

Abstract
Redox active tryptophans are important in biological electron transfer and redox biochemistry. Proteins can tune the electron transfer kinetics and redox potentials of tryptophan via control of the protonation state and the hydrogen bond strength. We examine the local environment of two neutral tryptophan radicals (Trp108 on the solvent-exposed surface, Trp48 buried in the hydrophobic core) in two azurin variants. Ultra-high-field EPR spectroscopy at 700 GHz and 25 T allows complete resolution of all principal components of the g tensors of the two radicals and reveals significant differences in the g tensor anisotropies. The spectra together with 2H ENDOR spectra and supporting density-functional theory calculations show that the g tensor anisotropy is directly diagnostic of the presence and the absence as well as the strength of a hydrogen bond to the indole nitrogen. The approach is a powerful one for identifying and characterizing hydrogen bonds that are critical in the regulation of tryptophan-assisted electron transfer and tryptophan-mediated redox chemistry in proteins.
Redox-active tryptophans play important roles in proteins. They serve as relays in long-range electron transfer, e.g. in photolyases1 and cryptochromes2, as well as in engineered proteins3–5. In cytochrome c peroxidase and related enzymes, oxidized tryptophans located near the heme active sites provide a strong oxidation equivalent6,7. In some peroxidases with ligninolytic activity8, surface-exposed tryptophans are the active high-potential oxidants that enable degradation of the recalcitrant substrate lignin. This functionality can be engineered into other peroxidases9 by incorporating the tryptophan and its binding pocket.
The pH-dependent redox potential of tryptophan (TrpH) is about 1.0 V at pH 7, about 0.1 V above that of tyrosine10. Upon oxidation of TrpH, the pKa of the hydrogen at the indole nitrogen N1 drops from about 17 in TrpH to about 4 in the cation radical Trp·H+. As a consequence, oxidation in non-acidic environments is accompanied or followed by deprotonation11, resulting in the neutral radical Trp·. Proteins can adjust the reactivity of the Trp radical by controlling this deprotonation. Finer control over the thermodynamic and kinetic behavior of TrpH oxidation, which is especially important in electron transfer, can be accomplished by providing hydrogen bonding partners to N1 and varying the available H-bonds strength.
Unlike in tyrosyl radicals4,12–16, hydrogen bonds are difficult to observe directly in oxidized Trp17,18, and consequently little is known about their impact on Trp reactivity. Here, we show that the H-bond environment can be directly identified via the magnetic structure of the Trp radicals. We find that the anisotropy of the g tensors of the Trp radicals, resolved using ultra-high-field EPR, reveals the presence/absence as well as the strength of an H-bond to the indole nitrogen.
For this study, model tryptophan radicals photogenerated in two different variants of the copper protein azurin were used18. In a tyrosine-depleted azurin mutant (AzC), direct photoexcitation of the native Trp48 in the hydrophobic core of the protein induces intra-protein electron transfer over 10 Å to Cu(II) combined with deprotonation of the indole nitrogen and results in a Trp• neutral radical18. In another modified azurin (ReAzS), the surface-exposed non-native Trp108 is situated in a hydrophilic environment and can be oxidized via a Re(I) phototrigger attached to His10718,19. After deprotonation, a Trp· neutral radical is generated. The two radicals are located in different environments, which affect their spectroscopic properties.
Figure 1 shows the X-band EPR spectra of the two Trp radicals. Both saturate easily, and lower temperatures or higher microwave power levels lead to broadening (see SI). The spectra are rich in hyperfine structure and can be simulated as neutral radicals Trp·. The hyperfine parameters compare well with those from a previous report18 and those of Trp· in RNR mutants17,20,21 and versatile peroxidases22–24. The radicals are not cationic Trp·H+, as observed in cytochrome c peroxidase6 and in acidic aqueous solution25,26, since the spectra show only minuscule changes upon 1H/2H exchange, and the 1H ENDOR spectra lack peaks due to a proton at N1 (see SI). According to the hyperfine couplings, the spin density is mostly concentrated on C3 (about 0.5), N1 (about 0.3), C5 and C7. The difference between the two spectra is largely due to a 10° difference in the dihedral angle χ2,1 (Cα-Cβ-C3-C2). The spectra are slightly asymmetric due to unresolved small g tensor anisotropy corresponding to about 0.2 mT.
Figure 1.
