEC that are adjacent to RCC correlate with infiltrating Mø, but not CD3+ T cells.

A. We evaluated the presence of CD68+ and CD3+ leukocytes in formalin-fixed sections by immunostaining. We confirmed staining specificity using isotype control Abs. Representative photomicrographs are shown (magnification 20×, enlargement 40×). B. Correlations of CSF-1R and CSF-1 expression with CD68+ and CD3+ leukocytes in RCC and the TEC in areas adjacent and distant to RCC. Values are the mean ± SEM.
Co-expression of CSF-1 and CSF-1R in RCC correlates with TEC proliferation
CSF-1 engaging with CSF-1R on the same or adjacent TEC induces proliferation, thereby replenishing necrotic and apoptotic TEC following transient ischemia (17). To determine whether the CSF-1R and CSF-1 co-expression of TEC in RCC has an impact on TEC proliferation resulting in an increase in RCC, we assessed CSF-1, CSF-1R and proliferation in TEC in sequential kidney sections from patients with RCC. The magnitude of CSF-1 and CSF-1R expression was greatest in proliferating (Ki67+) TEC in RCC and TEC adjacent to RCC (Figure 3). Moreover, we determined that CSF-1R+ TEC expressed the tyrosine phosphorylated CSF-1R, indicating that the CSF-1R is signally the TEC (Figure 3). Taken together, this suggests that co-expression of CSF-1 and CSF-1R on RCC and TEC adjacent to RCC promotes proliferation and is thereby responsible for promoting the expansion of this tumor.
Figure 3. CSF-1 and CSF-1R co-expression on RCC promotes survival/proliferation, thereby leading to increased RCC growth.

Using serial sections of kidney biopsy specimens from patients with RCC, we probed for CSF-1 and CSF-1R expression, CSF-1R phosphorylation at Y723 and proliferation in tubules. CSF-1R and CSF-1 are co-expressed on TEC and CSF-1R and tyrosine phosphorylated CSF-1R are co-expressed on TEC (immunostaining). Note, proliferating Ki67+ TEC in tubules co-expressing CSF-1 and CSF-1R. CSF-1R and CSF-1 Ab specificity was verified using peptide pre-absorption, Ki67 and phospho-Y723 CSF-1R Ab specificity was verified using rabbit IgG (right panels). Representative photomicrographs and correlation graphs. Magnification 20×, enlargement 40×. Values are means ± SEM; n=8–9/group.
We next investigated whether co-expression of CSF-1R, phospho CSF-1R and CSF-1 on proliferating TEC correlated with clinical disease. The criteria for clinical disease included tumor morphology (stage and grade/dedifferentiation) and patient survival. We evaluated 22 patients with RCC (average age of 64 years, 60% male and 40% female). We found CSF-1R and CSF-1 were associated with enhanced Ki67 expression and with accelerated tumor progression (tumor stage and grade and reduced survival of the patients, Supplemental Figure 1). Moreover, as the phospho CSF-1R expression was similarly associated with tumor progression this indicates that the CSF-1R was functional (Supplemental Figure 1). This suggests that co-expression of CSF-1 and CSF-1R on RCC hastens the progression of RCC by promoting proliferation and differentiation.
Mediators predominantly released from RCC, increase CSF-1R and CSF-1 expression on adjacent TEC
To test the hypothesis that increased expression of CSF-1R and CSF-1 on TEC adjacent to RCC is induced by mediators released by RCC and/or TAMS, we performed a series of in vitro experiments. Incubating the TEC cell line (HK2) with RCC (786-0) supernatant led to a concentration (1:1–1:8) dependent rise in CSF-1 protein (Supplemental Figure 2) and transcripts (Figure 4, bottom panel) as compared with TEC not incubated with RCC supernatant. Following RCC stimulation (TNF-α) the concentration dependent rise in CSF-1 expression rose even higher as compared with unstimulated RCC (Figure 4, top panel). Baseline expression of RCC before co-incubation with TEC is displayed as a dotted line. By comparison, incubating TEC with increasing concentrations of Mø (HL60) supernatant modestly increased CSF-1 expression on TEC (Figure 4, top panel, Supplemental Figure 2). Furthermore, we detected a similar pattern of CSF-1R transcript expression on TEC following incubation with supernatant generated by RCC and Mø (Figure 4, bottom panel). This suggests that mediators predominantly released by RCC, induce CSF-1 and CSF-1R expression on TEC adjacent to RCC.
Figure 4. CSF-1 and CSF-1R are increased on TEC by mediators released from RCC and, to a lesser degree, Mø.
