

Abstract
Purpose
Mortality of patients with head and neck squamous cell carcinoma (HNSCC) is primarily driven by tumor cell radioresistance leading to locoregional recurrence (LRR). In this study, we use a classification of TP53 mutation (disruptive vs. nondisruptive) and examine impact on clinical outcomes and radiation sensitivity.
Experimental Design
Seventy-four patients with HNSCC treated with surgery and postoperative radiation and 38 HNSCC cell lines were assembled; for each, TP53 was sequenced and in vitro radioresistance measured using clonogenic assays. p53 protein expression was inhibited using shRNA and over-expressed using a retrovirus. Radiation-induced apoptosis, mitotic cell death, senescence, and ROS assays were performed. The effect of the drug metformin on overcoming mutant p53-associated radiation resistance was examined in vitro as well as in vivo, using an orthotopic xenograft model.
Results
Mutant TP53 alone was not predictive of LRR; however, disruptive TP53 mutation strongly predicted LRR (p=0.03). Cell lines with disruptive mutations were significantly more radioresistant (p<0.05). Expression of disruptive TP53 mutations significantly decreased radiation-induced senescence, as measured by SA-beta-gal staining, p21 expression, and release of reactive oxygen species (ROS). The mitochondrial agent metformin potentiated the effects of radiation in the presence of a disruptive TP53 mutation partially via senescence. Examination of our patient cohort showed that LRR was decreased in patients taking metformin.
Conclusions
Disruptive TP53 mutations in HNSCC tumors predicts for LRR, due to increased radioresistance via the inhibition of senescence. Metformin can serve as a radiosensitizer for HNSCC with disruptive TP53, presaging the possibility of personalizing HNSCC treatment.
Keywords: p53, radiation, senescence, mitotic catastrophe, metformin
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the 6th leading cause of cancer worldwide, with an estimated 49,260 diagnoses each year in the United States alone (1). Although multimodality therapy is important in the management of HNSCC, the eradication of locoregional disease in the primary or postoperative setting is primarily achieved by external beam radiation. As the vast majority of patient deaths from HNSCC are due to locoregional recurrence (LRR), the survival of the patient largely depends upon the radiosensitivity the tumor. Unfortunately, there are few biomarkers to determine radioresistance in HNSCC.
One candidate biomarker is TP53, which encodes the p53 protein and is the most commonly altered gene in HNSCC, as demonstrated most recently by our group and others through whole exomic sequencing of a large panel of HNSCC tumors (2). TP53 has been previously investigated as a biomarker for LRR following radiotherapy in HNSCC with mixed results (3–6). A possible explanation for this finding is mutation-specific functionality of the p53 protein. One large study classifying TP53 mutations in HNSCC described TP53 mutation as “disruptive” or “nondisruptive” based upon alteration of DNA binding (7). Any mutation in either the L2 or L3 loop of the DNA binding domain, resulting in a polarity change within the protein, or any stop codon was classified as disruptive. In that study, the presence of a disruptive mutation was associated with poor survival in a heterogeneously treated patient population. Other studies have examined alternative classifications of TP53, which were also prognostic of survival in HNSCC (8; 9). Despite these results, there has been little study of the relationship between TP53 classification and LRR following radiotherapy.
The role of TP53 in in vitro radiosensitivity is also an area of active investigation much of the prior studies producing contradictory results (10–16). Radiation is known to result in p53 activation and apoptosis; however, the contribution of radiation-induced apoptosis to tumor response is not clear, and there are strong arguments that, for most tumor cells, apoptosis plays a minimal role in radiation response (17; 18). In contrast, it has been suggested that mitotic death is the primary mechanism of radiation-induced cell death (19). In this process, abnormal cell division due to DNA damage leads to the formation of large cells with multiple micronuclei (20). It is unclear whether this form of cell death is associated with TP53 (21–24). Furthermore, emerging data suggest that senescence may play a role in the radiation response, which in some models is dependent upon wild type TP53 (25). Regardless, the role of TP53 in radiosensitivity is far from clear.
To investigate the role of TP53 in radiosensitivity, the current study was performed to determine if a classification of TP53 (wild type, nondisruptive, or disruptive mutation) predicts for radiosensitivity in vitro as well as in the clinical setting. Furthermore, we wished to determine the mechanisms involved in the observed radiation response in vitro and use this knowledge to preferentially radiosensitize HNSCC cells.
Methods
Clinical samples
All clinical studies reported in this study have been approved by the University of Texas MD Anderson Cancer Center Institutional Review Board. Seventy-four snap-frozen, pre-treatment tumor samples were collected from patients with HNSCC at high risk for LRR treated with surgical resection and postoperative radiotherapy from 1992–2003. DNA was extracted from tumor samples, and TP53 was sequenced (exon 2–12) using Sanger sequencing as described previously (Ow et al, submitted). Mutations were then classified as according to the method of Poeta and colleagues (7). Patients were evaluated every 2–3 months for 1 year following treatment, every 3–4 months the following year, and every 6 months thereafter.
Cell culture and constructs
HNSCC cells were cultured in a 37° C incubator at 5% CO2 for this study as described previously (26). Cells stably expressing short hairpin RNA specific for p53 (shp53) were generated as described previously (27). Briefly, after infection with GFP tagged empty lentiviral vector (LVTHM) or encoding a short-hairpin RNA against p53 (LVUH-shp53) (Addgene, Cambridge, MA), cells were cultured for several passages, and then sorted using flow cytometry. GFP-positive cells were cultured normally. TP53 constructs (C176F, R282W, R175H and E336X) were generated by extracting RNA from cell lines known to express these mutants. RT-PCR was then performed using TP53 specific primers. The resulting product was purified and inserted into a pBabe retroviral vector containing a puromycin resistance insert (pBabe-puro) (Addgene, Cambridge, MA) using standard cloning techniques. The resulting vectors were verified by Sanger sequencing at the MD Anderson Cancer Center DNA core facility. After transfection and packaging in 293T cells, viral supernatant was centrifuged at 1,000 rpm to remove cellular debris and added to UMSCC1 cells in combination with polybrene. After one passage, the cells underwent selection with puromycin. All cell lines used in this study have been authenticated against the parental recipient cell line via STR analysis.