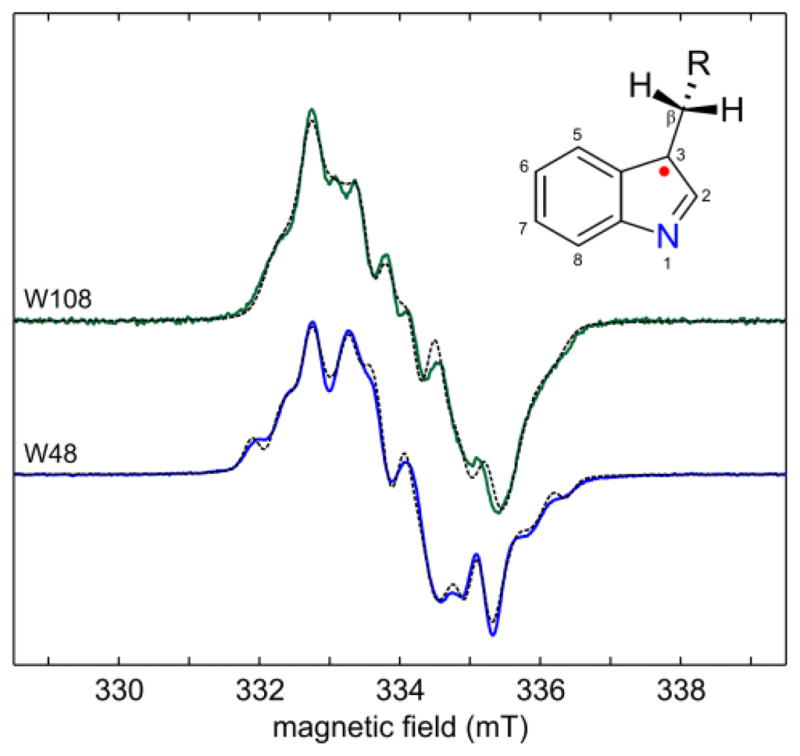
X-band EPR spectra of Trp radicals in azurin mutants in 1H2O, 20% glycerol. (top) Trp108 in ReAzS, (bottom) Trp48 in AzC. Recorded at 9.38 GHz, 200 K, 2 μW. Solid: experimental spectra, dashed: simulations (see SI for details).
The g tensor anisotropy can be resolved using high-field EPR. For tyrosyl radicals with their larger g anisotropy, 95 GHz/3.3 T is sufficient27, but Trp radical g tensors are less anisotropic and are only partially resolved even at 285 GHz/10.2 T9,19,28. The separation of the features corresponding to the three g principal values is complete only at ultra-high fields and frequencies (25 T, 700 GHz)29–31, as shown in Fig. 2. The values, calibrated against an atomic hydrogen standard32, are (gx, gy, gz) = (2.00346, 2.00264, 2.00216) for ReAzS-Trp108 and (2.00361, 2.00270, 2.00215) for AzC-Trp48. The Trp48 g tensor is similar to the earlier one derived from a partially resolved 285 GHz spectrum19. The high-resolution spectra shown in Fig. 2 are measured at the highest field and frequency yet achieved for organic radicals. The hyperfine structure is lost due to magnetic field inhomogeneity.
Figure 2.

Ultra-high-field EPR spectra of Trp radicals in azurin mutants in 1H2O, 20% glycerol. (top) Trp108 in ReAzS, (bottom) Trp48 in AzC. Recorded at 687–695 GHz, 24.8 T, 5 K. Solid: average of experimental spectra, dashed: simulations. The absolute error for the g values is 0.00010, and the error of differences such as gx − gz is 0.00003.
What causes the difference in the g tensors? DFT indicates that the dihedral angle χ2,1 can affect the g tensor by modulating the spin density at N1 (see SI). However, χ2,1 of the two radicals are similar and this explanation can only account for about a sixth of the observed difference in gx−gz. The other major structural determinant in the microenvironment is the presence or absence of a hydrogen bond to the indole nitrogen. The crystal structure of ReAzS (pdb 1R1C) shows partial presence of a water oxygen atom 2.3 Å from N1 of non-oxidized Trp108. Assuming an N-H distance of 1.0 Å, this implies an (N)H…O distance of 1.3 Å, indicative of a strong H-bond33. Whether this H-bond persists after oxidation is unknown. Its presence in the radical form (as N…HX) in solution was inferred from the frequency of the W17 mode of the radical as measured by resonance Raman spectroscopy18. Also, upon 1H/2H exchange, the X-band EPR spectrum of Trp108 sharpens slightly, and its 1H ENDOR spectrum shows distinctive loss of signal around the 1H Larmor frequency (see SI), both suggesting the presence of an H-bond. However, the aforementioned facts constitute only indirect evidence for an H-bond.