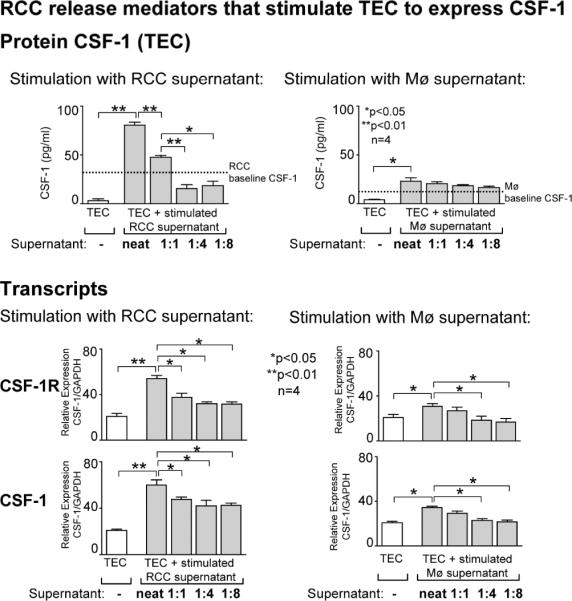
Top panel: CSF-1 expression of cultured TEC (HK2 cell line) following incubation with supernatant of stimulated RCC (786-0 cell line) or Mø (HL60 cell line). Representative data is shown for stimulation with TNF-α, stimulations with TNF-α/LPS and Phorbol 12-Myristate 13 Acetate (TPA) showed comparable results (data not shown). Supernatants were added to the cultured TEC cell line (HK2) undiluted, diluted 1:1, 1:4 and 1:8 with media from stimulated RCC cells (786-0) or Mø (HL60). Bottom panel: CSF-1 and CSF-1R transcript expression of cultured TEC (HK2 cell line) following incubation with supernatant of stimulated RCC or Mø. Results are representative of 3 separate experiments. Values are means ± SEM.
CSF-1 engaging with CSF-1R on RCC promotes survival/proliferation, thereby, fostering RCC growth
We hypothesized that CSF-1 engaging with CSF-1R on TEC promotes survival/ proliferation in RCC. To test this hypothesis we first established that human RCC cell lines, 786-0 (Figure 5) and Caki-1 (=Caki) (wild-type van Hippel-Lindau (VHL) and Caki-2 (deficient for VHL) express CSF-1R and CSF-1 transcripts and protein. We detected more robust CSF-1R transcript and protein (immunofluorescence) expression on TEC in RCC lines than on a human proximal tubule line (HK2) and a breast carcinoma line (MCF-7). And CSF-1R transcript expression rose when RCC line was stimulated with Phorbol 12-Myristate 13 Acetate (PMA) (Figure 5, Supplement Figure 3). In parallel with CSF-1R expression, we detected an up-regulation of CSF-1 transcripts and protein expression following stimulation with TNF-α expressed in tumor associated Mø (35) along with LPS, a conventional stimulant, in cultured RCC cell lines. CSF-1 up-regulation by TNF-α/LPS was even more robust than in HK2 and MCF-7 cells (Figure 5B, Supplement Figure 3B). Taken together, CSF-1R and CSF-1 are expressed and robustly up-regulated after stimulation in RCC lines.
Figure 5. CSF-1R and CSF-1 are expressed on RCC lines.

A. Left panel: CSF-1R transcript expression in RCC cells (786-0) determined by real-time PCR. Normal human TEC (HK2) human breast carcinoma line, (MCF-7) and the human leukemic cell line (HL60) stimulated with TPA served as positive controls. TPA stimulation of the human T cell line (JURKAT) served as a negative control. Results are representative of 3 separate experiments; means ± SEM. Right panel: RCC lines (786-0), HK2 cell line and MCF-7 lines stimulated with TPA and stained with anti-CSF-1R and control rabbit IgG. Results are representative of 3 separate experiments; means ± SEM. B. Left panel: CSF-1 transcript expression in RCC cells determined by real-time PCR. HK2, MCF-7 lines stimulated with TNF-α/LPS served as positive controls. Results are representative of 3 separate experiments; means ± SEM. Right panel: Stimulation of RCC cells increases secretion of CSF-1 protein. Following exposure of RCC cells to increasing concentrations of TNF-α/LPS for 48h, we analyzed CSF-1 in the supernatant by ELISA. Results are representative of 3 separate experiments. Values are means ± SEM.
As CSF-1R and CSF-1 are expressed on RCC lines, we tested the hypothesis that CSF-1 engaging with CSF-1R on TEC stimulates survival/proliferation in RCC lines. For this purpose, we stimulated human RCC cell lines, 786-0 (Figure 6) and Caki (data not shown) with increasing concentrations of CSF-1 and evaluated survival/proliferation (MTT assay). Increasing concentrations of CSF-1 augmented the RCC mass (Figure 6). CSF-1 dependent stimulation was specific as blocking the CSF-1R suppressed the CSF-1 dependent rise in the RCC mass (Figure 6A). Note, replacing the CSF-1R Ab with a control (rabbit IgG) did not suppress CSF-1 stimulation. Thus, stimulation with CSF-1 increases survival/proliferation of human RCC lines. Moreover, using a silencing approach of the CSF-1R by transient inhibition of CSF-1R expression by siRNA and stable inactivation of the CSF-1R expression in RCC and HK2 cells by shRNA suppressed CSF-1 mediated tumor growth (Supplement Figure 4).
Figure 6. CSF-1 engaging with CSF-1R on RCC promotes survival/proliferation, and suppresses RCC apoptosis thereby leading to increased RCC growth.