Clonogenic assay
HNSCC cells were seeded in 12 well plates at predetermined densities for different radiation doses to allow for an approximately equal number of resultant colonies. The next day, cells were irradiated using a high dose rate 137Cs irradiator (4.5 Gy/min) and cultured for 10–14 days to allow for colony formation. Cells were then fixed in a 3% crystal violet/10% formalin solution. Colonies of >50 cells were then counted and survival fraction was determined. All treatments were in triplicate or greater. For metformin (Sigma-Aldrich, St. Louis, MO) or z-Vad-fmk (BD Pharmingen, San Diego, CA) treatment, cells were pre-treated for 2h prior to radiation with either drug or vehicle (PBS and DMSO respectively) and drug treatment continued overnight. For N-acetyl cysteine (NAC) treatment, the cells were treated starting 2 hours following radiation and drug treatment continued overnight. The cells were then washed and cultured in fresh media for the remainder of the experiment.
Immunofluorescence
HNSCC cells were plated on coverslips and treated as indicated. Cells were washed twice with PBS and fixed using a 1:1 mixture of methanol and acetone for 10 minutes. Cells were then washed in TBS-Tween (TBST) and permeabilized using 0.1% Triton-X for 5 minutes. Cells were again washed with TBST and incubated with FITC-Phalloidin for 1 hour at room temperature. Cells were washed with TBST and mounted on standard glass slides using DAPI-Vectashield (Vector Labortories, Burlingame, CA). For assessment of mitotic death, the number of cells with one or more micronucleus per high power field (hpf) were counted and reported as a percentage of total cells per field. Images were taken from five quadrants, in duplicate, and pooled.
SA-beta-gal staining
Senesence Associated (SA)-beta-gal staining was performed per the manufacturer’s instructions (Cell signaling, Danvers, MA). Briefly, HNSCC cells were plated in 6 well plates were irradiated with 4–6 Gy on the next day. Cells were cultured normally until the indicated times post treatment. Cells were then fixed for 10 minutes and stained overnight for SA-beta-gal activity at 37° C. Blue staining cells were scored as senescent and reported as a percentage of all the cells observed per high power field.
Annexin V staining
Annexin V stain was performed per the manufacturer’s instructions (BD Pharmingen, San Diego, CA). Briefly, HNSCC cells were cultured normally and treated as indicated. Media was collected, and adherent cells were briefly trypsinized and added to the previously collected media. The cells were then centrifuged, supernatant was removed, and the cells were washed in PBS. The cells were then re-suspended in Annexin V binding buffer, and Annexin V and 7-AAD was added. Samples were mixed gently and incubated at RT. Samples were then analyzed for Annexin V and 7-AAD staining using a Beckman Coulter XL 4 color cytometer (Beckman Coulter, Brea, CA), and the data were analyzed using Flo-Jo software (FloJo, Ashland, OR). Early apoptosis was determined by the proportion of cells that were Annexin V positive and 7-AAD negative.
Cell cycle analysis
HNSCC cells were seeded in a 6 well plate and incubated for the indicated time. Media was collected; cells were washed twice in PBS and trypsinized. The resulting cell suspension was washed in PBS and fixed with 70% ethanol at room temperature for 30 minutes. Cells were then washed in PBS and stained with propidium iodide. Cell cycle detection was then performed using a Beckman Coulter XL 4 color cytometer, (Beckman Coulter, Brea, CA) and the data were analyzed using Flo-Jo software (FloJo, Ashland, OR).
Reactive oxygen species (ROS) measurement
Intra-cellular ROS levels were measured according to previously published protocols using 5-(and-6)-carboxy-2′,7′-dichlorofluorescein (CM-H2DCFDA) dye(28). Briefly, cells were loaded with CM-H2DCFDA for 60 minutes in culture media. Media was changed prior to irradiation to remove excess dye. Fluorescence was measured at various times following irradiation using a standard spectrophotometer, normalized to the control condition and total amount of DNA (29). For flow cytometric experiments, cells were incubated with CM-H2DCFDA dye as described and trypsinized, and fluorescence was analyzed using flow cytometry as described above.
Immunoblotting
Cells were treated as indicated and washed three times with cold PBS. Standard lysis buffer was then added to each plate and incubated on ice for 15 minutes. Cells were then collected using a plastic scraper and centrifuged at 14,000 rpm at 4° C for 10 minutes. The supernatant was removed, and total protein concentration was then calculated using Bio-Rad protein assay (Bio-Rad, Hercules, CA). Immunoblot analysis was performed as described previously (26). Membranes were blocked for 1 hour at RT using 1% powdered milk in 0.1% Tween 20 in Tris-buffered saline, then incubated overnight with either anti-p53 DO-1 (Santa Cruz Biotechnology, CA), anti-β actin (Cell signalling, Danvers, MA), or anti-p21 (BD Pharmingen, San Diego, CA) at 4° C. The following day, membranes were washed with 0.1% Tween 20 in Tris-buffered saline and incubated for 1 hour at RT with species-specific secondary antibody. For p53 and actin, fluorescently conjugated secondary antibodies (Invitrogen, Carlsbad, CA) were used and signal was analyzed using an odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE) and associated software (v3.0). For p21, horseradish peroxidase conjugated anti-rabbit immunoglobulin G (Santa Cruz Biotechnology, Santa Cruz, CA) was used and signal was generated using the SuperSignal West chemiluminescent system (Pierce Biotechnology, Rockford, IL).
p21 transcription
Induction of p21 transcription was measured via luciferase reporter activity using a vector containing the 2.4 kb p21 promoter and firefly luciferase (pWWP-Luc)(30) (Addgene, Cambridge, MA). UMSCC1 (p53 null) cells expressing various TP53 constructs were co-transfected with pWWp-Luc and a constitutively active Renilla luciferase construct using Liopfectamine 2000 (Introgen, Carlsbad, CA). Cells were irradiated the following day at the indicated doses and incubated for 24 hours prior to collection. Luciferase activity was measured using the Promega Dual-Luciferase assay system (Madison, WI).