Direct evidence for the H-bond to the Trp108 radical is seen in the 2H ENDOR spectrum of a sample prepared in 2H2O (Fig. 3, top) that shows a signal from a fairly strongly coupled 2H. Simulation gives a 2H quadrupole coupling constant e2qQ/h = 0.16(1) MHz, smaller than in 2H2O ice (0.2134 MHz)34, but similar to 2H-bonded semiquinone or tyrosyl radicals35–39. The coupling constant is consistent with the deuteron participating in an H-bond to N1. The asymmetry of the quadrupole tensor, η = 0.10(2), is typical for water (2H2O: 0.112) and suggests water as the H-bond partner. The 2H hyperfine tensor with principal values (aiso − T, aiso − T, aiso + 2T) is approximately axial with an isotropic component aiso = 0.03(2) MHz and a dipolar component T = 0.36(3) MHz. The very small aiso indicates about 0.03*6.511/1420 ≈ 0.1% spin population on the hydrogen, again consistent with a non-covalent H-bond. From T and a typical spin population of about 0.3 on the indole nitrogen, a distance N…H of about 2 Å can be estimated.
Figure 3.
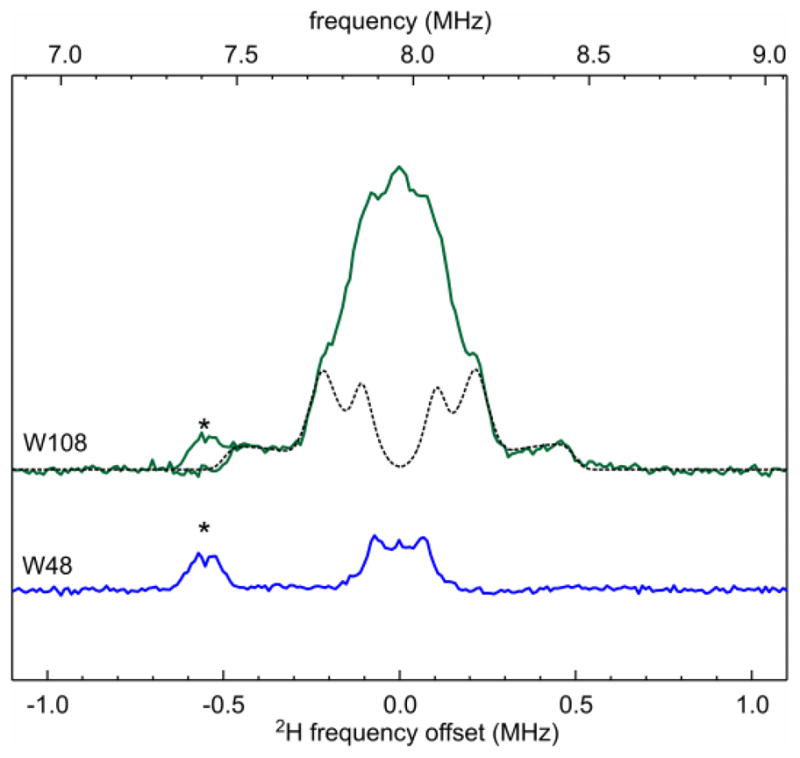
Q-band 2H Mims ENDOR spectra of ReAzS-Trp108 and AzC-Trp48, both in 2H2O, recorded at 20 K and 1.218 T. Microwave pulse lengths 36 ns, rf pulse length 75 μs, τ = 600 ns, repetition time 1 second. Solid: experimental, dashed: simulation. The zero of the frequency offset denotes the 2H Larmor frequency, which is 1/6.511 the 1H Larmor frequency. The asterisk * indicates the 7th harmonic of the 1H spectrum.
In contrast to Trp108, the 2H ENDOR spectrum of Trp48 (Fig. 3, bottom) lacks features that would indicate deuterons close to the Trp radical. The total width of 0.3 MHz of the observed 2H matrix peak implies T < 0.08 MHz, meaning that the closest deuteron is at least 3 Å away from the indole nitrogen. Consequently, Trp48 is not H-bonded. We cannot logically eliminate the possibility that the 1H released by Trp48 after oxidation remains H-bonded to N1 and does not exchange with the solvent in the time before the freeze-quench (ca. 1 min). However, this is unlikely since (a) there are no potential hydrogen bond partners in the immediate vicinity of the Trp48 indole nitrogen, and (b) the shifts in several Raman modes compared to Trp108 corroborate the absence of an H bond18. Collectively, the 2H ENDOR data show that the significant difference in the two g tensors correlates with the absence and presence of an H-bond.