A. CSF-1 stimulates survival/proliferation (MTT assay) of RCC (786-0) and blocking with anti-CSF-1R Ab suppresses CSF-1 mediated RCC proliferation. HK2 and MCF-7 lines served as positive controls. Results are representative of 4 separate experiments; means ± SEM. B. RCC were incubated with TNF-α/LPS in the presence and absence of anti-CSF-1R Ab for 72h prior to determination of cell mass by MTT assay. HK2 and MCF-7 lines served as positive controls. Results are representative of 3 separate experiments. Values are means ± SEM. C. TNF-α /LPS stimulated RCC are self-protective through a CSF-1-dependent mechanism that dampens RCC apoptosis. RCC were cultured for 72h in the absence of CSF-1. Blocking the CSF-1R increases TNF-α/LPS-induced TEC apoptosis. Means ± SEM; n=6–8/group.
As CSF-1 expression increases in injured (TNF-α/LPS, actinomycin D, cysplatin) normal mouse TEC and, in turn, promotes TEC survival/proliferation, (17) we hypothesized that an increase in CSF-1 and CSF-1R displayed on RCC leads to tumor growth. To test this hypothesis we blocked the CSF-1R along with TNF-α/LPS stimulation to increase CSF-1 expression and evaluated survival/proliferation (as above). We detected a decrease in RCC mass following blockade of the CSF-1R (by CSF-1R Ab, transient and stable silencing by siRNA and shRNA), enhanced in RCC with prior TNF-α/LPS stimulation (Figure 6B,C, data not shown for siRNA and shRNA). Thus, RCC generates sufficient CSF-1 to trigger CSF-1R signaling on RCC resulting in enhanced RCC survival/proliferation, thereby increasing the tumor growth.
Since inactivating mutations in the von Hippel-Lindau (VHL) tumor suppressor gene are associated with RCC (36), we investigated whether CSF-1 and CSF-1R expression in RCC is dependent on VHL. We did not detect a difference in CSF-1R and CSF-1 expression comparing the Caki (wild-type VHL) to Caki-2 (VHL-deficient) cell line (data not shown) or the 786-0 RCC line with and without VHL expression (786-0-VHL+/786-0-VHL−). Furthermore, CSF-1 stimulated a similar increase in proliferation/ survival in the Caki/Caki-2 and 786-0-VHL+/786-0-VHL− cell lines (Supplemental Figure 5). Thus, the CSF-1R and CSF-1 expression is not dependent on the VHL mutation.
CSF-1 engaging with CSF-1R on RCC suppresses apoptosis, thereby, increasing RCC growth
CSF-1 dependent dampened apoptosis contributes to renal tubular repair, following transient ischemic injury (17). Therefore, we hypothesized that CSF-1 mediates an increase in RCC mass via CSF-1 dependent suppression of apoptosis in RCC. To test this hypothesis we exposed RCC lines to increasing concentrations of TNF-α/LPS to increase CSF-1 and CSF-1R and evaluated the magnitude of RCC apoptosis. We detected a rise in the magnitude of apoptotic RCC cells with increasing concentrations of TNF-α/LPS (Figure 6C). Moreover, blocking signaling through the CSF-1R on RCC lines using anti-CSF-1R Ab led to a rise in apoptotic cells indicating that eliminating the CSF-1R signaling suppresses apoptosis (Figure 6C). Taken together, CSF-1 dependent RCC autocrine/paracrine mechanisms promote survival/proliferation and dampen apoptosis, thereby, fostering tumor growth.
Blocking CSF-1R signaling in vivo inhibits RCC growth
We hypothesized that blocking the CSF-1R suppresses RCC growth. To test this hypothesis we evaluated the impact of a CSF-1R tyrosine kinase inhibitor on human RCC in vitro. We stimulated a RCC line with increasing concentrations of CSF-1 and added the CSF-1R tyrosine kinase inhibitor. The CSF-1R tyrosine kinase inhibitor suppressed CSF-1 dependent proliferation and survival (data not shown) of the RCC line. Thus, the CSF-1R tyrosine kinase inhibitor effectively dampens renal tumor cell growth.
To determine whether blocking the CSF-1R on RCC inhibits RCC growth in vivo, we constructed a RCC model in BALB/c mice. We implanted human RCC (786-0) under the renal capsule in athymic nu/nu BALB/c mice, as these cells will not be rejected. To block the CSF-1R we compared mice injected with a CSF-1R tyrosine kinase inhibitor (28,37) and PBS for 21 days (Figure 7). We implanted human TEC (HK2) under the renal capsule to serve as a negative control. We detected a dramatic decrease in local tumor growth following CSF-1R tyrosine kinase inhibitor compared to PBS treatment (Figure 7). However, provision of the CSF-1R tyrosine kinase inhibitor, did not reduce the RCC mass to baseline (level of implanted normal human TEC) (Figure 7). Thus, CSF-1R tyrosine kinase blockade suppresses human RCC growth. To determine whether the reduced tumor cell expansion is a result of reduced RCC proliferation, we evaluated the number of proliferating (Ki67+) RCC. We detected fewer proliferating RCC in the CSF-1R tyrosine kinase inhibitor treated mice as compared with PBS injected mice (Figure 7). This further supports the concept that RCC are stimulated to proliferate via a CSF-1/CSF-1R autocrine/paracrine feed-back loop. Mø have been implicated in the promotion of human carcinomas (15). Thus, we evaluated the number of kidney infiltrating Mø within the implant site. The number of Mø (CD68+) decreased in CSF-1R tyrosine kinase treated mice comparable to the mice implanted with HK2 cells (Figure 7). In fact, the magnitude of Mø in CSF-1R tyrosine kinase inhibited mice was reduced to the level of those implanted with non-malignant HK2 cells. Taken together, blocking the CSF-1R effectively suppresses the growth of RCC.