Orthotopic mouse model
All animal experimentation was approved by the University of Texas MD Anderson Cancer Center Animal Care and Use Committee (ACUC). The orthotopic mouse model has been previously described (31). Briefly, mice were injected with 100,000 HN 31 cells in the anterior half of the oral tongue on Day 0. Once tumor growth was noted, tumors were treated with radiation using a Co60 irradiator and custom lead blocks (5 Gy) on Day 8 post-injection and/or metformin (250mg/kg daily, intra-peritoneal). Each treatment group consisted of a total of 10 mice. Mice received a total of 8 daily metformin treatments over the experimental period. Tumor measurements were obtained on Days 6, 13, 16, and 20 post-injection. Tumor volume was calculated as previously described (31).
Statistics
LRR and overall survival (OS) for the patient population was calculated using the Kaplan-Meier method, and comparisons between groups were determined using log rank statistics. Multivariate analysis for LRR was performed using forward step-wise Cox regression analysis. Variables included: tumor and nodal stage, surgical margin status or extracapsular extension, site, gender, smoking history, and TP53 status. ANOVA with post-hoc analysis or student’s t-tests were performed to analyze in vitro data and tumor volume. All p values are 2-sided. P values less than 0.05 were considered significant.
Results
Disruptive TP53 mutations are associated with increased LRR following PORT
Disruptive TP53 is associated with decreased survival in HNSCC, which we hypothesized was due to higher rates of LRR following PORT. To explore this hypothesis, we sequenced TP53 in HNSCC tumors from 74 patients treated with PORT. Patient characteristics were similar between different TP53 classification groups (Supplemental Table 1). Median follow-up of surviving patients was 154 months (range 82–185). No significant difference in LRR was found between patients with wild type TP53 and any mutant TP53 (Figure 1A). However, classification of TP53 as disruptive, nondisruptive, or wild type, demonstrated that disruptive TP53 is an independent predictor of LRR on multivariate analysis (p=0.022). Patients with disruptive TP53 had a 5 year freedom from LRR of 41% compared to 64% and 76% in patients with wild type and nondisruptive TP53, respectively (p=0.03, Figure 1B). No significant difference in LRR was seen between patients with wild type and nondisruptive TP53 (p=0.48, Figure 1B). Similarly, the OS rates for patients in this study were predicted by TP53 classification. Specifically, 5 year OS for patients with disruptive TP53 mutations was 19% compared to 52% in patients with wild type TP53 and 41% in patients with nondisruptive TP53 (p=0.031).
Figure 1. Locoregional Recurrence (LRR) occurs more frequently in patients with tumors expressing TP53 disruptive mutations.

(A) LRR in the study population in patients with either wild type TP53 or any TP53 mutation (p=0.9). (B) LRR in the same group of patients with the indicated TP53 status.
Disruptive TP53 mutations are associated with p53-mediated radioresistance
The finding that disruptive TP53 mutations are associated with a high rate of LRR led to the hypothesis that HNSCC tumor cells with disruptive TP53 are intrinsically more radioresistant. To test this hypothesis, we evaluated the radiosensitivity of a panel of 38 HNSCC cell lines of known TP53 status (Supplemental Table 3) and found that cell lines harboring disruptive TP53 mutations were significantly more radioresistant than those with either nondisruptive or wild type TP53 (Figure 2A).
Figure 2. In vitro radiosensitivity is modulated by p53 expression depending on TP53 classification.

(A) A panel of 38 HNSCC cell lines was sequenced for TP53 status and evaluated for radiosensitivity using standard clonogenic assays. Each cell line was then grouped by TP53 status, and the average clonogenic survival data for each group is shown. (B) The effect of stable inhibition of wild type p53 expression (shp53) on radiosensitivity in HN 30 and UMSCC1 17A cells. (C) The effect of forced expression of wild type, R175H, R282W, or C176F TP53 in p53 null UMSCC1 cells on radiosensitivity. (D) The effect of stable inhibition of p53 expression (shp53) on radiosensitivity in FADU and HN 31 cells (disruptive TP53).
To determine if this observed correlation was due to p53 expression, p53 was silenced using stably expressed short hairpin RNA (shp53) in the following HNSCC cell lines: i) UMSCC1 17A and HN 30 (wild type TP53), ii) Detroit (R175H, nondisruptive TP53), and iii) HN 31 (C176F) and FADU (R248L) (disruptive TP53). Inhibition of p53 expression in cell lines expressing wild type or nondisruptive TP53 rendered cells more radioresistant (Figure 2B & Supplemental Table 2). However, inhibition of p53 expression in cell lines expressing disruptive TP53 rendered these cells more radiosensitive (Figure 2D & Supplemental Table 2). Conversely, UMSCC1 cells, which have no endogenous p53, were engineered to express either: i) wild type TP53, ii) nondisruptive TP53 (R175H, R282W), or iii) disruptive TP53 (C176F, E336X). Disruptive TP53 expressing UMSCC1 cells were found to be radioresistant relative to wild type TP53 and nondisruptive TP53 expressing cells (Figure 2C & Supplemental Table 2).