Fig. 4 compares the two measured g tensors to DFT-predicted g tensors of the isolated neutral, the H-bonded neutral and the isolated cation radical of 3-methylindole (see SI)41. The experimentally observed trend is reproduced: In the absence of a hydrogen bond, the g tensor is most anisotropic with the largest span, gx−gz, and the smallest skew, (gx−gy)/(gx−gz). With an H-bond, the span shrinks substantially due to decreasing gx values. DFT predicts that in the protonated cation radical, the span would be smallest and the skew largest. The agreement between theory and experiment indicates that the H-bond is responsible for the decrease in g anisotropy in Trp108 compared to Trp48 and that other local effects (from χ2,1, interactions with the π spin density etc.) on the g tensor are most likely insignificant. Note that in contrast to 2H ENDOR, the g tensor anisotropy can reveal the absence of an H-bond, and not only its presence. Fig. 4 includes the few other (less accurately) known Trp g tensors9,17,20,22,23,28,40, which have gx − gz values between 0.0012 and 0.0014. These values are similar to the span of Trp108 and clearly smaller than Trp48, indicating that all of these Trp radicals are H-bonded, with possible exception of the one in KatG. Among H-bonded radicals, the variation in gx − gz might report on the strength of the H-bond.
Figure 4.

Comparison of known g tensors of Trp radicals. W48, W108: this work; W108 (Miller): earlier W108 value19; RNR-W77 and RNR-W111: ribonucleotide reductase mutants17; BaVP: B. adusta versatile peroxidase22; PeVP: P. eryngii versatile peroxidase23; CcO: cytochrome c oxidase40; KatG: catalase-peroxidase28; CiP: C. cinereus peroxidase mutant9) (see SI for a list). Values for neutral and cation radical of 3-methylindole (MI) calculated by DFT in vacuo are included as well. The dashed line visually indicates the span/skew correlation.
The trend in Fig. 4 can be rationalized based on a per-atom breakdown of the DFT-predicted g tensors42. On each atom, the g shifts Δgx,y,z = gx,y,z − ge depend on the spin population and the spin-orbit coupling and originate from excitations of electrons between filled and half-filled orbitals perpendicular to the magnetic field axis17,27. In the isolated neutral radical, the gx shift Δgx = gx − ge is dominated by the contribution from N1 (80%), with a small contribution (10%) from C3, the other atom with large spin population. Upon hydrogen bonding, the N1 contribution to Δgx decreases, whereas the C3 contribution remains unchanged. H-bonding lowers the energy of the non-bonding MO that contains the inplane lone pair orbital on N1, resulting in an increased energy gap to the SOMO and thus to a decreased g shift along a direction, denoted x, perpendicular to both the N1 lone pair orbital and the SOMO. The computed N1 spin populations do not vary significantly with H-bonding, although the experimental N1 hyperfine couplings indicate that the N1 spin density in Trp108 is lower than in Trp48. The value of Δgy is less sensitive to H-bonding. It is a sum over several atomic contributions (from N1, C2, C3, C5, C7) with similar magnitudes but different signs. Upon variation of the H-bonding, these contributions all undergo small changes mostly due to shifts in the spin density distribution, giving in toto only a small change in Δgy. The deviations of gz from ge are negligibly small. The g tensor anisotropies of all Trp radicals are much smaller than those of tyrosyl radicals (TyrO·), which have gx − gz = 0.004 to 0.007, depending on the presence of H-bonds to the phenoxy oxygen16,27. This is a consequence of the smaller charge, and spin-orbit coupling, of the nitrogen nucleus in Trp· compared to oxygen in TyrO·.
To summarize: (1) By employing EPR at ultra-high field and frequency, we could completely resolve the very small g anisotropies of two model Trp radicals. The two g tensors differ substantially in their anisotropies. (2) 2H ENDOR shows the presence of a hydrogen bond to Trp108, but none to Trp48. (3) The experimental findings, in tandem with DFT calculations, established a clear correlation between the g tensor and the H-bond properties of Trp radicals: smaller g anisotropy indicates an H-bond via the indole nitrogen, whereas larger g anisotropy indicates the absence of an H-bond. (4) We expect this correlation to be helpful in identifying H-bonds and in studying their role in the regulation of tryptophan-assisted electron transfer in proteins and tryptophan-mediated redox chemistry, which are both crucial in a variety of natural processes such as DNA repair and the biodegradation of lignin. A clear understanding of H-bonding to tryptophans is also essential for deploying them as components in engineered proteins and enzymes.