Figure 7. Blocking CSF-1R signaling in vivo inhibits RCC growth.

A. CSF-1 stimulates proliferation (MTT assay) of RCC (786-0) and blocking with CSF-1R tyrosine kinase inhibitor suppresses CSF-1 mediated RCC proliferation. HK2 line served as a positive control. Results are representative of 2 separate experiments; means ± SEM. B. RCC (786-0) and HK2 cells were implanted under the renal capsule of athymic nu/nu Balb/c mice. CSF-1R tyrosine kinase inhibitor was injected beginning 1 day after surgery for 21 days. Mice were sacrificed 22 days following surgery. Growth (mass), proliferation (Ki67) and infiltration of CD68+ leukocytes in RCC was suppressed in mice treated with the CSF-1R tyrosine kinase inhibitor compared with PBS treated controls. Proliferating cells and CD68+ leukocytes are circled. Representative photomicrographs (T= tumor). Values are means ± SEM; n=4–5/group.
EGF mediates CSF-1-dependent RCC increased proliferation and suppressed apoptosis
Breast carcinoma cells generate CSF-1 that, in turn, stimulates Mø to express EGF. And EGF stimulates breast carcinoma cells to express CSF-1 (4,15). Thus, we hypothesized that EGF stimulates RCC to express CSF-1, thereby promoting proliferation. To test this hypothesis, we incubated RCC with EGF and measured CSF-1 in the supernatant and cell mass. We detected a progressive rise in the level of CSF-1 released in the supernatant (Figure 8A), accompanied by increased proliferation of RCC in response to EGF (Figure 8B). As expected, blocking the EGF receptor (EGFR) suppressed EGF mediated CSF-1-stimulated TEC proliferation. Moreover, inhibiting EGFR along with CSF-1R additively suppressed RCC proliferation. In conclusion, the interaction of CSF-1 and EGF enhances the expansion of RCC by promoting RCC proliferation (Supplemental Figure 6).
Figure 8. EGF mediates CSF-1-dependent enhanced proliferation and suppressed apoptosis of RCC.

A. EGF stimulates RCC cells to express CSF-1. CSF-1 measured in the supernatant of RCC cells (786-0 and Caki) by ELISA. Results are representative of 3 separate experiments. Values are means ± SEM. B. EGF stimulates proliferation of RCC cells and anti-EGFR Ab or anti CSF-1R Ab suppresses EGF dependent proliferation of RCC cells. HK2 cell line served as a positive control. Proliferation assessed by the MTT assay. Values are means ± SEM. Results are representative of 4 separate experiments. C. EGF mediated CSF-1 expression suppresses RCC apoptosis. RCC were cultured for 72h in the absence of CSF-1. Cells were stimulated with concentrations of TNF-α and LPS. EGF stimulation dampens apoptosis, and blocking the CSF-1R with anti-CSF-1 Ab or blocking the EGFR with anti-EGFR Ab increases TNF-α/LPS-induced TEC apoptosis. Means ± SEM; n=6/group. D. RCC and Mø release mediators that stimulate TEC to express EGF. RCC (786-0) and Mø were stimulated with TNF-α. Supernatants were added to the cultured TEC cell line (HK2) undiluted, diluted 1:1, 1:4 and 1:8 with media from stimulated RCC cells (786-0) or Mø (HL60). EGF expression was measured by ELISA. Note, comparable results stimulating RCC and Mø with TNF-α/LPS and TPA (data not shown).
To determine if EGF stimulated CSF-1 suppresses apoptosis of RCC cells, we stimulated RCC with TNF-α/LPS. Adding EGF dampened apoptosis of RCC. Dampened RCC apoptosis is specific for EGF as blocking with anti-EGFR Ab restored apoptosis and replacing the EGFR Ab with a control (mouse IgG2b) did not restore apoptosis. To determine whether EGF stimulates CSF-1 and thereby promotes proliferation and dampens apoptosis in RCC, we repeated the studies in Figure 8B and 8C and blocked the CSF-1R. Adding anti-CSF-1R Ab to the EGF stimulated TEC decreased proliferation (Figure 8B) and restored apoptosis (Figure 8C), thereby supporting the concept that EGF mediates proliferation and apoptosis via a CSF-1R dependent mechanism. Thus, EGF induces CSF-1 dependent increased RCC proliferation and reduced apoptosis.