Alteration of p53 expression does not affect radiation-induced mitotic death or apoptosis
To explore the means by which disruptive TP53 mutation confers radioresistance in HNSCC cells, we evaluated several modes of cellular response to radiation. Initially we examined mitotic death, thought to be one of the primary modes of cell death following radiation(19). Mitotic death, as measured by the presence of micronuclei, was significant following radiation, with all HNSCC cell lines tested exhibiting mitotic death in 20–30% of irradiated cells. However there was no correlation between micronuclei formation, TP53 expression, and relative radiosensitivity (Supplemental Figure 2).
Furthermore, when either annexin V or sub-G1 staining were analyzed, neither of these measures of apoptosis were related to TP53 status (Supplemental Figure 3). In fact, only a very small proportion of apoptotic cells were observed (~1–5%), even in those cells shown to be radiosensitive in the clonogenic survival assay. As radiation-induced apoptosis is thought to be caspase-dependent(32), we also treated cells with a pan-caspase inhibitor (Z-vad-fmk) to determine the role of apoptosis in this model. Inhibition of caspase activity had almost no effect on clonogenic survival following radiation (Supplemental Figure 3), supporting to the hypothesis that apoptosis does not play a major role in the radiation response.
Radiation robustly induces SA-beta-gal activity in HNSCC cells which is strongly associated with TP53 status and correlates with clonogenic survival
In cells with wild type or nondisruptive TP53, treatment with radiation resulted in a decrease in the proportion of cells in S phase and a less prominent G1 arrest (Supplemental Figure 4). Furthermore, inhibition of p53 expression in HN 30 (wild type TP53) cells partially abrogated the observed inability to progress through S phase. Because this phenomenon has been previously linked to cellular senescence, we hypothesized a role for altered senescence in the observed differences in radiosensitivity. To evaluate for cellular senescence, cells were treated with radiation and stained for the SA-beta-gal. As shown in Figure 3, radiation treatment led to significant levels of SA-beta-gal activity in cell lines expressing either wild type (HN 30) or nondisruptive mutant TP53 (Detroit, R175H). Furthermore, in these cells with SA-beta-gal staining, the morphologic characteristics of cellular senescence were observed, specifically a large, flattened cell with the classic “fried egg” appearance on microscopy. Inhibition of p53 expression in both of these cell lines significantly reduced levels of radiation-induced SA-beta-gal activity. However, in HN 31 (C176F, disruptive TP53) cells, which have disruptive TP53 and are otherwise isogenic with the HN 30 (wild type TP53) cell line, inhibition of p53 expression was associated with elevated levels of radiation-induced SA-beta-gal activity. Forced expression of wild type, as well as two nondisruptive TP53 mutations (R175H and R282W) in p53 null cells, increased radiation-induced SA-beta-gal activity; conversely, expression of disruptive TP53 mutations (C176F and E336X) led to decreased SA-beta-gal activity compared to empty vector control (Figure 3B). Furthermore, in experiments comparing micronuclei formation and SA-beta-gal staining in the same cell, the main differences between cells of different TP53 statuses appear to be within the cells staining positive for SA-beta-gal alone (Figure 3D).
Figure 3. TP53 modulates radiation-induced senescence, which correlates with radiosensitivity.

(A) Representative light microscopy showing SA-Gal staining. (B) Percentage of SA-Gal positive cells per total number of cells in a high power field after 4 Gy of radiation for the times indicated in UMSCC1 cells expressing representative wild type and mutant TP53 constructs and HN 30 (WT), Detroit (R175H) and HN 31 (C176F) where p53 is inhibited. (C) Representative microscopy showing SA-Gal staining, DAPI fluorescence, and GFP. (D) Percentage of cells either SA-Gal positive (S alone), exhibiting micronuclei (M alone) or both at 4 days after 4 Gy of radiation in HN 30 and HN 31 cells where p53 expression is inhibited and UMSCC1 cells expressing representative wild type and mutant TP53 constructs. * - significantly elevated over baseline (p<0.05), + - significantly different from null at the indicated time point (p<0.05), # - significantly different from HN 30 control.
The effects of TP53 status on radiation-induced SA-beta-gal activity are correlated with p21 expression and mediated by ROS production
In addition to SA-beta-gal activity, senescence has been previously linked to induction of p21 expression and ROS production, both of which are believed to be necessary for maintenance of the senescent phenotype (33–36). To determine if radiation-induced p21 expression correlates with TP53 classification, we assayed p21 protein levels following radiation in cell lines expressing representative TP53 mutations. In cells expressing wild type TP53 or a nondisruptive TP53 mutation, p21 protein and reporter activity were induced by radiation treatment (Figure 4A). In contrast, cells expressing disruptive TP53 (C176F) had no induction of p21.
Figure 4. Radiation-induced senescence is associated with p21 expression and dependent on prolonged ROS production.

(A) p21 protein expression and p21 luciferase reporter activity in UMSCC1 cells expressing the indicated TP53 constructs treated with the indicated doses of radiation for 24h unless otherwise stated. (B) ROS production measured as indicated in the methods after 2 Gy of radiation. (C) Percentage of cells either SA-Gal positive (S alone), exhibiting micronuclei (MC alone) or both at 4 days after 4 Gy of radiation. Cells were treated with the indicated dose of NAC starting 2 h after radiation and treatment continued overnight. (D) Clonogenic survival after 2 Gy of radiation. Cells were treated with NAC in an identical manner to (C). * - significantly increased over unirradiated control (p<0.05), + - significantly different from UMSCC1 wild type in the same group (p<0.05). # - significantly increased compared to non-NAC treated group (p<0.001).