Supplementary Material
Acknowledgments
This work was supported by the NSF (CHE-0911766, J.E.K.), NIH (GM-73789, R.D.B.) and DOE (DE-FG02-09ER16117, R.D.B.). A portion of this work was performed at the National High Magnetic Field Laboratory, which is funded by the NSF (DMR-0654118), the State of Florida, and the U.S. Department of Energy. H.S.S. was supported by a NSF Graduate Research Fellowship. We thank Dr. Stephen Hill for the microwave frequency measurements and Dr. Brian Leigh for the synthesis of the Re compound.
Footnotes
Supporting Information. Materials and methods; EPR power saturation and room temperature spectra; EPR and 1H ENDOR after 1H/2H exchange; EPR simulation details; published Trp g tensors; details on DFT prediction of g tensors; spin density distribution. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brettel K, Byrdin M. Curr Opin Struct Biol. 2010;20:693. doi: 10.1016/j.sbi.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Langenbacher T, Immeln D, Dick B, Kottke TJ. Am Chem Soc. 2009;131:14274. doi: 10.1021/ja901628y. [DOI] [PubMed] [Google Scholar]
- 3.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Jr, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 4.Yeh HC, Gerfen GJ, Wang JS, Tsai AL, Wang LH. Biochemistry. 2009;48:917. doi: 10.1021/bi801382v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray HB, Winkler JR. Biochim Biophys Acta. 2010;1797:1563. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Huyett JE, Doan PE, Gurbiel R, Houseman ALP, Sivaraja M, Goddin DB, Hoffman BMJ. Am Chem Soc. 1995;117:9033. [Google Scholar]
- 7.Jasion VS, Polanco JA, Meharenna YT, Li H, Poulos TL. J Biol Chem. 2011;286:24608. doi: 10.1074/jbc.M111.230524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammel KE, Cullen D. Curr Opin Plant Biol. 2008;11:349. doi: 10.1016/j.pbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Smith AT, Doyle WA, Dorlet P, Ivancich A. Proc Nat Acad Sci USA. 2009;106:16084. doi: 10.1073/pnas.0904535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harriman A. J Phys Chem. 1987;91:6102. [Google Scholar]
- 11.Zhang MT, Hammarström L. J Am Chem Soc. 2011;133:8806. doi: 10.1021/ja201536b. [DOI] [PubMed] [Google Scholar]
- 12.Engström M, Himo F, Gräslund A, Minaev B, Vahtras O, Ågren H. J Phys Chem A. 2000;104:5149. [Google Scholar]
- 13.Faller P, Coussias C, Rutherford AW, Un S. Proc Nat Acad Sci USA. 2003;100:8732. doi: 10.1073/pnas.1530926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svistunenko DA, Cooper CE. Biophys J. 2004;87:582. doi: 10.1529/biophysj.104.041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka H, Shen JR, Kawamori A, Nishiyama K, Ohba Y, Yamauchi S. J Am Chem Soc. 2011;133:4655. doi: 10.1021/ja2000566. [DOI] [PubMed] [Google Scholar]
- 16.Bennati M, Stubbe J, Griffin RG. Appl Magn Reson. 2001;21:389. [Google Scholar]
- 17.Bleifuss G, Kolberg M, Pötsch S, Hofbauer W, Bittl R, Lubitz W, Gräslund A, Lassmann G, Lendzian F. Biochemistry. 2001;40:15362. doi: 10.1021/bi010707d. [DOI] [PubMed] [Google Scholar]
- 18.Shafaat HS, Leigh BS, Tauber MJ, Kim JE. J Am Chem Soc. 2010;132:9030. doi: 10.1021/ja101322g. [DOI] [PubMed] [Google Scholar]
- 19.Miller JE, Grdinaru C, Crane BR, Di Bilio AJ, Wehbi WA, Un S, Winkler JR, Gray HB. J Am Chem Soc. 2003;125:14220. doi: 10.1021/ja037203i. [DOI] [PubMed] [Google Scholar]