CSF-1 and CSF-1R expression on TEC adjacent to RCC is mediated, in part, by EGF
CSF-1 and the CSF-1R are upregulated on TEC adjacent, not distant, by mediators released primilary from RCC (Figure 4). Therefore, we tested the hypothesis that EGF is released by RCC and is responsible for inducing CSF-1 and CSF-1R on TEC adjacent to RCC. Stimulating RCC and TEC lines with EGF increases CSF-1 expression (Figure 8A). Thus, we hypothesized that the TEC adjacent to the RCC are stimulated by RCC and TAMS to release EGF. Incubating the TEC cell line (HK2) with supernatant of TNF-α stimulated RCC (786-0) or Mø (HL60) led to a concentration dependent rise in EGF expression (Figure 8D, Supplemental Figure 2, bottom panel) as compared with TEC that were not incubated with RCC or Mø supernatant. This suggests that RCC and TAMS release mediators that induce EGF expression on TEC adjacent to RCC that, in turn, up-regulate CSF-1/CSF-1R expression and lead to a rise in TEC proliferation and a reduction in TEC apoptosis.
Discussion
We now report the novel finding that autocrine CSF-1 dependent RCC mechanisms are central to the growth of RCC. While recent findings indicate that the CSF-1R is expressed on RCC (5,16) our study clarifies the CSF-1 mediated mechanisms central to promoting RCC. In summary, CSF-1R and CSF-1 are abundantly co-expressed in RCC and in intra-renal TEC adjacent, but not distant, to RCC. The engagement of CSF-1 with its sole cognate receptor, CSF-1R, promotes RCC survival and proliferation and reduces apoptosis. This suggested that signaling via the CSF-1R promotes RCC growth. Moreover, blocking the CSF-1R with a CSF-1R tyrosine kinase inhibitor in human RCC xenografts in mice decreases the survival and proliferation of the RCC and the infiltration of Mø, thereby leading to a dramatic reduction in tumor mass. Furthermore, EGF induces CSF-1 and the CSF-1R on RCC, that in turn, promoting tumor cell proliferation and dampening tumor cell apoptosis. RCC has an impact on the kidney microenvironment adjacent to the tumor, as it releases mediators that induce CSF-1, CSF-1R and EGF expression on these TEC. Taken together, we suggest that targeting the CSF-1R is a potential therapeutic approach for human RCC.
Our findings highlight the CSF-1 dependent autocrine mechanism that fosters RCC growth. A wealth of evidence has detailed the involvement of CSF-1R bearing TAMS in tumor progression and metastasis (11,38). The paracrine interactions between tumor cells and TAMS facilitate the spread of a tumor in the host by promoting tumor cell migration, invasion and metastasis (4,15). As CSF-1 generated by the tumor is essential in regulating TAMS, CSF-1 and CSF-1R expressing Mø are key components in the tumor microenvironment regulating the host's fate. To identify CSF-1 dependent TAMS mediated mechanisms central to tumor progression, investigators studied tumors lacking the CSF-1R, thereby eliminating CSF-1 dependent tumor autocrine mechanisms (34,39). For example, CSF-1 blockade in human colon, devoid of CSF-1R, indicate that CSF-1 dependent Mø-mediated molecular mechanisms regulate tumor growth (40). However, malignant tumors such as breast and ovary co-express CSF-1 and the CSF-1R (5,7,9). Thus, CSF-1 and CSF-1R co-expression on breast and ovarian carcinoma prompted investigations to uncover a CSF-1 dependent autocrine loop that contributes to tumor invasiveness and metastasis (4,15,34) and tumor size (41). Moreover, heightened proliferation is evident in mammary glands of CSF-1 transgenic mice that result in mammary tumors (42). Our findings uniquely highlight the pivotal position of a CSF-1 dependent autocrine mechanism in the most common kidney tumor in humans, RCC. While a recent report indicates that the CSF-1R is expressed in RCC (5,16), using sequential sections from patients with RCC our findings illustrate CSF-1 and CSF-1R co-expression on proliferating TEC and establish that the CSF-1R is phosphorylated. And we detect enhanced tumor cell proliferation and dampened tumor cell apoptosis, proportional to CSF-1 expression in RCC lines, thereby indicating that RCC growth is mediated by CSF-1. Moreover, an increase in TAMS in the RCC and kidney adjacent to RCC suggests that, as in other cancers (38), CSF-1R expressing Mø may contribute to the progression of RCC. Taken together, CSF-1 dependent mechanisms are central to RCC.
Mediators released by the RCC maybe instrumental in altering the TEC adjacent to the RCC. We determined that CSF-1 and the CSF-1R are up-regulated on the TEC adjacent, but not distant, to RCC. As TAMS are more abundant in the kidney adjacent to RCC this suggests that mediators generated by the RCC and/or TAMS induce CSF-1 and CSF-1R on the TEC closest to the tumor. We determined that the RCC, and to a far lesser extent Mø, release mediators, including EGF, that induce CSF-1 and the CSF-1R on TEC. There are several possible consequences of up-regulating CSF-1 and the CSF-1R co-expression on TEC adjacent to RCC. It is possible that RCC injuries the TEC closest to the tumor and as we previously reported CSF-1 and CSF-1R co-expression triggers TEC proliferation and dampens TEC apoptosis, thus replenishing damaged tubules (17). On the other hand, up-regulated CSF-1 and the CSF-1R on TEC may be harmful. For example, over-expression of EGFR, promotes proliferation and in mutated EGFR leads to oncogenesis (43,44). Thus, up-regulated CSF-1 and CSF-1R co-expression on TEC adjacent to RCC enhances autocrine TEC proliferation, and along with exposure to additional triggers, may lead to malignant transformation of TEC. Alternatively, our findings indicate that CSF-1, generated by TEC adjacent to RCC, contribute to Mø recruitment. Thus, it is possible that these Mø are instrumental in promoting tumor growth. Despite the consequence of up-regulating these molecules in TEC adjacent to RCC, it is clear that RCC releases mediators that trigger CSF-1 and CSF-1R expression in the kidney microenvironment.