ROS also play a key role in senescence and radiation effect. In the current study, radiation-induced ROS levels correlated with p21 expression, SA-beta-gal activity, and relative radiosensitivity. Specifically, ROS were most highly induced in cells which had greater levels of radiation induced SA-beta-gal activity (wild type and R175H), with little or no ROS induction was seen in cells with disruptive TP53 (C176F) (Figure 4B). Inhibition of ROS using N-acetyl cysteine (NAC) 2 hours following radiation treatment, designed to inhibit secondary ROS production following the initial radiation insult, dramatically decreased senescence in cell lines with normally high levels of radiation-induced SA-beta-gal, but had no effect on cells with disruptive TP53 (Figure 4C). These results closely correlated with the observed effects of NAC on clonogenic survival following radiation (Figure 4D).
Metformin preferentially potentiates the effects of radiation in HNSCC cells partially via increased senescence
Based on the combined morphologic appearance of irradiated cells, the presence of SA-beta-gal staining, the characteristic induction of p21 and ROS, and the lack of apoptosis, cells with either wild type or non-disruptive TP53 mutation appear to be undergoing radiation-induced senescence, while cells with a disruptive TP53 mutation are not. Because the induction of senescence may be clinically beneficial, we wished to investigate drug based therapies designed to this end. As there are no specific senescence inducing agents currently available, we examined metformin, an anti-diabetic agent with minimal clinical toxicity, that has been shown to both induce ROS in some cellular contexts (37) as well preferentially target cell lines with mutant TP53 (38). In the current study, concurrent radiation and metformin treatment was active against HNSCC cell lines that harbored disruptive TP53 (Supplemental Figure 5). Conversely, no effect of metformin was seen in cells expressing wild type TP53 (HN 30 and MCF-7). Also, the addition of metformin dramatically increased ROS in HN 31 cells, which have a disruptive (C176F) TP53 mutation, but had much less effect in HN 30 cells, their wild type TP53 isogenic counterpart (Supplemental Figure 5). Similarly, metformin decreased clonogenic survival following radiation and increased radiation-induced SA-beta-gal activity in HN 31 cells (C176F, disruptive TP53), but had little effect in HN 30 cells (wild type TP53) (Figure 5A & B). Furthermore, after inhibition of wild type p53 expression, metformin was found to potentiate SA-beta-gal activity and decrease clonogenic survival (Figure 5A & B).
Figure 5. Metformin selectively radiosensitizes cells with disruptive TP53 mutations, partially due to altered senescence.
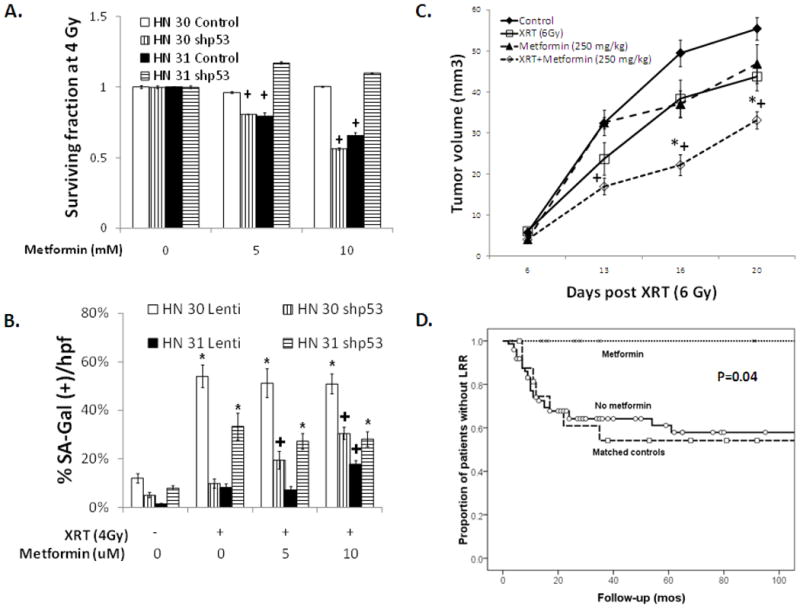
(A) Clonogenic survival after treatment with radiation at the indicated doses along with metformin for 24 hours in HN 30 and HN 31 cells in which p53 expression is inhibited using shRNA (shp53). (B) Percentage of SA-beta-gal positive cells per total number of cells in a high power field 4 days following 4 Gy of radiation and the indicated doses of metformin. * - significantly different from unirradiated control (p<0.05), + - significantly changed compared to no metformin treatment (p<0.05). (C) Tumor volume in mice with orthotopic tumors derived from HN 31 cells (C176F) after treatment with radiation (6 Gy), metformin (250 mg/kg, intraperitoneal) or both. * - significantly different from radiation alone (p<0.05), + -significantly different from metformin treatment alone (p<0.05). (D) LRR in patients taking metformin during treatment compared to the remainder of the study population as well as patients matched for tumor and nodal stage, surgical margin status, and TP53 status to the metformin treated group (p=0.04).
Given the above findings, we examined the effect of metformin treatment on tumor response to radiation in an orthotopic model of HNSCC described previously (26; 31). As seen in Figure 5C, the addition of metformin to radiation dramatically decreased tumor growth compared to either radiation alone (p=0.015) or metformin alone (p=0.008).
To further investigate the clinical utility of metformin, a cohort of patients treated with PORT for HNSCC was identified and the impact of concurrent metformin use was investigated. In addition to the patients in the current study, an additional 30 patients with similar disease characteristics and treatment as well as known TP53 status were evaluated. Although only 10 patients were taking metformin at the time of radiation, these patients had a dramatically lower LRR rate compared to controls matched for tumor and nodal stage, surgical margin status, and TP53 status (p=0.04, Figure 5D). On multivariate analysis controlling for TP53 classification, tumor stage, and surgical margin positivity, metformin use was significantly associated with decreased LRR (p=0.04) as well as improved OS (p=0.01). Specifically, 5 year OS was 87% in the patients taking metformin compared to 41% in the remaining patients (p=0.04).