- 20.Lendzian F, Sahlin M, MacMillan F, Bittl R, Fiege R, Pötsch S, Sjöberg BM, Gräslund A, Lubitz W, Lassmann G. J Am Chem Soc. 1996;118:8111. [Google Scholar]
- 21.Pötsch S, Lendzian F, Ingemarson R, Hörnberg A, Thelander L, Lubitz W, Lassmann G, Gräslund A. J Biol Chem. 1999;274:17696. doi: 10.1074/jbc.274.25.17696. [DOI] [PubMed] [Google Scholar]
- 22.Pogni R, Baratto MC, Giansanti S, Teutloff C, Verdin J, Valderrama B, Lendzian F, Lubitz W, Vazquez-Duhalt R, Basosi R. Biochemistry. 2005;44:4267. doi: 10.1021/bi047474l. [DOI] [PubMed] [Google Scholar]
- 23.Pogni R, Baratto MC, Teutloff C, Giansanti S, Ruiz-Dueñas FJ, Choinowski T, Piontek K, Martínez AT, Lendzian F, Basosi R. J Biol Chem. 2006;281:9517. doi: 10.1074/jbc.M510424200. [DOI] [PubMed] [Google Scholar]
- 24.Pogni R, Teutloff C, Lendzian F, Basosi R. Appl Magn Reson. 2007;31:509. [Google Scholar]
- 25.Kiryutin AS, Morozova OB, Kuhn LT, Yurkovskaya AV, Hore PJ. J Phys Chem B. 2007;111:11221. doi: 10.1021/jp073385h. [DOI] [PubMed] [Google Scholar]
- 26.Connor HD, Sturgeon BE, Mottley C, Sipe HJ, Jr, Mason RP. J Am Chem Soc. 2008;130:6381. doi: 10.1021/ja0780277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoll S. In: In Electron Paramagnetic Resonance. Gilbert BC, Chechik V, Murphy DM, editors. Vol. 22. Royal Society of Chemistry; Cambridge: 2011. p. 107. [Google Scholar]
- 28.Colin J, Wiseman B, Switala J, Loewen PC, Ivancich A. J Am Chem Soc. 2009;131:8557. doi: 10.1021/ja901402v. [DOI] [PubMed] [Google Scholar]
- 29.Konovalova TA, Krzystek J, Bratt PJ, van Tol J, Brunel LC, Kispert LD. J Phys Chem B. 1999;103:5782. [Google Scholar]
- 30.Bratt PJ, Ringus E, Hassan AK, van Tol J, Maniero AL, Brunel LC, Rohrer M, Bubenzer-Hange C, Scheer H, Angerhofer A. J Phys Chem B. 1999;103:10973. [Google Scholar]
- 31.Bratt PJ, Heathcote P, Hassan A, van Tol J, Brunel LC, Schrier J, Angerhofer A. Chem Phys. 2003;294:277. [Google Scholar]
- 32.Stoll S, Ozarowski A, Britt RD, Angerhofer A. J Magn Reson. 2010;207:158. doi: 10.1016/j.jmr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steiner T. Angew Chem Int Ed. 2002;41:48. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 34.Edmonds DT, Mackay AL. J Magn Reson. 1975;20:515. [Google Scholar]
- 35.van Dam PJ, Willems JP, Schmidt PP, Pötsch S, Barra AL, Hagen WR, Hoffman BM, Andersson KK, Gräslund A. J Am Chem Soc. 1998;120:5080. [Google Scholar]
- 36.Bar G, Bennati M, Nguyen HHT, Ge J, Stubbe J, Griffin RG. J Am Chem Soc. 2001;123:3569. doi: 10.1021/ja003108n. [DOI] [PubMed] [Google Scholar]
- 37.Flores M, Isaacson RA, Calvo R, Feher G, Lubitz W. Chem Phys. 2003;294:401. [Google Scholar]
- 38.Flores M, Isaacson R, Abresch E, Calvo R, Lubitz W, Feher G. Biophys J. 2007;92:671. doi: 10.1529/biophysj.106.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kessen S, Teutloff C, Kern J, Zouni A, Bittl R. Chem-PhysChem. 2010;11:1275. doi: 10.1002/cphc.200901019. [DOI] [PubMed] [Google Scholar]
- 40.Wiertz FGM, Richter OMH, Ludwig B, de Vries S. J Biol Chem. 2007;282:31580. doi: 10.1074/jbc.M705520200. [DOI] [PubMed] [Google Scholar]
- 41.Un S. Magn Reson Chem. 2005;43:S229. doi: 10.1002/mrc.1660. [DOI] [PubMed] [Google Scholar]
- 42.Stone A. J Proc Roy Soc London. 1963;271:424. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.