Is the CSF-1R a therapeutic target for human RCC? We now report that blocking the CSF-1R on RCC dramatically suppresses RCC growth. CSF-1R tyrosine kinase inhibitor, known to block the CSF-1R in vivo (28, 37) delivered into a human RCC xenograft mouse (human RCC cells implanted under the kidney capsule of athymic nude mice) dramatically reduced RCC mass. Our findings are consistent with other therapeutic approaches (anti-sense, interfering RNA, antibodies) that target the CSF-1R in other forms of cancer (colon, mammary) and lead to tumor suppression (40,45,46). However, our findings are in contrast to the failure of a mouse anti-CSF-1R Ab to inhibit tumor growth in subcutaneous mesothelioma and Lewis lung carcinoma models (47). Recalling that Mø, and some malignant tumors, express the CSF-1R, blocking the CSF-1R may suppress tumor expansion via CSF-1 dependent autocrine and paracrine mechanisms. While these tumors (mesothelioma and lung) are similar to RCC as they originate from cells of epithelial origin, CSF-1R expression on these tumors was not explored. And despite decreasing TAMS using this anti-CSF-1R Ab, the CSF-1 dependent mechanisms that may regulate these particular tumors have not been detailed. Thus, CSF-1R blockade may be a promising therapeutic approach for some, but not all, malignant tumors. It is worth noting that Sunitinib, a multi-targeted receptor tyrosine kinase inhibitor, that targets the CSF-1R along with many other receptor tyrosine kinases, is a first-line therapy for advanced RCC (1,48,49). Taken together, our findings clearly indicate that targeting the CSF-1R is a potential therapeutic for human RCC. Detailing the relative contribution of autocrine CSF-1 dependent and the unexplored potential Mø mediated paracrine mechanisms will be central to tailoring therapeutic strategies to halt human RCC.
CSF-1 and EGF expression and functions are closely linked in the kidney during inflammation. Following acute renal injury heparin-binding (Hb) EGF is up-regulated on TEC and promotes TEC proliferation, findings that parallel CSF-1 expression and function (50–52). Moreover, an increase in CSF-1R and EGFR expression correlates with poor prognosis of human tumors (43, 44). And in mammary tumors, blocking EGFR and CSF-1R inhibits carcinoma invasiveness (53). Finally, CSF-1 and EGF are components of a positive feedback loop. EGF generated by Mø induces CSF-1 expression in breast carcinoma cells and, in turn, CSF-1 generated by breast carcinoma cells induces EGF expression in Mø (15). Thus, we hypothesized that EGF induces CSF-1 in RCC. We determined that EGF stimulates CSF-1 expression in RCC lines and promotes RCC cell proliferation and dampens RCC cell apoptosis. As blocking EGFR and CSF-1R reverses these EGF incited findings, we suggest that EGF induces CSF-1 in RCC. In addition, we determined that RCC, and to a lesser degree Mø, release mediators that induce EGF expression, and in turn CSF-1/CSF-1R expression on TEC and thereby modify the kidney microenvironment.
In conclusion, we have identified CSF-1 dependent autocrine mechanisms that are instrumental in fostering RCC growth. Taken together, an autocrine CSF-1 dependent mechanism, intended to promote tubular repair following renal injury, has been subverted and now drives tumor expansion. Our findings suggest that CSF-1R is a potential therapeutic target for human RCC.
Supplementary Material
Acknowledgements
This work was supported by NIH grants DK-36149 (VRK), Alliance for Lupus Research (VRK) and the Deutsche Forschungsgemeinschaft (ME-3194/1-1) (JM). We thank Michaela Blanfeld for excellent technical support.