Discussion
In HNSCC, the majority of patient deaths result from LRR, with up to 60–75% dying of their disease (39). The primary method of treating advanced HNSCC involves radiation, either in the definitive or postoperative setting. To date, there are few clinically useful predictive indicators of LRR following radiotherapy. In the current study, we examined LRR in HNSCC and, for the first time, demonstrated that the disruptive classification of TP53 is predictive of poor response to a specific therapy, namely PORT. Interestingly, mutant TP53 alone was not predictive for LRR, arguing for the use of a TP53 mutation classification scheme that can demonstrate differential function between TP53 mutants in regards to radioresistance.
Because the goal of PORT is to eradicate microscopic residual disease, we hypothesized that LRR would primarily result from intrinsically radioresistant cells. To investigate this, we utilized an in vitro approach, combining silencing of endogenous p53 expression with genetically engineered expression of different TP53 mutations; this approach demonstrated that HNSCC cells with disruptive TP53 exhibited a radioprotective effect of p53 expression, arguing for a “gain of function” (GOF) in regards to radiosensitivity. These GOF mutations in TP53 have been shown previously for other cellular processes; however, this report is one of the first to do so for in vitro radiosensitivity. Furthermore, we found that two types of cell death most commonly associated with radiation, namely apoptosis and mitotic death, were either not present and/or unaffected by TP53 status. In fact, the main response to radiation affected by TP53 status in this model was cellular senescence.
Senescence is a form of cell arrest in which cells remain metabolically active, but lack replicative potential. Several recent studies have linked response to chemotherapy and radiation to this process; however, the exact role of senescence in the response to radiation is far from clear (17; 40; 41). In the current study, we found radiation-induced senescence correlated strongly with in vitro radiosensitivity and was inhibited in the presence of a disruptive TP53 mutant, again arguing for a GOF by at least a subset of TP53 mutations within the disruptive category, driving resistance to radiotherapy.
Induction of both p21 and ROS appear to be critical mediators of cellular senescence. Induction of p21 alone can lead to increased ROS production (34), and ROS inhibition can reverse senescence in this context. Conversely, ROS have been linked to induction of p21, and a feedback loop between ROS and p21 is proposed to be required for DNA damage-induced senescence (35; 42; 43).
In the current model, we found radiation-induced ROS and p21 only in HNSCC cells with wild type and nondisruptive TP53. Specifically, p21 expression was increased in response to radiation in the context of the nondisruptive TP53 mutation R175H. Previously, R175H has been shown to have little remnant p53 driven transcription (44; 45). Furthermore, in one report, the disruptive mutation, C176F, has been shown to have some p53 dependent transcriptional activity in a yeast based assay (45); however, this has not been observed in other studies (46; 47). Nevertheless, in the current model, both protein expression and transcription of p21 is induced by radiation in cells expressing either wild type or R175H TP53. This could be due to p53 transcription independent induction of p21 similar to that seen in other models (48; 49) or it could result from residual p53 transcriptional activity. It has been shown recently that only a small subset of p53 transcriptional targets are necessary for induction of senescence; thus, even a small remnant of transcriptional function may be sufficient (50). Further studies are in progress to discern the mechanism behind this phenomenon.
Radiation-induced ROS production was also inhibited by disruptive TP53. In our study, inhibition of secondary ROS production several hours after the initial cellular insult by radiation had a dramatic effect on cellular senescence. This argues for prolonged ROS production following radiation playing a key role in the maintenance of the senescent phenotype. These ROS are most likely derived from the mitochondria and, in fact, we have previously observed decreased baseline mitochondrial complex activity in cells expressing C176F compared to wild type TP53 (27). This argues for a suppressive effect on the mitochondria of disruptive TP53, which is relieved by inhibition of p53 expression. This offers a possible link between disruptive TP53 mutations GOF and the inhibition of radiation-induced ROS and senescence. Theoretically, disruptive p53 could interact with known targets of wild type p53 involved in mitochondrial function, exerting an inhibitory function as opposed to the stimulation seen with wild type or some nondisruptive forms of TP53.
Our data strongly suggest that disruptive TP53 portends poor clinical outcome in HNSCC associated with increased radioresistance. Parallel work in our laboratory has demonstrated that cells with disruptive TP53 exhibit a therapeutically exploitable decrease in metabolic flexibility (27). One agent that can target this process is metformin. Recently, it has been shown that patients taking metformin have improved responses following neoadjuvant chemotherapy (51) and that metformin is a radiosensitizer in vitro (52). Metformin is hypothesized to affect mitochondrial function, and several studies have shown that it induces ROS in certain cellular backgrounds (37; 38). Also, it has been hypothesized that metformin may induce senescence in cells that are “senescence prone” (53). Interestingly, when we examined the effects of metformin in vitro, radiation-induced senescence and toxicity were potentiated only in cells expressing disruptive TP53, an effect that was modulated by altering p53 expression. This dramatic effect on the efficacy of radiation was also seen in our in vivo orthotopic model, as well as in patients taking metformin at the time of PORT. Although this only included 10 patients in the metformin group, the reduction in LRR was profound and argues for the possibility of a dramatic clinical benefit with targeting of radiation-induced senescence.
The current study is limited by its retrospective nature, although the patient cohort includes a homogenous group of patients treated uniformly with surgery and PORT. Furthermore, the in vitro studies are limited by the analysis of a subset of disruptive and nondisruptive TP53 mutations, and the possibility exists that other disruptive TP53 mutations may behave differently. However, examination of a large panel of HNSCC cell lines confirmed the in vitro predictive value of the disruptive classification. Also, clinical data showing dramatically increased LRR in patients with disruptive TP53 mutations supports the overall hypothesis. Although the disruptive category is empiric in nature, the fact that senescence appears to be strongly inhibited by these mutations allows further refinement. Specifically, testing of TP53 mutants’ ability to induce senescence could be performed, refining an already clinically predictive model. Finally, although the clinical data regarding the benefit of metformin is compelling, only a small number of patients were treated with the drug at the time of radiation. Despite the intriguing in vitro, in vivo, and clinical finding of radiation potentiation by metformin, these findings do not provide a definitive conclusion, but rather are hypothesis-generating and require additional validation.