References
- 1.Cohen HT, McGovern FJ. Renal-cell carcinoma. The New Engl J Med. 2005;353(23):2477–90. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 2.Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, et al. The Heidelberg classification of renal cell tumours. The Journal of pathology. 1997;183(2):131–3. doi: 10.1002/(SICI)1096-9896(199710)183:2<131::AID-PATH931>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Gossage L, Eisen T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nature reviews. 7(5):277–88. doi: 10.1038/nrclinonc.2010.42. [DOI] [PubMed] [Google Scholar]
- 4.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer research. 2009;69(24):9498–506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soares MJ, Pinto M, Henrique R, Vieira J, Cerveira N, Peixoto A, et al. CSF1R copy number changes, point mutations, and RNA and protein overexpression in renal cell carcinomas. Mod Pathol. 2009;22(6):744–52. doi: 10.1038/modpathol.2009.43. [DOI] [PubMed] [Google Scholar]
- 6.Ide H, Seligson DB, Memarzadeh S, Xin L, Horvath S, Dubey P, et al. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14404–9. doi: 10.1073/pnas.222537099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers SK, Kacinski BM, Ivins CM, Carcangiu ML. Overexpression of epithelial macrophage colony-stimulating factor (CSF-1) and CSF-1 receptor: a poor prognostic factor in epithelial ovarian cancer, contrasted with a protective effect of stromal CSF-1. Clin Cancer Res. 1997;3(6):999–1007. [PubMed] [Google Scholar]
- 8.Sapi E, Kacinski BM. The role of CSF-1 in normal and neoplastic breast physiology. Proc Soc Exp Biol Med. 1999;220(1):1–8. doi: 10.1046/j.1525-1373.1999.d01-1.x. [DOI] [PubMed] [Google Scholar]
- 9.Kacinski BM. CSF-1 and its receptor in breast carcinomas and neoplasms of the female reproductive tract. Molecular reproduction and development. 1997;46(1):71–4. doi: 10.1002/(SICI)1098-2795(199701)46:1<71::AID-MRD11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 10.Komohara Y, Hasita H, Ohnishi K, Fujiwara Y, Suzu S, Eto M, et al. Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer science. 102(7):1424–31. doi: 10.1111/j.1349-7006.2011.01945.x. [DOI] [PubMed] [Google Scholar]
- 11.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–70. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douglass TG, Driggers L, Zhang JG, Hoa N, Delgado C, Williams CC, et al. Macrophage colony stimulating factor: not just for macrophages anymore! A gateway into complex biologies. International immunopharmacology. 2008;8(10):1354–76. doi: 10.1016/j.intimp.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Hemmerlein B, Markus A, Wehner M, Kugler A, Zschunke F, Radzum HJ. Expression of acute and late-stage inflammatory antigens, c-fms, CSF-1, and human monocytic serine esterase 1, in tumor-associated macrophages of renal cell carcinomas. Cancer Immunol Immunother. 2000;49(9):485–92. doi: 10.1007/s002620000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, et al. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood. 1998;92(12):4778–91. [PubMed] [Google Scholar]
- 15.Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer research. 2005;65(12):5278–83. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 16.Toma MI, Grosser M, Herr A, Aust DE, Meye A, Hoefling C, et al. Loss of heterozygosity and copy number abnormality in clear cell renal cell carcinoma discovered by high-density affymetrix 10K single nucleotide polymorphism mapping array. Neoplasia (New York, NY. 2008;10(7):634–42. doi: 10.1593/neo.08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menke J, Iwata Y, Rabacal WA, Basu R, Yeung YG, Humphreys BD, et al. CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. The Journal of clinical investigation. 2009;119(8):2330–42. doi: 10.1172/JCI39087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aharinejad S, Abraham D, Paulus P, Zins K, Hofmann M, Michlits W, et al. Colony-stimulating factor-1 transfection of myoblasts improves the repair of failing myocardium following autologous myoblast transplantation. Cardiovascular research. 2008;79(3):395–404. doi: 10.1093/cvr/cvn097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, et al. Biology and action of colony--stimulating factor-1. Molecular reproduction and development. 1997;46(1):4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 20.Roth P, Stanley ER. The biology of CSF-1 and its receptor. Curr Top Microbiol Immunol. 1992;181:141–67. doi: 10.1007/978-3-642-77377-8_5. [DOI] [PubMed] [Google Scholar]
- 21.Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell. 1985;41(3):665–76. doi: 10.1016/s0092-8674(85)80047-7. [DOI] [PubMed] [Google Scholar]
- 22.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. The Journal of clinical investigation. 2008;118(11):3522–30. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menke J, Hsu MY, Byrne KT, Lucas JA, Rabacal WA, Croker BP, et al. Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Fas(lpr) mice. J Immunol. 2008;181(10):7367–79. doi: 10.4049/jimmunol.181.10.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HT, Emala CW. Adenosine attenuates oxidant injury in human proximal tubular cells via A1 and A2a adenosine receptors 10.1152/ajprenal.00195.2001. Am J Physiol Renal Physiol. 2002;282(5):F844–52. doi: 10.1152/ajprenal.00195.2001. [DOI] [PubMed] [Google Scholar]
- 25.Bar-Shavit Z, Teitelbaum SL, Stricklin GP, Eisen AZ, Kahn AJ, Welgus HG. Differentiation of a human leukemia cell line and expression of collagenase inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(16):5380–4. doi: 10.1073/pnas.82.16.5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kikawada E, Lenda DM, Kelley VR. IL-12 Deficiency in MRL-Fas(lpr) Mice Delays Nephritis and Intrarenal IFN-gamma Expression, and Diminishes Systemic Pathology. J Immunol. 2003;170(7):3915–25. doi: 10.4049/jimmunol.170.7.3915. [DOI] [PubMed] [Google Scholar]