Despite these limitations, we have demonstrated for the first time that disruptive TP53 mutation predicts both LRR following PORT and in vitro response to radiation in HNSCC. Additionally, we have demonstrated that this effect is due to altered senescence and can be overcome using metformin, which appears to have dramatic clinical benefit.
Supplementary Material
Translational Relevance.
The lack of clinically utilized biomarkers in head and neck cancer is a significant problem in moving toward more individualized therapy. TP53, the gene that encodes the protein p53, is by far the most commonly mutated gene found in head and neck cancer. However, this finding has not been clinically useful in guiding therapy. We have used a method of grading TP53 mutation based upon predicted functionality of the resultant protein and shown that a particular class of mutation has a much higher rate of locoregional failure following radiotherapy as well as a much higher rate of in vitro radioresistance by inhibiting radiation induced cellular senescence. Furthermore, we show that these resistant cells can be selectively radiosensitized via the use of the anti-diabetic drug metformin. Finally, we show significant decreases in locoregional failure following radiation as well as improved survival in patients taking metformin. Thus, we provide a common marker for poor outcome in head and neck cancer as well as a novel means by which to target these aggressive tumors.
Acknowledgments
The authors thank Mei Zhou and Samar Jasser for their assistance with basic laboratory work and logistics. The authors also thank Christina Dodge for her assistance with the in vivo experiments.
Funding
This work was partially supported by an American Society of Radiation Oncology Resident Seed Award; the U.T. M.D. Anderson Cancer Center PANTHEON program; National Institutes of Health Specialized Program of Research Excellence Grant P50CA097007; RO1 DE14613 National Institutes of Health; National Research Science Award Institutional Research Training Grant T32CA60374; and National Institutes of Health Program Project Grant CA06294 and Cancer Center Support Grant CA016672.
Glossary
- ROS
reactive oxygen species
- PORT
postoperative radiation therapy
- LRR
locoregional recurrence
- HNSCC
head and neck squamous cell carcinoma
- NAC
N-acetyl cysteine
Bibliography
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011 Apr;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011 Aug;333(6046):1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsner J, Sørensen SB, Overgaard J. TP53 mutation is related to poor prognosis after radiotherapy, but not surgery, in squamous cell carcinoma of the head and neck. Radiother Oncol. 2001 May;59(2):179–185. doi: 10.1016/s0167-8140(01)00301-2. [DOI] [PubMed] [Google Scholar]
- 4.Koch WM, Brennan JA, Zahurak M, Goodman SN, Westra WH, Schwab D, et al. p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J Natl Cancer Inst. 1996;88(21):1580–6. doi: 10.1093/jnci/88.21.1580. [DOI] [PubMed] [Google Scholar]
- 5.Thames HD, Petersen C, Petersen S, Nieder C, Baumann M. Immunohistochemically detected p53 mutations in epithelial tumors and results of treatment with chemotherapy and radiotherapy. A treatment-specific overview of the clinical data. Strahlenther Onkol. 2002 Aug;178(8):411–421. doi: 10.1007/s00066-002-0923-x. [DOI] [PubMed] [Google Scholar]
- 6.Fallai C, Perrone F, Licitra L, Pilotti S, Locati L, Bossi P, et al. Oropharyngeal squamous cell carcinoma treated with radiotherapy or radiochemotherapy: prognostic role of TP53 and HPV status. Int J Radiat Oncol Biol Phys. 2009 Nov;75(4):1053–1059. doi: 10.1016/j.ijrobp.2008.12.088. [DOI] [PubMed] [Google Scholar]
- 7.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357(25):2552–61. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrone F, Bossi P, Cortelazzi B, Locati L, Quattrone P, Pierotti MA, et al. TP53 mutations and pathologic complete response to neoadjuvant cisplatin and fluorouracil chemotherapy in resected oral cavity squamous cell carcinoma. J Clin Oncol. 2010 Feb;28(5):761–766. doi: 10.1200/JCO.2009.22.4170. [DOI] [PubMed] [Google Scholar]
- 9.Erber R, Conradt C, Homann N, Enders C, Finckh M, Dietz A, et al. TP53 DNA contact mutations are selectively associated with allelic loss and have a strong clinical impact in head and neck cancer. Oncogene. 1998 Apr;16(13):1671–1679. doi: 10.1038/sj.onc.1201690. [DOI] [PubMed] [Google Scholar]
- 10.Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Russell J, et al. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol. 2008 Apr;84(4):253–264. doi: 10.1080/09553000801953342. [DOI] [PubMed] [Google Scholar]
- 11.Cuddihy AR, Bristow RG. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004 Dec;23(3–4):237–257. doi: 10.1023/B:CANC.0000031764.81141.e4. [DOI] [PubMed] [Google Scholar]
- 12.Gupta N, Vij R, Haas-Kogan DA, Israel MA, Deen DF, Morgan WF. Cytogenetic damage and the radiation-induced G1-phase checkpoint. Radiat Res. 1996 Mar;145(3):289–298. [PubMed] [Google Scholar]
- 13.Kawashima K, Mihara K, Usuki H, Shimizu N, Namba M. Transfected mutant p53 gene increases X-ray-induced cell killing and mutation in human fibroblasts immortalized with 4-nitroquinoline 1-oxide but does not induce neoplastic transformation of the cells. Int J Cancer. 1995 Mar;61(1):76–79. doi: 10.1002/ijc.2910610113. [DOI] [PubMed] [Google Scholar]
- 14.Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994 Oct;79(2):329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 15.Pardo FS, Su M, Borek C, Preffer F, Dombkowski D, Gerweck L, et al. Transfection of rat embryo cells with mutant p53 increases the intrinsic radiation resistance. Radiat Res. 1994 Nov;140(2):180–185. [PubMed] [Google Scholar]
- 16.Biard DS, Martin M, Rhun YL, Duthu A, Lefaix JL, May E, et al. Concomitant p53 gene mutation and increased radiosensitivity in rat lung embryo epithelial cells during neoplastic development. Cancer Res. 1994 Jul;54(13):3361–3364. [PubMed] [Google Scholar]