- 27.Menke J, Bork T, Kutska B, Byrne KT, Blanfeld M, Relle M, et al. Targeting transcription factor Stat4 uncovers a role for interleukin-18 in the pathogenesis of severe lupus nephritis in mice. Kidney international. 79(4):452–63. doi: 10.1038/ki.2010.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conway JG, Pink H, Bergquist ML, Han B, Depee S, Tadepalli S, et al. Effects of the cFMS kinase inhibitor 5-(3-methoxy-4-((4-methoxybenzyl)oxy)benzyl)pyrimidine-2,4-diamine (GW2580) in normal and arthritic rats. The Journal of pharmacology and experimental therapeutics. 2008;326(1):41–50. doi: 10.1124/jpet.107.129429. [DOI] [PubMed] [Google Scholar]
- 29.Sapi E, Flick MB, Rodov S, Carter D, Kacinski BM. Expression of CSFI and CSF-I receptor by normal lactating mammary epithelial cells. J Soc Gynecol Investig. 1998;5(2):94–101. doi: 10.1016/S1071-5576(97)00108-1. [DOI] [PubMed] [Google Scholar]
- 30.Maher MG, Sapi E, Turner B, Gumbs A, Perrotta PL, Carter D, et al. Prognostic significance of colony-stimulating factor receptor expression in ipsilateral breast cancer recurrence. Clin Cancer Res. 1998;4(8):1851–6. [PubMed] [Google Scholar]
- 31.Li W, Stanley ER. Role of dimerization and modification of the CSF-1 receptor in its activation and internalization during the CSF-1 response. The EMBO journal. 1991;10(2):277–88. doi: 10.1002/j.1460-2075.1991.tb07948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goede V, Brogelli L, Ziche M, Augustin HG. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer. 1999;82(5):765–70. doi: 10.1002/(sici)1097-0215(19990827)82:5<765::aid-ijc23>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 33.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer research. 1996;56(20):4625–9. [PubMed] [Google Scholar]
- 34.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. The Journal of experimental medicine. 2001;193(6):727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang B, Wang J, Gao J, Guo Y, Chen X, Wang B, et al. Alternatively activated RAW264.7 macrophages enhance tumor lymphangiogenesis in mouse lung adenocarcinoma. Journal of cellular biochemistry. 2009;107(1):134–43. doi: 10.1002/jcb.22110. [DOI] [PubMed] [Google Scholar]
- 36.Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL−/− renal cancer. Cancer research. 2005;65(12):5221–30. doi: 10.1158/0008-5472.CAN-05-0169. [DOI] [PubMed] [Google Scholar]
- 37.Conway JG, McDonald B, Parham J, Keith B, Rusnak DW, Shaw E, et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16078–83. doi: 10.1073/pnas.0502000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer research. 2007;67(11):5064–6. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 40.Aharinejad S, Abraham D, Paulus P, Abri H, Hofmann M, Grossschmidt K, et al. Colony-stimulating factor-1 antisense treatment suppresses growth of human tumor xenografts in mice. Cancer research. 2002;62(18):5317–24. [PubMed] [Google Scholar]
- 41.Toy EP, Lamb T, Azodi M, Roy WJ, Woo HH, Chambers SK. Inhibition of the c-fms proto-oncogene autocrine loop and tumor phenotype in glucocorticoid stimulated human breast carcinoma cells. Breast cancer research and treatment. doi: 10.1007/s10549-010-1247-7. [DOI] [PubMed] [Google Scholar]
- 42.Kirma N, Luthra R, Jones J, Liu YG, Nair HB, Mandava U, et al. Overexpression of the colony-stimulating factor (CSF-1) and/or its receptor cfms in mammary glands of transgenic mice results in hyperplasia and tumor formation. Cancer research. 2004;64(12):4162–70. doi: 10.1158/0008-5472.CAN-03-2971. [DOI] [PubMed] [Google Scholar]
- 43.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19(56):6550–65. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 44.Wells A. EGF receptor. The international journal of biochemistry & cell biology. 1999;31(6):637–43. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 45.Aharinejad S, Paulus P, Sioud M, Hofmann M, Zins K, Schafer R, et al. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer research. 2004;64(15):5378–84. doi: 10.1158/0008-5472.CAN-04-0961. [DOI] [PubMed] [Google Scholar]
- 46.Paulus P, Stanley ER, Schafer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer research. 2006;66(8):4349–56. doi: 10.1158/0008-5472.CAN-05-3523. [DOI] [PubMed] [Google Scholar]
- 47.MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 116(19):3955–63. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- 48.Grimaldi AM, Guida T, D'Attino R, Perrotta E, Otero M, Masala A, et al. Sunitinib: bridging present and future cancer treatment. Ann Oncol. 2007;18(Suppl 6):vi31–4. doi: 10.1093/annonc/mdm221. [DOI] [PubMed] [Google Scholar]
- 49.Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, et al. Sunitinib in patients with metastatic renal cell carcinoma. Jama. 2006;295(21):2516–24. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC. Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol. 2003;14(12):3147–54. doi: 10.1097/01.asn.0000098681.56240.1a. [DOI] [PubMed] [Google Scholar]
- 51.Hise MK, Salmanullah M, Liu L, Drachenberg CI, Papadimitriou JC, Rohan RM. Control of the epidermal growth factor receptor and its ligands during renal injury. Nephron. 2001;88(1):71–9. doi: 10.1159/000045962. [DOI] [PubMed] [Google Scholar]
- 52.Harris RC. Growth factors and cytokines in acute renal failure. Advances in renal replacement therapy. 1997;4(2 Suppl 1):43–53. [PubMed] [Google Scholar]
- 53.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer research. 2007;67(6):2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.