- 17.Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010 Aug;31(4):363–372. doi: 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 18.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005 Mar;5(3):231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 19.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008 Jul;15(7):1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 20.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001 Oct;4(5):303–313. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 21.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003 Feb;3(2):117–129. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 22.Castedo M, Perfettini J-L, Roumier T, Valent A, Raslova H, Yakushijin K, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004 May;23(25):4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- 23.Ianzini F, Bertoldo A, Kosmacek EA, Phillips SL, Mackey MA. Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. 2006;6:11. doi: 10.1186/1475-2867-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merritt AJ, Allen TD, Potten CS, Hickman JA. Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene. 1997 Jun;14(23):2759–2766. doi: 10.1038/sj.onc.1201126. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007 Mar;6(5):595–605. doi: 10.4161/cc.6.5.3901. [DOI] [PubMed] [Google Scholar]
- 26.Yigitbasi OG, Younes MN, Doan D, Jasser SA, Schiff BA, Bucana CD, et al. Tumor cell and endothelial cell therapy of oral cancer by dual tyrosine kinase receptor blockade. Cancer Res. 2004 Nov;64(21):7977–7984. doi: 10.1158/0008-5472.CAN-04-1477. [DOI] [PubMed] [Google Scholar]
- 27.Sandulache VC, Skinner HD, Ow TJ, Zhang A, Xia X, Luchak JM, et al. Individualizing anti-metabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer. 2011 Jun 20; doi: 10.1002/cncr.26321. e-Pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 29.Rago R, Mitchen J, Wilding G. DNA fluorometric assay in 96-well tissue culture plates using Hoechst 33258 after cell lysis by freezing in distilled water. Anal Biochem. 1990 Nov;191(1):31–34. doi: 10.1016/0003-2697(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 30.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993 Nov;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 31.Sano D, Matsumoto F, Valdecanas DR, Zhao M, Molkentine DP, Takahashi Y, et al. Vandetanib restores head and neck squamous cell carcinoma cells’ sensitivity to Cisplatin and radiation in vivo and in vitro. Clin Cancer Res. 2011 Apr;17(7):1815–1827. doi: 10.1158/1078-0432.CCR-10-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verheij M, Bartelink H. Radiation-induced apoptosis. Cell Tissue Res. 2000 Jul;301(1):133–142. doi: 10.1007/s004410000188. [DOI] [PubMed] [Google Scholar]
- 33.Catalano A, Rodilossi S, Caprari P, Coppola V, Procopio A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J. 2005 Jan;24(1):170–179. doi: 10.1038/sj.emboj.7600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macip S, Igarashi M, Fang L, Chen A, Pan Z-Q, Lee SW, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002 May;21(9):2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue T, Kato K, Kato H, Asanoma K, Kuboyama A, Ueoka Y, et al. Level of reactive oxygen species induced by p21Waf1/CIP1 is critical for the determination of cell fate. Cancer Sci. 2009 Jul;100(7):1275–1283. doi: 10.1111/j.1349-7006.2009.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anedda A, Rial E, González-Barroso MM. Metformin induces oxidative stress in white adipocytes and raises uncoupling protein 2 levels. J Endocrinol. 2008 Oct;199(1):33–40. doi: 10.1677/JOE-08-0278. [DOI] [PubMed] [Google Scholar]
- 38.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007 Jul;67(14):6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 39.Goodwin WJ. Salvage Surgery for Patients With Recurrent Squamous Cell Carcinoma of the Upper Aerodigestive Tract: When Do the Ends Justify the Means? The Laryngoscope. 2000 Mar;110(S93):1–18. doi: 10.1097/00005537-200003001-00001. [DOI] [PubMed] [Google Scholar]
- 40.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005 Mar;5(3):231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 41.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-Induced Senescence in Cancer. J Natl Cancer Inst. 2010 Oct;102(20):1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattera L, Courilleau C, Legube G, Ueda T, Fukunaga R, Chevillard-Briet M, et al. The E1A-Associated p400 Protein Modulates Cell Fate Decisions by the Regulation of ROS Homeostasis. PLoS Genet. 2010 Jun;6(6) doi: 10.1371/journal.pgen.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003 Nov;102(10):3765–3774. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- 44.Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci USA. 2009 Aug;106(34):14373–14378. doi: 10.1073/pnas.0903284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato S, Han S-Y, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA. 2003 Jul;100(14):8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jordan JJ, Inga A, Conway K, Edmiston S, Carey LA, Wu L, et al. Altered-function p53 missense mutations identified in breast cancers can have subtle effects on transactivation. Mol Cancer Res. 2010 May;8(5):701–716. doi: 10.1158/1541-7786.MCR-09-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dearth LR, Qian H, Wang T, Baroni TE, Zeng J, Chen SW, et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2006 Jul;28(2):289–298. doi: 10.1093/carcin/bgl132. [DOI] [PubMed] [Google Scholar]
- 48.Jung Y-S, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010 Jul;22(7):1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D, et al. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent inhibition of sirtuin 1. J Biol Chem. 2011 May 27;286(21):19100–8. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell. 2011 May;145(4):571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009 Jul;27(20):3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz J-C, et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010 Sep;78(1):221–229. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Menendez JA, Cufí S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressant Metformin: less is more. Aging (Albany NY) 2011 Apr;3(4):348–362. doi: 10.18632/aging.100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.